Toward highly efficient NLO chromophores: Synthesis and properties of heterocycle-based electronically gradient dipolar NLO chromophores†
Xiaohua
Ma
a,
Fei
Ma
b,
Zhenhua
Zhao
a,
Naiheng
Song
*a and
Jianping
Zhang
*b
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, Department of Polymer Science and Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, China. E-mail: nsong@pku.edu.cn; Fax: +86-10-62755644; Tel: +86-10-62755644
bDepartment of Chemistry, Renmin University of China, Beijing, 100872, People's Republic of China. E-mail: jpzhang@chem.ruc.edu.cn; Tel: +86-10-62516604
First published on 2nd February 2010
Abstract
To realize organic nonlinear optical (NLO) chromophores with optimized ground-state polarization and very large molecular optical nonlinearities, a novel series of heterocycle-based electronically gradient dipolar chromophores were designed and synthesized. These chromophores are featured by their same strong electron acceptor (i.e., 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran, TCF) and the same length of π-conjugation, but different electron donors (e.g., dialkylamine and dianisylamine), different (hetero)aromatics with varying electron densities (i.e., pyrrole, thiophene, and benzene) as the auxiliary donor, and electron-poor 1,3-heteroaromatic thiazole with different regiostructures (e.g., either electron-poor C2, “matched”, or electron-rich C5, “un-matched”, is connected to the acceptor) as the auxiliary acceptor, which allows for a systematic fine-tuning of the ground-state polarization. The gradient electronic structures and optical properties of these NLO chromophores were carefully characterized by 1H NMR, CV, UV-vis, and Hyper-Rayleigh scattering experiments. All the NLO chromophores exhibited very large static molecular first hyperpolarizabilities (β0) in the range of 450–960 × 10−30 esu, which showed significant dependence on the gradient electronic structures. Upon using electron-rich heteroaromatic cycle as the auxiliary donor, “matched” thiazole as the auxiliary acceptor, and/or dianisylamine as the electron donor, substantially enhanced β were obtained. Theoretical studies were carried out to understand the structure-property relationships, which showed that multiple states excitations contributed to the β values of this series of NLO chromophores. TGA investigations showed excellent thermal stability for most of the resulting NLO chromophores, with on-set temperatures for thermal decomposition higher than 250 °C. The very large β0 values coupled with the high thermal stability indicates good application potential of this series of NLO chromophores.
Introduction
Organic second-order nonlinear optical (NLO) chromophores are potentially useful in ultrafast electrooptic (EO) applications.1,2 In order to achieve large optical nonlinearities (e.g., as characterized by the large molecular first hyperpolarizability, β), a great research effort has been made to optimize the ground-state polarization of D-π–A type NLO chromophores by using different combinations of electron donor (D), electron acceptor (A), and π–conjugated bridge.3–9 Recently, a few NLO chromophores based on a novel strong electron acceptor (i.e., 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran, TCF) and its modified forms have been successfully developed to exhibit large β values and used to yield NLO polymers with very high EO coefficients (e.g., >300 pm/V).7–9 However, the full potential of NLO chromophores is not yet realized; and it is still highly desirable to develop novel highly efficient NLO chromophores with even enhanced β and excellent complementary properties such as thermal and photochemical stabilities for practical EO device applications.To realize such NLO chromophores, systematic and careful optimization of ground-state polarization is to be carried out.10 However, typical methods for modification of the electronic structures of dipolar NLO chromophores through, for example, using different electron donor and acceptor pairs with a delocalizable π–framework, may not be an efficient approach for the optimization of ground state polarization, due to the limited accessibility to electron donors and acceptors with systematically varying electron donating and accepting strength. To overcome such a problem, an electronically gradient π–bridge can be coupled with strong electron donor and acceptor pairs for a fine-tuning of ground-state polarization. Previous theoretical studies11,12 have shown that, upon introduction of electron-rich heterocycles to the donor end of the π–bridge as auxiliary donor and electron-poor heterocycles to the acceptor end as auxiliary acceptor, the donor and acceptor strength can be effectively enhanced. Thus, by a systematic variation of the electron density of the auxiliary donor and the auxiliary acceptor in the π–bridge as well as the donor and acceptor strength, optimization of the ground-state polarization of NLO chromophore can be expected, which will in turn lead to large β values.
To construct varying electronic gradients of NLO chromophores, heteroaromatic cycles with different electron densities are to be incorporated into the π–bridge. For this purpose, we have recently established an efficient synthetic approach for NLO chromophores containing electron-rich heteroaromatics (such as pyrrole and thiophene) as the auxiliary donor and electron-poor thiazole as the auxiliary acceptor.13 By systematically controlling the donor strength, the electron density of the auxiliary donor, and the regiostructure of the thiazole as the auxiliary acceptor (e.g., either the electron-poor C2 or the electron-rich C5 is connected to the acceptor), a series of NLO chromophores based on tricyanovinyl (TCV) as the electron acceptor and having fine-tuned gradient electronic structures were successfully prepared.13 Significantly enhanced β due to the electronic gradient structure was realized, which can be attributed to an optimized ground-state polarization of the NLO chromophores. However, in comparison with the β (e.g., β0.65eV ≈ 9800 × 10−30 esu) achieved with the so-called “TMC” series of zwitterionic chromophores developed by Marks et al.,14 there is still plenty of room for the improvement of NLO chromophores.
Thus, in order to achieve an advanced understanding of the structure-property relationship and highly efficient NLO chromophores, we have herein prepared a novel series of electronically gradient dipolar NLO chromophores based on the efficient TCF as the electron acceptor. Studies by Jen and Dalton et al.4,7 have shown that the replacement of TCV by TCF as the electron acceptor could lead to a substantial increase in optical nonlinearity. Thus, through careful control of the electronically gradient structure, even enhanced β values can be expected for the resulting NLO chromophores. In previous studies,13,15–17 distinctive effects of the regiostructures of thiazole as the auxiliary acceptor and triarylamine as the electron donor were observed on β values of NLO chromophores. In this study, careful examination of these structural effects was carried out through both experimental measurements and theoretical calculations. As expected, all the resulting TCF-based NLO chromophores show much larger β than previously synthesized TCV-based NLO chromophores. Owing to the electronically gradient structures, the resulting NLO chromophores also exhibit significantly enhanced β in relative to the state-of-the-art “FTC” chromophore developed by Dalton et al.7
Experimental section
Materials
2-Dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran (TCF),18 1-methyl-5-(piperidin-1-yl)-1H-pyrrole-2-carbaldehyde13 and 5-(piperidin-1-yl)thiophene-2-carbaldehyde13 were prepared according to the reported methods. Malononitrile was purchased from Beijing Chemical Reagent company and vacuum distilled before use. N,N-Dimethylformamide (DMF) was distilled over calcium hydride and stored over molecular sieves (pore size 3 Å). Anhydrous tetrahydrofuran, dioxane, and toluene were distilled over sodium prior to use. 3-Hydroxy-3-methylbutan-2-one (Alfa Aesar), potassium t-butyloxide (t-BuOK, Alfa Aesar), n-butyl lithium (Acros Organics), and p-nitroaniline (98%, Sinopharm Chemical Reagent Co., Ltd) were purchased and used as received. All other chemical reagents were purchased from Beijing Chemical Reagent Company and used as received.Instruments
1H NMR and 13C NMR spectra were recorded on a Varian 300 MHz or a Bruker ARX400 spectrometer using deuterated chloroform (CDCl3) or dimethylsulfoxide-d6 (DMSO-d6) as the solvent. Chemical shifts were reported in ppm scale with tetramethylsilane as the internal standard. Infrared (IR) spectra were measured on a Nicolet Magna 750 Fourier transform infrared spectrometer. UV-vis spectra were recorded with a PE spectrometer with the wavelength recorded in the range of 190 nm to 1100 nm. Thermogravimetric analysis (TGA) was performed on a TA TGA-DSC Q600 thermogravimetric analyzer with a heating rate of 20 °C/min. Melting points were measured with a SGW-X-4 microscopic melting point instrument. High and low resolution mass ESI spectroscopy was measured with a Bruker Apex IV FTMS mass spectrometer. Cyclic voltammetry (CV) was performed on a CHI 600C cyclic voltametric analyzer in a solution of tetrabutylammonium perchlorate (0.1 M) in acetonitrile at a scanning rate of 100 mV/s. The working electrode was a glassy carbon disk (diameter 2 mm, freshly polished) for voltammetry. A platinum stick (1 mm thick) was used as the counter-electrode. The reference electrode was AgCl/Ag. All potentials reported here are potentials measured versus the AgCl/Ag electrode. Before each measurement, the cell was deoxygenated with argon for 20 min.Computational method
All calculations were carried out using the Gaussian 03 program package.19 Ground-state geometric structures of the NLO chromophores were optimized by density hybrid function (B3LYP) method with 6–31G as the basis set20,21 without imposing any symmetry constraints. Dipole moments of the NLO chromophores at excited states were calculated using the single-excitation configuration interaction (RCIS) method,22 and transition dipole moments and transition energies were calculated using the time-dependent density functional theory (TD-DFT).23Hyper-Rayleigh Scattering (HRS) measurements
HRS measurements of the NLO chromophores were performed at the excitation wavelength of 1000/1100 nm. A third harmonic wave at 355 nm with pulse duration of 7 ns (repetition rate, 10 Hz) was supplied by a Q-switched Nd3+:YAG laser, and was used to drive an optical parametric oscillator (MOPO-SL/MOPO-PO, Spectra-Physics) to deliver the light pulses at 1000/1100 nm. The laser beam was passed through a Pelin–Broca prism and other steering optics, and focused into the sample cell with a lens (f = 200 mm). The pulse energy measured in front of the sample cell was 2 mJ/pulse. The HRS signal collected with a camera lens (f/1.4) was further focused via a lens (f = 150 mm) onto the entrance slit of a triplet spectrometer (Trivista, SP2500i, Princeton Instruments/Acton), and was detected by an intensified CCD detector (ICCD PI-MAX, Princeton Instruments/Acton) operated with a gate width of 10 ns. For each HRS measurement, the sample was exposed to 1000 laser shots and the accumulated harmonic signal at 500/550 nm was extracted via spectral deconvolution from the background fluorescence induced by two-photon absorption.Results and discussion
Synthesis
The synthesis and the chemical structures of the NLO chromophores are shown in Scheme 1. All the NLO chromophores were designed to have the same strong electron acceptor (i.e., TCF) and the same length of π-conjugation, but have different electron donors, auxiliary electron donors and auxiliary electron acceptors. By systematically varying the donor ability (e.g., dialkylamine and diarylamine), the electron density of the auxiliary donors (i.e., benzene, thiophene, and pyrrole), and the nature and the regiostructure of the auxiliary acceptors (e.g., thiophene and thiazole with “matched” and “un-matched” configurations), a series of NLO chromophores with fine-tuned electronically gradient structures were obtained. Herein, the “matched” and “un-matched” configurations of 1,3-heteroaromatic thiazole are defined by the relative orientations between dipoles of thiazole and the NLO chromophore as a whole. For “matched” configuration, the electron-rich C5 of thiazole is at the donor end and the electron-poor C2 at the acceptor end so that the dipole of thiazole is in the same direction as that of the NLO chromophore; for “un-matched” configuration, the opposite is true. To further understand the relationship between the regiostructure of thiazole and optical nonlinearities, a NLO chromophore (i.e., T-Ti4-TCF) with a “neutral” connecting configuration of thiazole, i.e., C2 at the acceptor end and C4 at the donor end, where the dipole of thiazole is nearly perpendicular to the dipole of the NLO chromophore, was also prepared for NLO studies. Structurally speaking, this series of NLO chromophores have similarities to the state-of-the-art, the so-called FTC chromophores (e.g., 2-dicyanomethylen-3-cyano-4-{2-[E-(4-N,N-di(2-acetoxyethyl)amino)phenylene-(3,4-dibutyl)thien-5]-E-vinyl}-5,5-dimethyl-2,5-dihydrofuran)7 that were developed by Dalton et al. Thus, for comparison purposes, the dimethylamino analogue of FTC chromophores (i.e., B-T-TCF, Scheme 1) was also prepared in this study.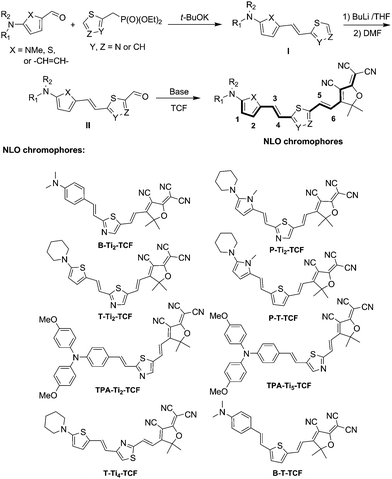 | ||
| Scheme 1 Synthesis and chemical structures of the NLO chromophores | ||
As shown in Scheme 1, the NLO chromophores were prepared through a three-step synthesis, i.e., the Horner–Wadsworth–Emmons (HWE) coupling between amino-substituted aryl aldehydes and heteroaromatic cycle (e.g., thiophene and thiazole)-based HWE reagents at −78 °C in the presence of t-BuOK as a base; the aldehydation of the coupled products (I) through lithiation using n-butyl lithium followed by quenching with DMF; and finally the Knoevenagel condensation reactions between the aldehyde precursors (II) and TCF using triethylamine as the base. The purity and the chemical structures of the key intermediates and the NLO chromophores were demonstrated by a combination of characterization techniques including NMR spectroscopy, IR spectroscopy and high-resolution mass spectroscopy. Since proton NMR spectroscopy showed only large coupling constants (e.g., 3JHH = 15–16 Hz) attributable to trans alkene protons, the resulting NLO chromophores are expected to take exclusively trans alkene configurations.
The NLO chromophores were obtained as blue or dark green powders and showed moderate to good solubility in common organic solvents such as chloroform, dichloromethane, tetrahydrofuran and DMF. In comparison, NLO chromophores containing “un-matched” thiazole as the auxiliary acceptor (e.g., B-Ti2-TCF, T-Ti2-TCF and P-Ti2-TCF) showed poorer solubility (<1 mg/mL) than other chromophores in less polar solvents such as chloroform and tetrahydrofuran. The triarylamine-based NLO chromophores (i.e., TPA-Ti2-TCF and TPA-Ti5-TCF) appeared to have the best solubility in the series. They were readily soluble in almost all the solvents tested including the non-polar solvents such as toluene and dioxane.
Electronic structure analysis
Due to the introduction of electron-rich and electron-poor heteroaromatics to the electron donor and acceptor ends, respectively, as the auxiliary donor and acceptor, the resulting NLO chromophores are expected to exhibit electronically gradient structures that have different ground-state polarizations and charge transfer (CT) properties, which should in turn lead to different nonlinear optical properties.Table 1 summarizes the chemical shifts of protons of the NLO chromophores. It can be noted that the introduction of electron-rich heteroaromatic cycles (e.g., thiophene and pyrrole) to the donor end shifted most protons on the NLO chromophores (e.g., H-1–H-5, see Scheme 1) to higher fields (e.g., T-Ti2-TCF and P-Ti2-TCF vs. B-Ti2-TCF), particularly for protons at the donor end (e.g., H-1 and H-2), indicating an increased electron density as well as an increased electronic gradient from the donor to the acceptor for the NLO chromophores. In comparison, the pyrrole-based NLO chromophore P-Ti2-TCF appear to have higher electron density than its thiophene analogue (i.e., T-Ti2-TCF), as shown by the higher fields of protons H-1 and H-2. In contrast, the introduction of the electron-poor thiazole with “un-matched” configuration to the acceptor end (e.g., B-Ti2-TCF vs. B-T-TCF and P-Ti2-TCF vs. P-T-TCF) shifted all protons to lower fields, particularly for protons close to the thiazole ring (e.g., H-4 and H-5), indicating a decreased electron density. The “matched” thiazole has a different effect from the “un-matched” thiazole on the electronic structures of the NLO chromophores, as shown by the different chemicals shifts between TPA-Ti5-TCF and TPA-Ti2-TCF. The “neutral” thiazole-based T-Ti4-TCF has a higher electron density at the donor end than T-Ti2-TCF, which can be attributed to the unfavorable resonance structure of the T-Ti4-TCF that prohibits an efficient charge transfer from the donor to the acceptor. The dialkylamino and dianisylamino donors have also different effect on the electronic structures of the NLO chromophores, but mainly at the donor end. For example, the differential chemical shifts between protons H-1 and H-2 is much smaller for TPA-Ti2-TCF (Δδ = 0.26 ppm) than B-Ti2-TCF (Δδ = 0.77 ppm), suggesting a better electron delocalization within the phenyl ring for TPA-Ti2-TCF.
| NLO chromophore | 1H NMR (ppm) a | |||||
|---|---|---|---|---|---|---|
| H-1 | H-2 | H-3 | H-4 | H-5 | H-6 | |
| a Proton NMR measurements carried out in deuterated chloroform at 400 MHz. The numbering scheme of protons is shown in Scheme 1. b Not identifiable due to overlapping, δ in the range of 6.87–6.93 ppm. | ||||||
| B-Ti2-TCF | 6.72 | 7.50 | 6.57 | 7.57 | 7.11 | 7.77 |
| T-Ti2-TCF | 6.02 | 7.09 | 6.47 | 7.65 | 6.62 | 7.76 |
| P-Ti2-TCF | 5.83 | 6.77 | 6.47 | 7.53 | 6.88 | 7.74 |
| P-T-TCF | 5.80 | 6.61 | 6.55 | 6.96 | 6.87 | 7.70 |
| T-Ti4-TCF | 5.96 | 6.89 | 6.56 | 7.57 | 7.39 | 7.68 |
| TPA-Ti2-TCF | —b | 7.38 | 6.58 | 7.54 | 7.12 | 7.78 |
| TPA-Ti5-TCF | 7.03 | 7.30 | 6.86 | 7.10 | 7.19 | 7.67 |
| B-T-TCF | 6.70 | 7.41 | 6.56 | 7.10 | 7.01 | 7.77 |
The redox properties of the NLO chromophores were investigated by cyclic voltammetry in acetonitrile containing tetrabutylammonium perchlorate (0.1 M) as the supporting electrolyte. All the NLO chromophores showed irreversible redox processes except for the triarylamine-based NLO chromophores (i.e., TPA-Ti2-TCF and TPA-Ti5-TCF) that exhibited reversible oxidation-reduction properties in positive scans. Table 2 lists the cathodic and anodic peak potentials for the reduction and oxidation processes, respectively. In comparison, the introduction of “un-matched” thiazole as the auxiliary acceptor (e.g., B-Ti2-TCF vs. B-T-TCF and P-Ti2-TCF vs. P-T-TCF) could significantly increase the oxidation potentials while decreasing the reduction potentials; the introduction of electron-rich thiophene or pyrrole as the auxiliary donor led to decreases in the oxidation potential (e.g., T-Ti2-TCF and P-Ti2-TCF vs. B-Ti2-TCF). These agree with the previous 1H NMR analysis that showed increased electron densities for thiophene or pyrrole-containing NLO chromophores and decreased electron densities for thiazole-containing NLO chromophores. In comparison, the “matched” thiazole-based NLO chromophore (i.e., TPA-Ti5-TCF) exhibited a lower reduction potential but a similar oxidation potential relative to its “un-matched” isomer (i.e., TPA-Ti2-TCF), indicating a smaller bandgap for the former. The bandgap energies of the NLO chromophores were derived from the redox potentials and displayed in Table 2. All benzene-based NLO chromophores (e.g., B-Ti2-TCF, B-T-TCF, TPA-Ti2-TCF, and TPA-Ti5-TCF) showed relatively large bandgaps in the range of 1.28–1.42 eV. The introduction of the auxiliary donors and auxiliary acceptors affects the bandgaps to a great extent. As the increase of electron density of the auxiliary donor (e.g., from benzene to thiophene to pyrrole), a decrease in the bandgap was observed in the order of B-Ti2-TCF, T-Ti2-TCF, and P-Ti2-TCF. On the other hand, the introduction of electron-poor thiazole as the auxiliary acceptor resulted in an increase of the bandgap energy (e.g., B-Ti2-TCF vs. B-T-TCF and P-Ti2-TCF vs. P-T-TCF).
| NLO chromophore | λ max (nm)a | Δ λb (nm) | ε (×10 4) | E red (V) | E ox (V) | E ge (eV) | BLAg (Å) | |
|---|---|---|---|---|---|---|---|---|
| CHCl3 | dioxane | |||||||
| a Maximal charge-transfer absorption wavelength. b Solvatochromism from dioxane to chloroform. c Extinction coefficient in chloroform. d Cathodic and anodic peak potentials vs. Ag/Ag+ reference electrode for irreversible reduction and oxidation processes; scan rate 0.1 V s−1. e Half potential for reversible oxidation processes. f Bandgap in eV. g Bond-length-alteration calculated based on B3LYP/6-31g optimized geometric structures. | ||||||||
| B-Ti2-TCF | 619 | 572 | 47 | 4.64 | −0.47 | 0.95 | 1.42 | 0.028 |
| T-Ti2-TCF | 696 | 621 | 75 | 4.70 | −0.47 | 0.73 | 1.20 | 0.030 |
| P-Ti2-TCF | 684 | 628 | 56 | 5.05 | −0.39 | 0.58 | 0.97 | 0.027 |
| P-T-TCF | 725 | 660 | 65 | 6.53 | −0.42 | 0.49 | 0.91 | 0.025 |
| T-Ti4-TCF | 604 | 565 | 39 | 0.28 | −0.50 | 0.43 | 0.93 | 0.052 |
| TPA-Ti2-TCF | 638 | 593 | 45 | 4.80 | −0.48 | 0.84 (0.78e) | 1.32 | 0.033 |
| TPA-Ti5-TCF | 671 | 612 | 59 | 6.45 | −0.35 | 0.83 (0.77e) | 1.28 | 0.035 |
| B-T-TCF | 650 | 604 | 46 | 4.96 | −0.50 | 0.86 | 1.36 | 0.026 |
The UV-vis spectra of the NLO chromophores have been obtained in chloroform and are displayed on Fig. 1. All the NLO chromophores showed a strong and broad charge-transfer (CT) absorption band peak in the spectral range of 620–725 nm except for T-Ti4-TCF that displays a much less intense absorption band at around 614 nm. From Table 2, it can be noted that the maximal absorption wavelength (λmax) for this CT band blue shifted (>30 nm) for “un-matched” thiazole-based NLO chromophores (e.g., B-Ti2-TCF vs. B-T-TCF and P-Ti2-TCF vs. P-T-TCF) and red shifted significantly (>70 nm in chloroform) for chromophores containing electron-rich heteroaromatics (i.e., thiophene or pyrrole) as the auxiliary donor (e.g., T-Ti2-TCF and P-Ti2-TCF vs. B-Ti2-TCF). These agree with the bandgap calculations based on the redox properties (Table 2), which show increased bandgaps for “un-matched” thiazole-based NLO chromophores and decreased bandgaps for thiophene and pyrrole-based NLO chromophores. The thiophene-based T-Ti2-TCF exhibits a larger λmax than the pyrrole-based P-Ti2-TCF, due probably to the twisted structure of P-Ti2-TCF resulted from the steric hindrance of the 1-methyl group on the pyrrole ring. The triarylamine-based NLO chromophore TPA-Ti2-TCF exhibited a red-shifted λmax (19 nm) relative to its N,N-dimethylaniline-based analogue (i.e., B-Ti2-TCF), suggesting a stronger π-donor strength of N,N-dianisylaniline than N,N-dimethylaniline. The regiostructure of thiazole as the auxiliary acceptor also has a significant effect on the electronic absorption property of NLO chromophores. For example, the “matched” thiazole-based TPA-Ti5-TCF exhibited a distinctively red-shifted λmax (33 nm) with respect to its “un-matched” isomer TPA-Ti2-TCF.
 | ||
| Fig. 1 Normalized typical UV-vis absorption spectra of the NLO chromophores in chloroform: (a) B-Ti2-TCF; (b) T-Ti2-TCF; (c) P-Ti2-TCF; (d) TPA-Ti2-TCF; (e) TPA-Ti5-TCF; and (f) T-Ti4-TCF. | ||
Noticeably, all the resulting NLO chromophores also exhibited a high-energy CT absorption band at ca. 400–500 nm (Fig. 1). In comparison, triarylamine-based NLO chromophores (i.e., TPA-Ti2-TCF and TPA-Ti5-TCF) exhibited stronger high-energy CT bands than the other NLO chromophores. T-Ti4-TCF showed a strong absorption band at 400 nm but weak CT absorption at 614 nm, which can be attributed to the regiostructure of thiazole, whose resonant structures do not allow for an efficient charge transfer from the electron donor to the TCF acceptor.
To further understand the ground-state electronic structures, the BLA of the resulting NLO chromophores were calculated on geometric structures optimized at the B3LYP/6-31G level. All C–C double bonds outside the aromatic rings were set to be in trans configuration. The differences between the averaged lengths of C–C single and double bonds of the NLO chromophores, as shown by the bold bonds in Scheme 1, were calculated to give the BLA values in the range of 0.025–0.052 Å (Table 2). Among which, T-Ti4-TCF shows the largest BLA of 0.052 Å; all other chromophores exhibited BLA values smaller than 0.04 Å, which was predicted to be the optimal BLA for the maximized β,10 indicating strong CT effect of these NLO chromophores and the large contribution of zwitterionic resonance forms to the ground-state electronic structures. According to the BLA model proposed by Marder et al.,10 for NLO chromophores with BLA smaller than 0.04 Å, larger β can be expected as the BLA increases. However, no clear dependence of BLA on the structures of the NLO chromophores was revealed in this study. NLO chromophores based on different auxiliary donors (e.g., B-Ti2-TCF, T-Ti2-TCF and P-Ti2-TCF) were found to have similar BLAs in the range of 0.027–0.030 Å. Nevertheless, it is interesting to note that triarylamine-based NLO chromophores (i.e., TPA-Ti2-TCF and TPA-Ti5-TCF) have very different BLA values from the other chromophores, e.g., their N,N-dimethylaniline-based analogue B-Ti2-TCF, and are more close to 0.04 Å (i.e., 0.033 Å for TPA-Ti2-TCF and 0.035 Å for TPA-Ti5-TCF), suggesting larger β values for these two chromophores.
Nonlinear optical properties
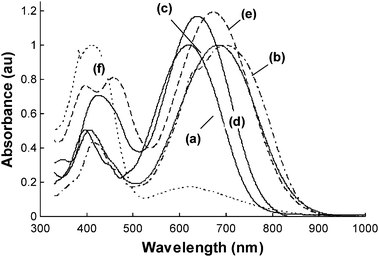
The molecular optical nonlinearities of the resulting NLO chromophores were evaluated by solvatochromism, Hyper-Rayleigh scattering (HRS), and theoretical calculations. Table 2 summarizes the solvatochromism (Δλ) of the NLO chromophores between dioxane and chloroform solutions. All the NLO chromophores exhibited very large positive solvatochromism in the range of 39–75 nm, which are comparable to or larger than that of the FTC analgoue B-T-TCF (Δλ = 46 nm). Since solvatochromism has been positively correlated to the molecular optical nonlinearity,25 such large Δλ values suggest large β values of these NLO chromophores. In comparison, the thiophene and pyrrole-based NLO chromophores (i.e., T-Ti2-TCF and P-Ti2-TCF) showed much larger Δλ (56–75 nm) than the benzene-based analogue B-Ti2-TCF (Δλ = 47 nm). The regiostructures of thiazole affect significantly the Δλ. For example, the “matched” thiazole-based NLO chromophore TPA-Ti5-TCF exhibited much larger Δλ than its “un-matched” isomer TPA-Ti2-TCF; and the “un-matched” thiazole-based T-Ti2-TCF showed much larger Δλ than the “neutral” thiazole-based T-Ti4-TCF.Fig. 2 displays typical UV-vis spectra of P-Ti2-TCF in different solvents. It is interesting to note that as the solvent polarity goes beyond chloroform, the NLO chromophore exhibited a distinctive negative solvatochromism. For example, Δλ from chloroform to acetone is as large as −39 nm. All other NLO chromophores exhibited similar solvatochromic behavior. Such a phenomenon was reported by Davies et al.16 and was ascribed to the back electron transfer from the acceptor side to the donor side in polar solvents. The same explanation should also hold for the present series of NLO chromophores.
 | ||
| Fig. 2 UV-vis spectra of P-Ti2-TCF in different solvents. (a) dioxane, (b) toluene, (c) chloroform, (d) tetrahydrofuran, and (e) acetone. | ||
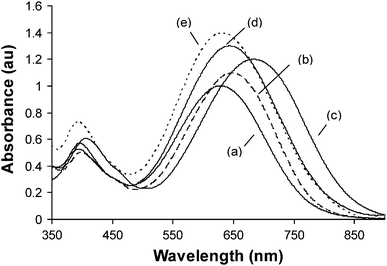
The β values of the resulting NLO chromophores were measured in chloroform by HRS at the excitation wavelengths of 1000 nm and 1100 nm. Because the NLO chromophores exhibited a minimum UV-vis absorption at the second harmoinc wavelengths (i.e., 500 nm and 550 nm), resonance effect on the HRS measurements can be diminished.26Fig. 3 shows the optical set-up for the HRS measurements. A third harmonic wave at 355 nm with pulse duration of 7 ns (repetition rate, 10 Hz) was supplied by a Q-switched Nd3+:YAG laser, and was used to drive an optical parametric oscillator to deliver the light pulses at 1000/1100 nm, which was passed through a Pelin–Broca prism and a series of steering optics and focused into the sample cell with an optical lens. The HRS signal generated was collected by a camera lens, further focused onto the entrance slit of a triplet spectrometer, and then detected by an ICCD with a gate width of 10 ns. For each measurement the sample was exposed to 1000 laser shots to reduce the variation effect of laser pulses.
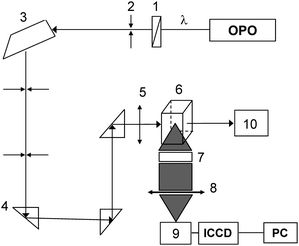 | ||
| Fig. 3 Schematic drawing of the HRS experimental setup. 1, Glan prism; 2, filter; 3, Pelim-Broca prisms; 4, turning prisms; 5, focusing lens, f = 200 mm; 6, sample cell; 7, camera lens (f/1.4); 8, focusing lens, f = 150 mm; 9, monochromator; 10, beam dump. | ||
The HRS experiments were carried out in dilute solutions with concentrations in the range of 10−5–10−4 mol/L. For each NLO chromophore, at least five sample solutions with different concentrations were prepared and measured for the HRS signal. A p-nitroaniline (p-NA) solution in dimethylsulfoxide (DMSO) was used as the external standard. The static β (β0) of p-NA in DMSO was calculated to be 11.5 × 10−30 esu from the known β value (28.8 × 10−30 esu)27 measured at 1064 nm according to the two-level model,28 and was used to yield the β at 1000 nm (34.5 × 10−30 esu) and 1100 nm (26.5 × 10−30 esu). Although two-photon fluorescences (TPF) were observed for all the resulting NLO chromophores, they can be readily removed by spectral deconvolution to yield the second harmonic signals.29Fig. 4 shows the typical HRS signals of P-Ti2-TCF solutions in chloroform with different concentrations; the inset shows a typical HRS signal after spectral deconvolution to remove the TPF signal. The data processing was referred to the literature methods.30 As the NLO chromophores are absorptive at the second harmonic wavelengths, the HRS signal intensities were calibrated using the Beer–Lambert law.
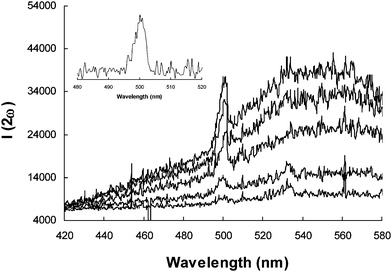 | ||
| Fig. 4 Experimental HRS signals of P-Ti2-TCF in chloroform with different concentrations (from bottom to top, c = 0.01, 0.02, 0.04, 0.06, 0.08 mM) at an excitation wavelength of 1000 nm. The inset shows a typical second harmonic HRS signal extracted from the two-photon fluorescence background by spectral deconvolution. | ||
Table 3 shows the β values obtained from the HRS measurements. Since the NLO chromophores have different UV-vis absorption properties, which give differently enhanced β values at the measurement wavelengths, static β (β0) were calculated using the two-level model for comparison. Although some discrepancies between β0 values from β1000 nm and β1100 nm are observable, the two series of measurements gave the same structure-NLO property relationship. In both cases, the experimentally derived β0 values of B-T-TCF (i.e., β0,1000 nm = 602 × 10−30 esu and β0,1100 nm = 451 × 10−30 esu) are comparable with the literature values of FTC (e.g., β0,1064 nm = 635 × 10−30 esu7 and β0,1900 nm = 445 ± 230 × 10−30 esu),16 indicating the validity of the HRS measurements.
| NLO chromophore | β 1000 nm | β 0 1 | β 1100 nm | β 0 2 | μ cal (D) | μ cal β 01 d | T d (°C) |
|---|---|---|---|---|---|---|---|
| a Molecular first hyperpolarizability (β, ×10−30 esu) measured in chloroform at 1000 nm and 1100 nm by Hyper-Rayleigh scattering experiment. b Static β values (β0, ×10−30 esu) calculated from the corresponding experimental values with the two-level model: β = β0×(1/λmax)4/{[(1/λmax2)−(4/λ2)]×[(1/λmax2) −(1/λ2)]}. c Dipole moment (μ, D) calculated from geometric structures optimized by B3LYP/6-31G. d Unit: ×10−48 esu. e Onset temperature for thermal decomposition measured by TGA at a heating rate of 20 °C/min in nitrogen. | |||||||
| B-Ti2-TCF | 1483 | 486 | 2915 | 531 | 13.4 | 6512 | 288 |
| T-Ti2-TCF | 1683 | 812 | 2276 | 821 | 13.4 | 10880 | 250 |
| P-Ti2-TCF | 1416 | 671 | 2223 | 773 | 12.3 | 8253 | 230 |
| P-T-TCF | 1174 | 614 | 1638 | 683 | 12.3 | 8227 | 210 |
| T-Ti4-TCF | — | — | — | — | 10.5 | — | 197 |
| TPA-Ti2-TCF | 2408 | 895 | 4141 | 948 | 14.2 | 12709 | 310 |
| TPA-Ti5-TCF | 2173 | 956 | 3142 | 964 | 12.6 | 12045 | 285 |
| B-T-TCF | 1514 | 602 | 1750 | 451 | 13.2 | 7946 | 265 |
From Table 3 it can be seen that all the resulting NLO chromophores exhibited very large β0 values in the range of 450–960 × 10−30 esu, except for the “neutral” thiazole-based T-Ti4-TCF whose HRS signal at the second harmonic wavelengths was not detected at the experimental conditions, due probably to a small molecular optical nonlinearity. The replacement of thiophene with “un-matched” thiazole as the auxiliary acceptor led pyrrole-based NLO chromophore P-Ti2-TCF to enhanced β0 values with respect to P-T-TCF. However, for benzene-based B-Ti2-TCF the effect of “un-matched” thiazole on β0 is not clear as B-Ti2-TCF shows smaller β0 at 1000 nm but larger β0 at 1100 nm than B-T-TCF. Replacement of benzene with electron-rich heteroaromatics (e.g., thiophene and pyrrole) as the auxiliary donor led to a significant increase in β0, as evidenced by the much higher β0 values of T-Ti2-TCF and P-Ti2-TCF (β0 = 670–820 × 10−30 esu) than that of B-Ti2-TCF (β0 = 500 × 10−30 esu). This observation agrees with our previous study on tricyanovinyl acceptor-based electronically gradient NLO chromophores13 and is in accordance with the theoretical predictions that a higher electron density of the auxiliary donor contribute to a larger molecular optical nonlinearity.11,12 It is interesting to note, however, that the pyrrole-based P-Ti2-TCF showed smaller β0 than the thiophene-based T-Ti2-TCF, although pyrrole is known to have a higher electron density than thiophene. Recall that P-Ti2-TCF has a blue-shifted maximal CT absorption wavelength, a smaller solvatochromism, and a smaller BLA relative to T-Ti2-TCF, which are all supportive to the smaller β0 of P-Ti2-TCF. The regiostructure of thiazole as the auxiliary acceptor showed a significant effect on β0. The “matched” thiazole-based TPA-Ti5-TCF exhibited larger β0 values than its “un-matched” isomer TPA-Ti2-TCF; and the “neutral” thiazole-based T-Ti4-TCF has a very small β that was not measureable at the HRS experimental conditions.
It should be noted that the triarylamine-based NLO chromophores (i.e., TPA-Ti2-TCF and TPA-Ti5-TCF) exhibited exceptionally large β0 values (e.g., 890–960 × 10−30 esu) although their solvatochromisms are moderate with respect to other NLO chromophores of the series. In comparison, the β0 values of TPA-Ti2-TCF are nearly double those of B-Ti2-TCF and are 10% higher than those of T-Ti2-TCF (Table 3). Recently, a few triarylamine-based NLO chromophores exhibiting large molecular optical nonlinearities and good thermal stabilities have been reported.16,17 We have also previously found such type of chromophores are unique in terms of their capability in giving rise to very large β0 values, which cannot simply be understood by the π–donor/acceptor strength.13Fig. 5 correlates the β0 values and the BLA of the NLO chromophores. A clear trend of increase of β0 with the increase of BLA can be observed. The triarylamine-based NLO chromophores (i.e., TPA-Ti2-TCF and TPA-Ti5-TCF) have larger BLAs than other chromophores and showed larger β0 values. However, the trend of β0versus BLA does not reflect a clear structure–β relationship. For “un-matched” thiazole-based NLO chromophores, the donor ability increase in the order of N,N-dimethylaniline ≈ triarylamine < piperidinylpyrrole ≤ piperidinylthiophene according to the previous electronic structure analysis, but the β0 values of the corresponding NLO chromophores did not vary according to such an order. In particular, the triarylamine-based TPA-Ti2-TCF exhibited exceptionally larger β0 values than the other NLO chromophores (i.e., B-Ti2-TCF, T-Ti2-TCF and P-Ti2-TCF).
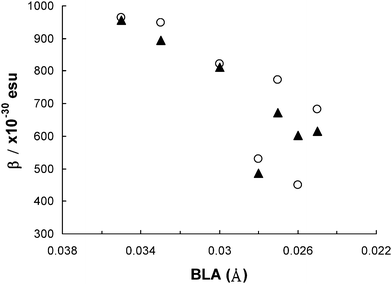 | ||
| Fig. 5 Correlation of BLA with experimental molecular first hyperpolarizability (β) of the NLO chromophores. | ||
In order to understand the structure-property relationship of the resulting series of NLO chromophores, the transition dipole moments (μeg) and bandgaps (ΔEeg) of the NLO chromophores were calculated with the time-dependent density function theory (TD-DFT) by using the previously optimized ground-state structures. Dipole moments of the NLO chromophores at excited states were calculated with the RCIS method. Ten excited electronic transition states were considered in the calculation, but only two with significant transition dipoles were taken into account for their contributions to β. By using the two-level model,28 β values associated with each excitation were calculated and are displayed in Table 4. An increase in the HOMO–LUMO bandgap and a decrease in the HOMO–LUMO transition dipole are observed upon replacement of thiophene by “un-matched” thiazole as the auxiliary acceptor, suggesting lower β values for B-Ti2-TCF and P-Ti2-TCF relative to B-T-TCF and P-T-TCF, respectively. With the introduction of electron-rich thiophene or pyrrole to the donor end as the auxiliary donor, the NLO chromophores T-Ti2-TCF and P-Ti2-TCF have enhanced HOMO–LUMO transition dipoles and lowered HOMO–LUMO bandgaps with respect to those of B-Ti2-TCF, and thus have larger β values. It is important to note that, for all the NLO chromophores, the second electronic transition contribute non trivially to the overall β (e.g., 4–65 × 10−30 esu), particularly for the triarylamine-based TPA-Ti2-TCF and TPA-Ti5-TCF whose second electronic transition contribute more than 10% to the overall β. This is in agreement with the stronger UV-vis absorption of TPA-Ti2-TCF and TPA-Ti5-TCF at around 500 nm than other NLO chromophores. From Table 3, it can be seen that although the HOMO–LUMO associated β values of the triarylamine-based NLO chromophores are not necessarily higher than the other NLO chromophores, the overall β of TPA-Ti2-TCF and TPA-Ti5-TCF are high due to the contribution from the second excited states.
| Chromophore | Excitation energy levelsa | Δμgeb | ΔEgec | μ ge | Δμge × μge2e | β0f |
|---|---|---|---|---|---|---|
| ΔEge2 | ||||||
| a Major excitations contributing to the nonlinear optical properties of the chromophores. “H” denotes the highest occupied molecular orbital (HOMO); “L” the lowest unoccupied molecular orbital (LUMO); “L + 1” the molecular orbital right above the LUMO. b Differential dipole moment between ground and excited states. c Energy gap between ground and excited states. d Transition dipole moments. e Contribution of each excitation to the static first hyperpolarizability (β0) calculated according the two-state model, unit in 10−30 esu. f Static first hyperpolarizability (β0) as a sum of the major contributions. | ||||||
| B-Ti2-TCF | H → L | 5.70 | 2.04 | 14.0 | 268.4 | 280.4 |
| H → L + 1 | 2.70 | 2.63 | 5.54 | 12.0 | ||
| T-Ti2-TCF | H → L | 6.00 | 2.01 | 15.4 | 352.2 | 355.8 |
| H → L + 1 | 1.10 | 2.59 | 4.71 | 3.6 | ||
| P-Ti2-TCF | H → L | 6.50 | 2.00 | 14.7 | 351.1 | 356.8 |
| H → L + 1 | 1.50 | 2.62 | 5.10 | 5.7 | ||
| P-T-TCF | H → L | 6.60 | 1.97 | 15.3 | 398.1 | 402.6 |
| H → L + 1 | 1.60 | 2.61 | 4.40 | 4.5 | ||
| TPA-Ti2-TCF | H → L | 5.10 | 1.74 | 13.4 | 302.5 | 367.7 |
| H → L + 1 | 3.90 | 2.47 | 10.1 | 65.2 | ||
| TPA-Ti5-TCF | H → L | 6.60 | 1.62 | 13.4 | 451.6 | 500.4 |
| H → L + 1 | 2.60 | 2.40 | 10.4 | 48.8 | ||
| B-T-TCF | H → L | 6.00 | 2.02 | 14.6 | 313.4 | 322.5 |
| H → L + 1 | 2.60 | 2.63 | 4.93 | 9.1 |
The thermal stability of the resulting NLO chromophores was evaluated by TGA in nitrogen. As shown in Table 3, most NLO chormophores exhibited excellent thermal stability with the onset temperature for decomposition (Td) above 250 °C, except for the pyrrole-based NLO chromophores (i.e., P-Ti2-TCF and P-T-TCF) that showed lower Td. In comparison, the “un-matched” thiazole-based NLO chromophores (e.g., B-Ti2-TCF and P-Ti2-TCF) showed an improved thermal stability by ca. 20 °C with respect to their thiophene analogues (i.e., B-T-TCF and P-T-TCF). On the other hand, with the increase of electron density of the auxiliary donor, i.e., from benzene to thiophene to pyrrole, the NLO chromophores (e.g., B-Ti2-TCF, T-Ti2-TCF and P-Ti2-TCF) showed a decreased thermal stability. The “matched” thiazole as the auxiliary acceptor led NLO chromophore TPA-Ti5-TCF to a significantly lower thermal stability than the “un-matched” thiazole-based TPA-Ti2-TCF. Nevertheless, both TPA-Ti2-TCF and TPA-Ti5-TCF appear to be very promising for EO applications because of their combination of very large β0 values and excellent stability.
Conclusions
A novel series of TCF-based, electronically gradient NLO chromophores with systematically tuned ground-state polarizations was successfully prepared by introduction of electron-rich heteroaromatics to the electron donor end as the auxiliary donor and introduction of electron-poor thiazole with different regiostructures to the acceptor end as the auxiliary acceptor. The electronic structures and NLO properties of the resulting NLO chromophores were carefully characterized and studied. Most of the NLO chromophores exhibited very large positive solvatochromism (e.g., Δλdioxane-chloroform = 45–75 nm) and static β values in the range of 450–960 × 10−30 esu. Correlation studies on β and structures revealed a clear dependence of β on the electronically gradient structure. With the increase of electron density of the auxiliary donor or upon the use of “matched” thiazole as the auxiliary acceptor, substantially enhanced β could be obtained. In accordance to previous studies, it was also found with this series of NLO chromophores that those based on dianisylamine as the electron donor showed very large β values. Theoretical studies showed that the second excited states of these NLO chromophores contributed significantly to β.Acknowledgements
We thank the financial support of the National Natural Science Foundation of China (Grant No.: 200504003).Notes and references
- L. Dalton, A. Harper, A. Ren, F. Wang, G. Todorova, J. Chen, C. Zhang and M. Lee, Ind. Eng. Chem. Res., 1999, 38, 8 CrossRef CAS; L. Dalton, Adv. Polym. Sci., 2002, 158, 1 CAS; F. Kajzar, K.-S. Lee and A. K.-Y. Jen, Adv. Polym. Sci., 2003, 161, 1 CAS.
- A. M. Sinyukov, M. R. Leahy, L. M. Hayden, M. Haller, J. Luo, A. K. Y. Jen and L. R. Dalton, Appl. Phys. Lett., 2004, 85, 5827 CrossRef CAS; A. Chen, H. Sun, A. Pyayt, X. Zhang, J. Luo, A. Jen, P. A. Sullivan, S. Elangovan, L. R. Dalton, R. Dinu, D. Jin and D. Huang, J. Phys. Chem. C, 2008, 112, 8072 CrossRef CAS; M. Lee, H. E. Katz, C. Erben, D. M. Gill, P. Gopalan, J. D. Heber and D. J. McGee, Science, 2002, 298, 1401 CrossRef CAS; Y. Shi, C. Zhang, H. Zhang, J. H. Bechtel, L. R. Dalton, B. H. Robinson and W. H. Steier, Science, 2000, 288, 119 CrossRef CAS.
- A. K.-Y. Jen, P. Rao, K. Y. Wong and K. J. Drost, J. Chem. Soc., Chem. Commun., 1993, 90 RSC; A. K.-Y. Jen, Y. Liu, L. Zheng, S. Liu, K. J. Drost, Y. Zhang and L. R. Dalton, Adv. Mater., 1999, 11, 452 CrossRef CAS; X. Wu, J. Wu, Y. Liu and A. K.-Y. Jen, J. Am. Chem. Soc., 1999, 121, 472 CrossRef CAS.
- A. K.-Y. Jen, Y. Cai, P. Bedworth and S. R. Marder, Adv. Mater., 1997, 9, 132 CrossRef CAS.
- J.-M. Raimundo, P. Blanchard, N. Gallego-Planas, N. Mercier, I. Ledoux-Rak, R. Hierle and J. Roncali, J. Org. Chem., 2002, 67, 205 CrossRef CAS; A. J. P. Akelaitis, B. C. Olbricht, P. A. Sullivan, Y. Liao, S. K. Lee, D. H. Bale, D. B. Lao, W. Kaminsky, B. E. Eichinger, D. H. Choi, P. J. Reid and L. R. Dalton, Opt. Mater., 2008, 30, 1504 CrossRef CAS; Y. Liao, B. E. Eichinger, K. A. Firestone, M. Haller, J. Luo, W. Kaminsky, J. B. Benedict, P. J. Reid, A. K.-Y. Jen, L. R. Dalton and B. H. Robinson, J. Am. Chem. Soc., 2005, 127, 2758 CrossRef CAS.
- A. J. Kay, A. D. Woolhouse, Y. Zhao and K. Clays, J. Mater. Chem., 2004, 14, 1321 RSC; A. M. R. Beaudin, N. Song, Y. Bai, L. Men, J. P. Gao, Z. Y. Wang, M. Szablewski, G. Cross, W. Wenseleers, J. Campo and E. Goovaerts, Chem. Mater., 2006, 18, 1709; M. Szablewski, P. R. Thomas, A. Thornton, D. Bloor, G. H. Cross, J. M. Cole, J. A. K. Howard, M. Malagoli, F. Meyers, J.-L. Brédas, W. Wenseleers and E. Goovaerts, J. Am. Chem. Soc., 1997, 119, 3144 CrossRef CAS.
- B. H. Robinson, L. R. Dalton, A. W. Harper, A. Ren, F. Wang, C. Zhang, G. Todorov, M. Lee, R. Aniszfeld, S. Garner, A. Chen, W. H. Steier, S. Houbrecht, A. Persoons, I. Ledoux, J. Zyss and A. K.-Y. Jen, Chem. Phys., 1999, 245, 35 CrossRef CAS.
- C. Zhang, L. R. Dalton, M.-C. Oh, H. Zhang and W. H. Steier, Chem. Mater., 2001, 13, 3043 CrossRef CAS.
- S. Liu, M. A. Haller, H. Ma, L. R. Dalton, S. H. Jang and A. K.-Y. Jen, Adv. Mater., 2003, 15, 603 CrossRef CAS; K. Schmidt, S. Barlow, A. Leclercq, E. Zojer, S.-H. Jang, S. R. Marder, A. K.-Y. Jen and J.-L. Brédas, J. Mater. Chem., 2007, 17, 2944 RSC; J. Luo, Y.-J. Cheng, T.-D. Kim, S. Hau, S.-H. Jang, Z. Shi, X.-H. Zhou and A. K.-Y. Jen, Org. Lett., 2006, 8, 1387 CrossRef CAS.
- S. R. Marder, B. Kippelen, A. K.-Y. Jen and N. Peyghambarian, Nature, 1997, 388, 845 CrossRef CAS; S. R. Marder, L. T. Cheng, B. G. Tiemann, A. C. Friedli and J. W. Perry, Science, 1994, 263, 511 CrossRef CAS; S. R. Marder, C. B. Gorman, B. G. Tiemann and L. T. Cheng, J. Am. Chem. Soc., 1993, 115, 2524 CrossRef CAS.
- D. L. Albert, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 1997, 119, 6575 CrossRef CAS.
- V. P. Rao, A. K.-Y. Jen, J. Chandrasekhar, I. N. N. Namboothiri and A. Rathna, J. Am. Chem. Soc., 1996, 118, 12443 CrossRef; P. Zhu, P. Wang and C. Ye, Chem. Phys. Lett., 1999, 311, 306 CrossRef CAS.
- X. Ma, F. Ma, Z. Zhao, N. Song and J. Zhang, J. Mater. Chem., 2009, 19, 2975 RSC; X. Ma, R. Liang, F. Yang, Z. Zhao, A. Zhang, N. Song, Q. Zhou and J. Zhang, J. Mater. Chem., 2008, 18, 1756 RSC.
- H. Kang, A. Facchetti, C. L. Stern, A. L. Rheingold, W. S. Kassel and T. J. Marks, Org. Lett., 2005, 7, 3721 CrossRef CAS; H. Kang, A. Facchetti, H. Jiang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Stern, Z. Liu, S.-T. Ho, E. C. Brown, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 3267 CrossRef CAS; H. Kang, A. Facchetti, P. Zhu, H. Jiang, Y. Yang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Z. Liu, S. Ho and T. J. Marks, Angew. Chem., Int. Ed., 2005, 44, 7922 CrossRef CAS.
- Y.-K. Wang, C.-F. Shu, E. M. Breitung and R. J. McMahon, J. Mater. Chem., 1999, 9, 1449 RSC; C.-F. Shu and Y.-K. Wang, J. Mater. Chem., 1998, 8, 833 RSC; E. M. Breitung, C.-F. Shu and R. J. McMahon, J. Am. Chem. Soc., 2000, 122, 1154 CrossRef CAS.
- J. A. Davies, A. Elangovan, P. A. Sullivan, B. C. Olbricht, D. H. Bale, T. R. Ewy, C. M. Isborn, B. E. Eichinger, B. H. Robinson, P. J. Reid, X. Li and L. R. Dalton, J. Am. Chem. Soc., 2008, 130, 10565 CrossRef CAS.
- Y.-J. Cheng, J. Luo, S. Hau, D. H. Bale, T.-D. Kim, Z. Shi, D. B. Lao, N. M. Tucker, Y. Tian, L. R. Dalton, P. J. Reid and A. K.-Y. Jen, Chem. Mater., 2007, 19, 1154 CrossRef CAS; B. K. Spraul, S. Suresh, T. Sassa, A. M. Herranz, L. Echegoyen, T. Wada, D. Perahia and D. W. Smith Jr., Tetrahedron Lett., 2004, 45, 3253 CrossRef CAS.
- L. Qiu, T. Zhang, J. F. Zai, Y. Q. Shen, CN Pat 1 752 079, 2006.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- G. Yang, T. Su, S. Shi, Z. Su, H. Zhang and Y. Wang, J. Phys. Chem. A, 2007, 111, 2739 CrossRef CAS; C. M. Isborn, A. Leclercq, F. D. Vila, L. R. Dalton, J. L. Bredas, B. E. Eichinger and B. H. Robinson, J. Phys. Chem. A, 2007, 111, 1319 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1998, 37, 785 CrossRef; A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- J. B. Foreman, M. Head-Gordon, J. A. Pople and M. J. Frisch, J. Phys. Chem., 1992, 66, 135 CrossRef; M. P. Fülscher, K. Andersson and B. O. Roos, J. Phys. Chem., 1992, 96, 9204 CrossRef CAS.
- X. Andrade, S. Botti, M. A. L. Marques and A. Rubio, J. Chem. Phys., 2007, 126, 184106 CrossRef; Y. Takimoto, C. M. Isborn, B. E. Eichinger, J. J. Rehr and B. H. Robinson, J. Phys. Chem. C, 2008, 112, 8016 CrossRef CAS.
- B. Narayana, K. K. Vijaya Raj, B. V. Ashalatha, N. Suchetha Kumari and B. K. Sarojini, Eur. J. Med. Chem., 2004, 39, 867 CrossRef CAS.
- B. G. Knöpfle, P. Prêtre and P. Günter, J. Appl. Phys., 1992, 71, 1594 CrossRef CAS.
- K. A. Firestone, D. B. Lao, D. M. Casmier, O. Clot, L. R. Dalton and P. J. Reid, Proc. SPIE., 2005, 5935, 59350.
- J. N. Woodford, M. A. Pauley and C. H. Wang, J. Phys. Chem. A, 1997, 101, 1989 CrossRef CAS.
- J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664 CrossRef CAS.
- K. Song, J. N. Woodford and C. H. Wang, J. Phys. Chem. A, 1997, 101, 3222 CrossRef CAS.
- M. A. Pauley, H.-W. Guan, C. H. Wang and A. K.-Y. Jen, J. Chem. Phys., 1996, 104, 7821 CrossRef CAS; M. A. Pauley, C. H. Wang and A. K.-Y. Jen, J. Chem. Phys., 1995, 102, 6400 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data and molecular modeling coordinates of the NLO chromophores. See DOI: 10.1039/b923185d |
| This journal is © The Royal Society of Chemistry 2010 |