Arsenic speciation with gradient hydride generation interfacing liquid chromatography and atomic absorption spectrometry†
Yong
Tian
a,
Ming-Li
Chen
a,
Xu-Wei
Chen
a,
Jian-Hua
Wang
*a,
Yoshihiro
Hirano
b,
Hideyuki
Sakamoto
b and
Ikumei
Setsu
b
aResearch Center for Analytical Sciences, Northeastern University, Box 332, Shenyang, 110004, China. E-mail: jianhuajrz@mail.neu.edu.cn; Fax: +86 24 83676698; Tel: +86 24 83688944
bHitachi High-Technologies Corporation, Hitachinaka, Ibaraki 312-8504, Japan
First published on 14th October 2009
Abstract
Arsenic speciation was performed based on liquid chromatographic separation followed by gradient hydride generation (GHG) and quartz atomizer atomic absorption spectrometric detection. The arsenic species, i.e., arsenate (As(V)), arsenite (As(III)), monomethylarsonic acid (MMA), dimethylarsinic acid (DMA) and trimethylarsine oxide (TMAO), were separated on C30-5 columns, and the concept of gradient hydride generation facilitates high conversion efficiency of the arsenic species into corresponding hydrides. The use of 2% L-cysteine in the GHG process gives rise to further improvements on the hydride generation efficiency of 13% to 32% for the arsenic species. The hydrides were separated in a unique design of gas–liquid separator, which not only ensures a complete separation but minimizes the dispersion of hydrides when delivering into the atomizer, resulting in a maximum of 13-fold improvement on the sensitivity of As(V) compared to previous studies. A separation process was finished within 800 s by injecting 100 µL sample solution, achieving detection limits of 0.9, 1.4, 1.4, 1.6, 1.5 µg/L, respectively, for As(V), As(III), MMA, DMA and TMAO. Precisions of less than 3% and 6% RSD were obtained for the five arsenic species at 100 µg/L and 20 µg/L, respectively. Three arsenic species, i.e., As(V), DMA and TMAO, were identified in Hijiki samples by this procedure.
Introduction
Arsenic in various sample matrixes causes a multitude of serious health problems.1 Arsenic in drinking water in South Asia has become one of the biggest environmental problems of the century.2 Generally, arsenic in environmental waters is almost exclusively inorganic,3 however, methylated arsenic species were frequently identified from biological samples and alga, e.g., Hijiki (a type of brown alga widely consumed in Asian countries, also called Hizikia fusiforme). Therefore, the total amount of arsenic present in a sample is no longer sufficient for providing comprehensive information about their toxicity or ease of removal among the various arsenic oxidation states, as both properties depend strongly on the chemical form of a specific arsenic species. Thus, the accurate identification of arsenic species has become one of the most important issues when evaluating the toxic effects of arsenic.4–6 Although a number of analytical protocols have been dedicated to arsenic speciation, however, there are no universal methodologies available providing standard procedures for such purposes. At this juncture, it is highly desirable to develop suitable and reliable approaches for arsenic speciation.So far, extensive efforts have been devoted to the hyphenation of LC separation with inductively coupled plasma mass spectrometry (ICP-MS).7–11 LC provides powerful separation for isolating the individual arsenic species, while the high sensitivity of ICP-MS and its flow through feature facilitate accurate and fast detection. However, in many circumstances, this approach is not suitable for routine analysis attributed to the high running cost of the instrumentation. For this purpose, studies have been dedicated to coupling LC with hydride generation and some other atomic spectrometric detectors, e.g., atomic absorption spectrometry (AAS)12–15 and atomic fluorescence spectrometry (AFS),16,17 for arsenic speciation.
The hyphenation of hydride generation with atomic absorption spectrometry is among the most frequently used techniques for the separation and quantification of arsenic species.18 There are, however, some intrinsic shortcomings of this technique when compared to those with detection by ICP-MS and AFS, e.g., the sensitivity of AAS can hardly compete with the latter systems. In order to be competitive, a significant improvement of the sensitivity is required with AAS as detector.12–15 When considering the inherent detection power of AAS limited by its intrinsic design, one of the most feasible approaches to improve the sensitivity is to ensure a complete capture of the analyte of interest at each single step of reaction. In arsenic speciation by using hydride generation, the conversion efficiencies of arsenic species into corresponding arsenic hydrides generally vary significantly. Although fixed experimental conditions might be well suitable for certain arsenic species, while the conversion efficiencies (and finally the detection capabilities) for some others could be significantly reduced.12–14 A pH-gradient hydride generation approach has been proposed for the improvement of hydride generation efficiency.19 On the other hand, as an indispensable component in the hyphenation of a hydride generation unit with an atomic spectrometric detector, the gas–liquid separator is an extremely important part when desiring to improve the sensitivity and stability. Recently, this issue has caught pertinent attentions.17,20–22 Generally, a sufficient chamber volume in the gas–liquid separator is required to provide a buffering for the hydride and the carrier gas in order to depress the noise signal. On the other hand, however, the chamber volume should be limited to the extent possible to minimize the dispersion of the hydride before it entering the atomization area.23 In this respect, it still deserves more efforts to be focused on this issue to further improve the performance of gas–liquid separation.
This work reports a gradient hydride generation protocol to improve the conversion efficiency of As(V), As(III), MMA, DMA and TMAO into their corresponding hydrides before AAS detection. In addition, a novel design of gas–liquid separator with limited dead volume was used providing minimum dispersion of the hydrides during their transportation from the separator to the detection point and thus facilitates high detection sensitivity. Arsenic speciation in Hijiki samples was performed by this procedure.
2. Materials and methods
Instrumentation
A L-2130 pump and a L-2300 column oven (Hitachi High-Technologies, Japan) with Wakopak Navi C30-5 columns (Φ6 × 250 mm and Φ6 × 150 mm, Wako Pure Chemical Ind., Ltd., Japan) were used for chromatographic separation of arsenic species. A six-port injection valve (Rheodyne, Cotati, CA, USA) with a 100 µL sample loop (1.0 mm i.d. PEEK tubing) was used for sample introduction. A HITACHI Z-2000 atomic absorption spectrophotometer (Hitachi High-Technologies, Japan) with a quartz atomizer (in addition to flame and electrothermal atomizers) was used for arsenic detection.A Minipuls evolution peristaltic pump (Gilson, France) was used for delivering HCl and NaBH4 solutions in the hydride generation process. The LC effluent, via a piece of PEEK tubing (0.13 mm i.d.), was introduced into the reaction system. A 60 °C water bath was used to improve the hydride generation efficiency of the arsenic species, followed by a cooling water bath to control the temperature of the reaction mixture before entering the gas–liquid separator. Four types of gas–liquid separator (Fig. 1) were employed and their separation efficiencies evaluated. The hydrides after separation were swept by argon to flow into the T-type quartz atomizer for arsenic quantification.
 | ||
| Fig. 1 The designs of gas–liquid separators used in the investigations. | ||
Fig. 2 illustrates the flow manifold of the liquid chromatographic separation linked to the gradient hydride generation-AAS detection system for arsenic speciation.
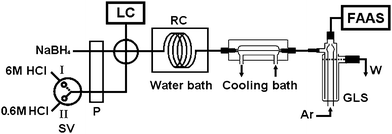 | ||
| Fig. 2 The flow manifold of the LC separation-gradient hydride generation-AAS system for arsenic speciation. LC: liquid chromatography; FAAS: quartz atomizer flame atomic absorption spectrometer; P: peristaltic pump; SV: selector valve; RC: reaction coil; GLS: gas–liquid separator; Ar: argon; W: waste; quartz atomizer.Valve position I: 6 mol/L HCl for the hydride generation of As(V), As(III) and MMA.Valve position II: 0.6 mol/L HCl for the hydride generation of DMA and TMAO. | ||
Reagents
All chemicals were at least of analytical-reagent grade, and de-ionized water of 18.2 MΩ cm was used throughout.Arsenic stock solutions of 1000 mg/L were prepared by dissolving appropriate amount of sodium arsenite, disodium hydrogen arsenate heptahydrate, monomethylarsonic acid (MMA), dimethylarsinic acid (DMA) and trimethylarsine oxide (TMAO) in water. Sodium arsenite and disodium hydrogen arsenate heptahydrate were purchased from Sinopharm Chemical Reagent Co., Ltd (China), while MMA, DMA and TMAO were received from Wako Pure Chemical Ind., Ltd (Japan).
The reagents used as mobile phase include sodium 1-butanesulfonate, malonic acid, tetramethylammonium hydroxide, methanol and ammonium tartrate. Sodium 1-butanesulfonate was received from Sigma-Aldrich (Switzerland). The other reagents were purchased from Sinopharm Chemical Reagent Co., Ltd (China), including sodium tetrahydroborate, hydrochloric acid and L-cysteine. In practice, NaBH4 solutions in 0.5% (m/v) NaOH were prepared daily.
Sampling and sample pretreatment
Two types of commercial Hijiki were purchased from supermarket (50 g packages). The Hijiki samples were carefully ground for homogenization. The homogenized samples were then stored in airproof flasks for future analysis.Total arsenic quantification
Electrothermal atomic absorption spectrometry (ETAAS) was used for the quantification of total arsenic with 10 µL Mg(NO3)2 solution (1000 mg/L) as matrix modifier. In practice, 50-fold and 500-fold dilutions with HNO3 (1%, v/v) were made when analyzing total arsenic in the wet acid digests with HNO3 and the individual arsenic species in the ultrasonic-assisted water leachate. Table S1† summarizes the ETAAS temperature program for total arsenic determination.LC separation-gradient hydride generation-AAS system for arsenic speciation
Fig. 2 illustrated the flow manifold and the operating parameters were summarized in Table S2†. The system was initiated as soon as 100 µL sample solution was injected into LC. The effluent from the LC system was directed to meet streams of NaBH4 and HCl solutions to initiate hydride generation, which was significantly accelerated when the reaction mixture flowed through the reaction coil in a heated water bath at 60 °C. After cooling down in a water bath, the reaction mixture was directed into the gas–liquid separator where the hydrides were isolated and further transported into the quartz atomizer for arsenic quantification. The gradient hydride generation was performed as such that a flow of 6 mol/L HCl was used for the hydride generation of As(V), As(III) and MMA, and when the signal of MMA was recorded, the selector valve was switched to introduce a stream of 0.6 mol/L HCl as hydride generation medium for the other arsenic species, i.e., DMA and TMAO. On the other hand, a fixed NaBH4 concentration of 1.0% (m/v) was used for the hydride generation of the 5 arsenic species.Results and discussion
The chromatographic separation process
Considering that the liquid chromatographic separation of arsenic species has been widely reported in the literature, the present study did not pay much attention to this point. The effect of each of the mobile phase constituents on the absorbance signal in addition to the retention time for each single arsenic species was thoroughly investigated. These include the concentrations of ion-pair reagent (sodium 1-butanesulfonate and tetramethylammonium hydroxide), methanol, malonic acid (buffer), chelating reagent (ammonium tartrate) and the acidity of the mobile phase. After careful evaluations, the conditions for liquid chromatographic separation as given in Table S2† were adopted.The hydride generation process
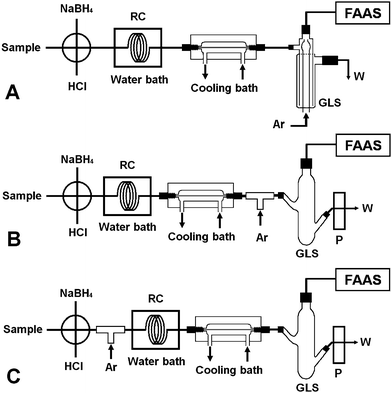 | ||
| Fig. 3 The various flow manifolds employed for evaluating the performance of the four types of gas–liquid separators. FAAS: quartz atomizer flame atomic absorption spectrometer; P: peristaltic pump; RC: reaction coil; GLS: gas–liquid separator; Ar: argon; W: waste; quartz atomizer. | ||
The experimental results for As(V) are summarized in Table 1. It is obvious that favorable analytical performance was achieved by incorporating separator I into the flow manifold of Fig. 3A, and this set-up was therefore used.
| Separator I | Separator II | Separator III | Separator IV | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| h | Y1/2 (s) | RSD (%) | h | Y1/2 (s) | RSD (%) | h | Y1/2 (s) | RSD (%) | h | Y1/2 (s) | RSD (%) | |
| a Note: HCl: 6 mol/L (with 2% L-cysteine), NaBH4: 1% (in 0.5% NaOH); A same flow rate of 1.1 mL/min was used for both HCl and NaBH4 solutions. The 100 µL sample solution contains 100 µg/L of As(V) at a sampling flow rate of 1.1 mL/min. The water bath was set at 60 °C. h: normalized peak height; Y1/2: peak half-width; RSD: relative standard deviation (n = 8). | ||||||||||||
| Flow manifold A | 1 | 6.9 | 2.5 | — | — | — | — | — | — | — | — | — |
| Flow manifold B | 0.96 | 6.3 | 3.4 | 0.93 | 6.4 | 3.4 | 0.60 | 11.0 | 4.6 | 0.50 | 17.9 | 4.1 |
| Flow manifold C | 0.65 | 7.2 | 5.4 | 0.77 | 7.0 | 5.2 | 0.55 | 11.6 | 7.5 | 0.46 | 17.0 | 3.6 |
Fig. 4 illustrates the dependency of hydride generation on HCl concentration. For As(V), As(III) and MMA, continual increase of the recorded signal was observed when enhancing the acid concentration within the whole range of 0.2–6 mol/L. For DMA and TMAO, however, a rapid increase of the signal was first encountered by increasing the acid concentration up to 0.6 mol/L where maximum signals were achieved, while a further increase led to decrease of the signal, especially for TMAO whose signal sharply dropped to a negligible level. These observations could be attributed to the protonation of a methyl-arsine, e.g., (CH3)2AsH from DMA and (CH3)3As from TMAO, which can readily occur in an acid medium and the produced water-soluble methyl-arsonium species in equilibrium with gaseous methyl-arsine reduces the hydride generation efficiency,24,25 corresponds to a rapid drop of the signal arising from DMA and TMAO. The much more significant drop of the signal from TMAO is explained by the favorable protonation of (CH3)3As than that of the (CH3)2AsH due to its more basic nature.
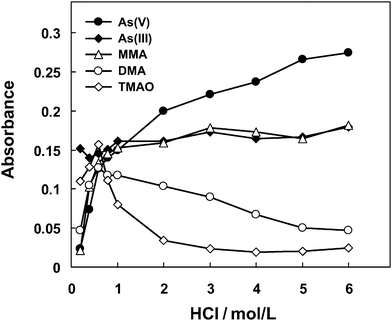 | ||
| Fig. 4 The dependency of hydride generation efficiencies on the concentration of HCl. A solution containing 100 µg/L of each arsenic species was employed. 2% L-cysteine (m/v) was included in the HCl solutions. A same flow rate of 1.1 mL/min was used for both HCl and NaBH4 solutions. Other experimental parameters are illustrated in Table S2†. The error estimation of the signals for each experimental point corresponds to standard deviation of RSD < 10% (n = 3). | ||
Fig. 4 indicates that each arsenic species requires a specific acid concentration for optimal efficiency of hydride generation.26,27 For a HCl concentration of 6 M at which maximum signals were recorded for As(V), As(III) and MMA, the signals from DMA and TMAO correspond to only 20% and 10% or less of the signal as obtained from a same amount of As(V). Thus, it is not possible to find a suitable acid concentration at which an identical or similar response could be achieved for the 5 arsenic species. In practice, a compromise acid concentration is generally selected which might be favorable for the hydride generation of some arsenic species, but inevitably sacrifices the hydride conversion efficiency for other species.12–14,17,28–30 In this respect, gradient hydride generation should be favorable to improve the efficiency of hydride generation for all the arsenic species. In the present case, gradient hydride generation is performed such that 6 M HCl was used for the hydride generation of As(V), As(III) and MMA, when the MMA signal was recorded, a stream of 0.6 mol/L of HCl as hydride generation medium was introduced for the other two arsenic species, i.e., DMA and TMAO. Fig. 5 clearly shows that the retention time interval between MMA and DMA is long enough for the gradient hydride generation process. During this process, a fixed concentration of NaBH4 at 1.0% (m/v) was used for the 5 arsenic species.
 | ||
| Fig. 5 The chromatograms recorded for arsenic speciation by the LC separation-hydride generation-AAS detection protocol. A and B were obtained by conventional hydride generation with a fixed HCl concentration for the 5 arsenic species, while C was achieved by a gradient hydride generation approach, i.e., 6.0 M HCl was used for the hydride conversion of As(V), As(III) and MMA, while 0.6 M HCl for DMA and TMAO. A solution containing 100 µg/L of each arsenic species was used. Other experimental parameters are illustrated in Table S2†. | ||
Fig. 5 illustrates the chromatograms recorded by gradient hydride generation employing a gradient of acid concentration, i.e., 6 M HCl for As(V), As(III) and MMA, while 0.6 M HCl for DMA and TMAO was used. As a comparison, those results achieved by a conventional hydride generation protocol with compromised HCl concentrations, i.e., 0.6 M and 6 M HCl for all the 5 arsenic species, were included. Much improved signals for the 5 arsenic species were recorded, ensuring both favorable hydride generation efficiency and detection sensitivity for these arsenic species. It is also worth mentioning that a maximum of 13-fold improvement on the detection sensitivity of As(V) was achieved in comparison with previous reports.12–15
L-cysteine has been widely used for the reduction of arsenic species prior to hydride generation. In this study, the introduction of L-cysteine (2%, m/v) in HCl solution results in further improvements on the hydride generation efficiency and the detection sensitivity for the 5 arsenic species, i.e., 16%, 19%, 24%, 13%, 32% for As(V), As(III), MMA, DMA, TMAO, respectively (Fig. S1†). These results confirmed the previous observations.31 A most likely explanation for this observation might be that in aqueous medium the formation of borane complexes of X-BH3− (X = SR), which are more active than BH4− in the hydride transfer to analytical substrates, is possible in the case of hydrolysis of BH4− in the presence of thiols in L-cysteine.32
Analytical performance of the system
The detailed performance data of the LC separation-gradient hydride generation-AAS system for the speciation of the 5 arsenic species were derived and summarized in Table S3†. By injecting 100 µL sample solution into the system, a complete analytical run could be finished within 13.5 min. The limits of detection for the 5 arsenic species, i.e., As(V), As(III), MMA, DMA and TMAO, were 0.9, 1.4, 1.4, 1.6 and 1.5 µg/L (As), respectively. In addition, relative standard deviations (RSD) of less than 3% and 6% were obtained at 100 µg/L and 20 µg/L.Table 2 summarized a comparison of the characteristic performance data of the present system with some of the relevant procedures based on LC separation hyphenated with detection by mass spectrometry and atomic spectrometry. A significant improvement on the overall sensitivities for the 5 arsenic species was achieved by using the present system incorporating gradient hydride generation when compared to the reported similar procedures with detection by AAS12–15 and ICPAES.29,30 Although the limits of detection of this procedure cannot compete with those achieved by ICPMS,11,33 it is sufficient for the requirement of routine arsenic speciation analysis in environmental water and seafood or seaweed samples.
| Method | LODs (µg/L) | RSD (%) | Sample volume (µL) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| As(V) | As(III) | MMA | DMA | TMAO | ||||
| LC-ICPMS | 0.1 | 0.02 | 0.04 | 0.06 | — | 10 | 200 | 11 |
| 0.3 | 0.1 | 0.2 | 0.1 | — | 2.7 | 50 | 33 | |
| LC-HG-AFS | 0.2 | 0.1 | 0.2 | 0.1 | — | 10.3 | 200 | 16 |
| 8.5 | 1.0 | 2.2 | 2.3 | — | 2.2 | 20 | 17 | |
| LC-HG-ICPAES | 20 | 5 | 10 | 20 | — | 4.8 | 100 | 29 |
| 15 | 10 | 30 | 65 | 5.9 | 200 | 30 | ||
| LC-HG-AAS | 2.6 | 2.4 | 2.4 | 2.3 | 1.1 | 6.4 | 100 | 12 |
| 8.4 | 1.5 | 3.3 | 4.3 | — | 3.7 | 100 | 13 | |
| 4.2 | 2.0 | 2.9 | 2.4 | — | 8.7 | 100 | 14 | |
| 12.0 | 7.8 | — | — | — | 12.1 | 200 | 15 | |
| 0.9 | 1.4 | 1.4 | 1.6 | 1.5 | 2.8 | 100 | This work | |
Applications of the procedure to arsenic speciation in real samples
Total arsenic concentrations in the wet acid digest with HNO3 and the ultrasonic-assisted water leachate of Hijiki were quantified by electrothermal atomic absorption spectrometry (ETAAS) after appropriate dilution (Table 3). The ultrasonic-assisted water extraction efficiencies of total arsenic in Hijiki 1 and Hijiki 2 were ca. 76.1% and 82.2% respectively. A longer extraction time has been used but no significant improvement on the extraction efficiencies was obtained. Thus, the low extraction efficiency might be attributed to the presence of some unleachable arsenic species in the Hijiki samples.| Species | Found Hijiki 1 (µg/g) | Found Hijiki 2 (µg/g) | Spiked (µg/L) | Spiking Recov. Hijiki 1 (%) | Spiking Recov. Hijiki 2 (%) |
|---|---|---|---|---|---|
| a Mean value ±95% confidence interval (number of replicates = 3). b n.d. = not detected. | |||||
| As(V) | 53.6 ± 9.2 | 60.8 ± 13.2 | 50 | 107.4 | 111.2 |
| As(III) | n.d.b | n.d. | 50 | 97.9 | 97.1 |
| MMA | n.d.b | n.d. | 50 | 106.9 | 104.9 |
| DMA | 2.7 ± 0.4 | 2.4 ± 0.5 | 50 | 112.5 | 105.7 |
| TMAO | 0.34 ± 0.12 | 0.15 ± 0.03 | 50 | 104.6 | 102.2 |
| Sum of the arsenic species determined | 56.6 ± 9.7 | 63.4 ± 13.7 | |||
| Total arsenic (water leachates) | 63.9 ± 3.5 | 63.8 ± 1.9 | |||
| Total arsenic (wet acid digest) | 84.0 ± 8.2 | 77.6 ± 4.2 |
The speciation of the 5 arsenic species of interest in the Hijiki samples was performed by employing the present LC separation-gradient hydride generation-AAS system. After appropriate dilutions of the ultrasonic-assisted water leachates, large amounts of As(V) were found from both Hijiki samples. The chromatograms were illustrated in Fig. 6 and the speciation results were given in Table 3. For Hijiki 2, a reasonable agreement was achieved between the sum of the three individual arsenic species identified (As(V), DMA and TMAO), i.e., 63.4 µg/g, and the total arsenic concentration quantified by ETAAS, i.e., 63.8 µg/g. However, a discrepancy was observed for Hijiki 1, which attributed probably to the presence of some arsenic species, e.g., arsenosugars, other than those investigated in the present work.34 In addition, favorable recoveries were obtained for both samples by spiking appropriate amount of the arsenic species of interest.
 | ||
| Fig. 6 The chromatograms for the analysis of arsenic species in Hijiki. A 5-fold dilution of the ultrasonic-assisted water leachates of the two Hijiki samples was used for analysis. The experimental parameters were identical as those illustrated in Table S2†. | ||
Acknowledgements
This work is supported by the Natural Science Foundation of China (20805004, key project 20635010, National Science Fund for Distinguished Young Scholars 20725517 and Major International Joint Research Project 20821120292). Hitachi High-Technologies is highly appreciated for technical assistance and financial support. We also thank Prof. P. K. Dasgupta, University of Texas at Arlington, for valuable discussions and suggestions.References
- R. K. Kwok, Toxicol. Appl. Pharmacol., 2007, 222, 344 CrossRef CAS.
- F. S. Islam, A. G. Gault, C. Boothman, D. A. Polya, J. M. Charnock, D. Chatterjee and J. R. Lloyd, Nature, 2004, 430, 68 CrossRef CAS.
- X.-W. Chen, A.-M. Zou, M.-L. Chen, J.-H. Wang and P. K. Dasgupta, Anal. Chem., 2009, 81, 1291 CrossRef CAS.
- X.-J. Wang, Z. Sun, W. Chen, K. E. Eblin, J. A. Gandolfi and D. D. Zhang, Toxicol. Appl. Pharmacol., 2007, 225, 206 CrossRef CAS.
- J. S. Petrick, F. Ayala-Fierro, W. R. Cullen, D. E. Carter and H. V. Aposhian, Toxicol. Appl. Pharmacol., 2000, 163, 203 CrossRef CAS.
- D. J. Thomas, M. Styblo and S. Lin, Toxicol. Appl. Pharmacol., 2001, 176, 127 CrossRef CAS.
- T. Nakazato and H. Tao, Anal. Chem., 2006, 78, 1665 CrossRef CAS.
- S. K. V. Yathavakilla, M. Fricke, P. A. Creed, D. T. Heitkemper, N. V. Shockey, C. Schwegel, J. A. Caruso and J. T. Creed, Anal. Chem., 2008, 80, 775 CrossRef CAS.
- R. Xie, W. Johnson, S. Spayd, G. S. Hallb and B. Buckley, J. Anal. At. Spectrom., 2007, 22, 553 RSC.
- Y. Morita, T. Kobayashi, T. Kuroiwa and T. Narukawa, Talanta, 2007, 73, 81 CrossRef CAS.
- F. A. Duarte, J. S. F. Pereira, M. F. Mesko, F. Goldschmidt, É. M. M. Flores and V. L. Dressler, Spectrochim. Acta, Part B, 2007, 62, 978 CrossRef.
- R. Sur and L. Dunemann, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2004, 807, 169 CrossRef CAS.
- M. B. Amran, F. Lagarde and M. J. F. Leroy, Microchim. Acta, 1997, 127, 195 CAS.
- W.-C. Tseng, G.-W. Cheng, C.-F. Lee, H.-L. Wu and Y.-L. Huang, Anal. Chim. Acta, 2005, 543, 38 CrossRef CAS.
- P. Niedzielski, Anal. Chim. Acta, 2005, 551, 199 CrossRef CAS.
- J. L. Gomez-Ariza, D. Sanchez-Rodas, I. Giraldez and E. Morales, Talanta, 2000, 51, 257 CrossRef CAS.
- C. J. Wei and J. X. Liu, Talanta, 2007, 73, 540 CrossRef CAS.
- F. A. Duarte, J. S. F. Pereira, J. S. Barin, M. F. Masko, V. L. Dressler, E. M. D. Flores and G. Knapp, J. Anal. At. Spectrom., 2009, 24, 224 RSC.
- R. A. Diaz-Bone and M. Hitzke, J. Anal. At. Spectrom., 2008, 23, 861 RSC.
- Y. Bohari, A. Astruc, M. Astruc and J. Cloud, J. Anal. At. Spectrom., 2001, 16, 774 RSC.
- P. Pohl, TrAC, Trends Anal. Chem., 2004, 23, 87 CrossRef CAS.
- M.-L. Chen, Y. Tian and J.-H. Wang, J. Anal. At. Spectrom., 2008, 23, 876 RSC.
- Z.-L. Fang, Flow Injection Analysis, Science Press, Beijing, 1999, p. 201 Search PubMed.
- P. Carrero, A. Malavé, J. L. Burguera, M. Burguera and C. Rondón, Anal. Chim. Acta, 2001, 438, 195 CrossRef CAS.
- S. A. Pergantis, W. Winnik, E. M. Heithmar and W. R. Cullen, Talanta, 1997, 44, 1941 CrossRef CAS.
- X.-C. Le, W. R. Cullen, K. J. Reimer and I. D. Brindle, Anal. Chim. Acta, 1992, 258, 307 CrossRef.
- M. H. Arbab-Zavar and A. G. Howard, Analyst, 1980, 105, 744 RSC.
- C.-G. Yuan, B. He, E.-L. Gao, J.-X. Lü and G.-B. Jiang, Microchim. Acta, 2007, 159, 175 CrossRef CAS.
- S. García Salgado, M. A. Quijano Nieto and M. M. Bonilla Simón, J. Chromatogr., A, 2006, 1129, 54 CrossRef CAS.
- R. T. Gettar, R. N. Garavaglia, E. A. Gautier and D. A. Batistoni, J. Chromatogr., A, 2000, 884, 211 CrossRef CAS.
- H. Chen, I. D. Brindle and X. C. Le, Anal. Chem., 1992, 64, 667 CrossRef CAS.
- E. Pitzalis, D. Ajala, M. Onor, R. Zamboni and A. D'Ulivo, Anal. Chem., 2007, 79, 6324 CrossRef CAS.
- Z.-L. Chen, K. F. Akter, M. M. Rahman and R. Naidu, Microchem. J., 2008, 89, 20 CrossRef CAS.
- A. Raab, P. Fecher and J. Feldmann, Microchim. Acta, 2005, 151, 153 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 The ETAAS temperature program for the determination of total arsenic; Table S2 Operating parameters of the liquid chromatographic separation-gradient hydride generation-AAS detection system; Table S3 The analytical performance of the liquid chromatographic separation-gradient hydride generation-AAS system for arsenic speciation; Fig. S1 The influence of L-cysteine on the hydride generation efficiency. See DOI: 10.1039/b913198a |
| This journal is © The Royal Society of Chemistry 2010 |