 Open Access Article
Open Access ArticleEnergy densification of levulinic acid by thermal deoxygenation†
Thomas J. Schwartz, Adriaan R. P. van Heiningen and M. Clayton Wheeler*
Department of Chemical and Biological Engineering and Forest Bioproducts Research Institute, 5737 Jenness Hall, Orono, ME 04469-5737, USA. E-mail: cwheeler@umche.maine.edu; Fax: +1 207 581 2323; Tel: +1 207 581 2280
First published on 23rd June 2010
Abstract
This communication presents a process for thermal deoxygenation (TDO) to upgrade levulinic acid whereby levulinic acid is neutralized with calcium hydroxide, and the resulting salt is heated to between 350 °C and 450 °C. The cyclic and aromatic products have low oxygen to carbon ratios, improved energy density, and may be candidates for upgrading to hydrocarbon fuels. The process may be useful for converting biomass-derived levulinic acid in an integrated forest products complex based on a kraft pulp mill which uses a lime cycle to regenerate calcium hydroxide.
World oil production is forecasted to peak within the next five years,1 which implies it is likely that the price of oil will increase significantly in the future. This is also evidenced by the fact that the price of oil was back to about $80/bbl at the end of 2009 after the worst economic recession since World War II. Furthermore, the combustion of fossil fuels is considered to be the major factor responsible for trends in global climate change, equaling about 82% of net green house gas emissions.2 In order to address these problems, there is a pressing need for the development of renewable fuels and energy derived from biomass, wind, geothermal heat and solar radiation in order to meet future economic and environmental requirements. Among these renewable resources, biomass is considered to be the only sustainable and carbon-neutral source for the production of liquid fuels.3 The US has the potential to sustainably produce biomass which can replace more than one-third of the 2004 US petroleum consumption.4
Levulinic acid has been identified as one of the top value-added, biomass-derived chemicals by the U.S. Department of Energy,5 and it can be produced from cellulose-containing biomass in high yields using the “Biofine” process.6 Levulinic acid esters such as ethyl levulinate have low energy content (26 MJ kg−1)7 compared to petroleum based fuels (46 MJ kg−1).8 While this relatively low energy density might be acceptable for ground transportation fuels it is unacceptable for aviation fuels. The energy density of the fuel impacts both the range and the carrying capacity of the aircraft. High-energy-density fuels can be synthesized from levulinic acid by hydrogenation to γ-valerolactone (GVL) then catalytic upgrading to hydrocarbons. Using solid acid catalysts, it has been demonstrated that GVL can be upgraded to long-chain, branched alkenes.9 GVL is also an intermediate in the synthesis of long-chain, branched alkanes using Pd/Nb2O5 as a catalyst.10
We demonstrate here that levulinic acid can be simultaneously condensed and deoxygenated using a method traditionally used for forming ketones from organic acids. For nearly 100 years, the reaction of acetic acid with calcium hydroxide, followed by thermal decomposition has been known to form acetone.11 Decomposition was shown to occur above 400 °C. The reaction has been extended to other salts of organic acids, most notably calcium butyrate and calcium propionate.12–17 While the chemistry of base-catalyzed ketonization of carboxylates is reasonably well-understood,18,19 the mechanism of thermal decomposition of dry carboxylate salts is not as well-documented. An advantage of using calcium as a reactant is that it can then be recycled by feeding the calcium carbonate byproduct to a lime kiln.
Organic acids other than levulinic can be derived from biomass and may also be suitable for upgrading via TDO. Many of the top value-added chemicals that can be produced from biomass5 are organic acids. Many of these acids have unique functionalities such as di-acid groups, alcohol groups, or carbonyl groups. We show that additional functionality leads to more complex reaction mechanisms which include aldol condensation and dehydration. When coupled with these other base-catalysed reactions we refer to the process as thermal deoxygenation (TDO) because of the significant loss of oxygen that can occur.
We find that TDO of levulinic acid results in formation of cyclic and aromatic molecules with very low oxygen to carbon ratios. The kinetics of the TDO of levulinic acid are reported. Both the composition and physical properties of the products of TDO were characterized and the broad product distribution may facilitate upgrading to hydrocarbon fuels with a potential emphasis on aviation turbine fuels.
Fig. 1 compares results of thermogravimetric analysis (TGA) of anhydrous calcium acetate to hydrated calcium levulinate. Mass lost below 200 °C is assumed to be water. Calcium acetate is known to decompose to acetone with nearly 100% yield (reaction 1) around 400 °C, and the mass loss at 700 °C is consistent with reaction 2.
 | (1) |
 | (2) |
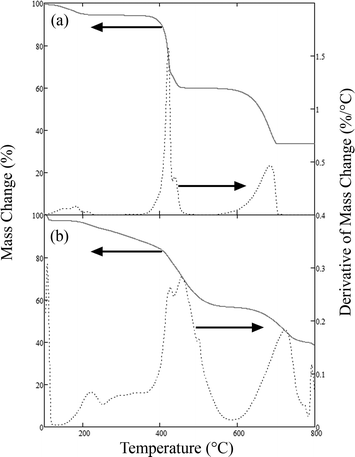 | ||
| Fig. 1 Comparison of thermal decomposition of calcium acetate (a) and calcium levulinate (b) in terms of mass loss on the left hand axis and rate of mass loss on the right hand axis (arrows denote which curve is associated with which axis). | ||
If calcium levulinate were to follow a similar ketonization mechanism to that proposed by Renz,18 we would expect:
 | (3) |
However, the TGA of calcium levulinate shows that decomposition occurs at temperatures ranging from 200 to 600 °C. This is much broader than calcium acetate, and suggests a more complicated reaction. The residual mass at 600 °C is not consistent with the formation of calcium carbonate as would be expected based on the TGA of calcium acetate. 11% of the mass of hydrated calcium levulinate is lost as water below 200 °C. Taking this into consideration, 37% of the anhydrous calcium levulinate is volatilized in the TGA. Calcium carbonate consists of an additional 37% of the mass of calcium levulinate. Thus, a further 26% by mass of the calcium levulinate remains in the residue. Total carbon analysis was performed on the char formed in the TGA at 600 °C, and the results indicated that 48% of the residue is carbon (organic or inorganic), consistent with formation of a carbonaceous char in addition to calcium carbonate. While the formation of char implies a decrease in energy efficiency of the TDO reaction itself, combustion of the char can provide energy required to regenerate calcium oxide from calcium carbonate in a lime kiln. It is also probable that char formation can be reduced by appropriate reactor design to improve mass transfer of the products out of the solid mass.
The rate of decomposition vs. temperature in the TGA between 200 and 600 °C indicates the existence of at least two reaction regimes. By calculating fractional conversion from the mass loss in the TGA, a kinetic analysis was performed using the distributed activation energy model (DAEM) presented in the ESI.† The DAEM was fit using a bimodal activation energy distribution, and average activation energies were calculated as 165 kJ/mol for the reactions in the 300–400 °C range and 212 kJ mol−1 for the reactions in the 400–600 °C range.
Proton and carbon NMR analysis of the products of TDO are not consistent with simple ketonization of levulinic acid (reaction 3). Table 1 presents the integration of the NMR spectra of the product of TDO at 450 °C. Both proton and 13C NMR show peaks in the range known to represent aromatic molecules. 40% of the carbon is contained in alkyl groups while 44% is contained in aromatic groups. The higher heating value of the liquid product generated at 450 °C was measured as 34.4 MJ kg−1.
GC/MS analysis of the products formed at both 350 °C and 450 °C is consistent with the NMR analysis. No long-chain, linear molecules were identified. Rather, the analysis revealed a variety of cyclic and aromatic compounds. Fig. 2 shows a comparison of two chromatograms, one for 350 °C and one for 450 °C, with the major peaks identified.
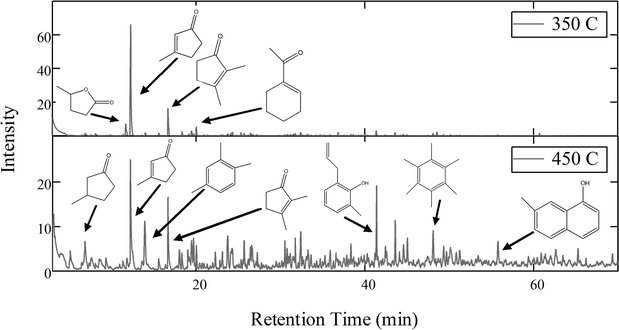 | ||
| Fig. 2 Comparison of GC/MS chromatograms of the hydrocarbon products of TDO. At 350 °C the product molecules tend toward lower molecular weight, substituted cyclic compounds. At 450 °C aromatic compounds are formed in addition to the cyclopentenones observed at 350 °C. | ||
The two temperature regimes explored have different product distributions. At the lower temperature, the majority of the identified products are substituted cyclopentenones. Formation of these products is consistent with intramolecular aldol condensation and dehydration within the ketone hypothesized in reaction 3 above. For example, the two most prominent products in the top chromatogram in Fig. 2 can be explained by Scheme 1.
 | ||
| Scheme 1 Possibilities for intramolecular aldol condensation and dehydration pathways resulting in molecules exhibiting significant deoxygenation identified in Fig. 2. | ||
The same aldol condensation and dehydration products are observed in the higher temperature chromatogram; however, there are also several higher carbon number molecules which cannot be explained by simple aldol condensation. Such molecules could be explained by a series of free-radical chain reactions which are likely to occur at high temperatures.
A mechanism for free radical formation and chain reaction has been proposed for the pyrolysis of ethylene–carbon monoxide copolymers.21 Ethylene–carbon monoxide copolymers have a structure which, when truncated, is identical to that of the ketone hypothesized in reaction (3). It would be possible, then, to form several small radicals which could feasibly result in the significantly deoxygenated C9–C12 aromatics found in the bottom chromatogram of Fig. 2.
To verify the importance of the calcium in TDO, a sample of pure levulinic acid was heated to 450 °C under a nitrogen atmosphere, and the products were condensed and analyzed by GC/MS. The major peak corresponded to levulinic acid, two minor peaks were identified corresponding to 5-methylhydrofuran-2-ones (cyclization and dehydration products), and a fourth peak was identified as 1,2-dimethylcyclopentane. Calcium, functioning as a base, must then facilitate the ketonic decarboxylation of levulinic acid, consistent with the mechanism described in a review by Renz.18 The resulting ketone is an intermediate in the overall reaction, and is further deoxygenated by aldol condensation and dehydration.
The energy yield of the products is estimated to be on the order of 80% of the heating value of the levulinic acid feed, and we estimate the energy efficiency that might be realized for the TDO process is on the order of 75%. The basis for calculation was production of 23% char, a product HHV of 35 MJ kg−1, and minimal CO2 formation. If char formation can be reduced, the energy yield could be nearly 100% with a process efficiency on the order of 85%. The process consists of four reaction steps: (1) slaking of calcium oxide, (2) neutralizing of levulinic acid, (3) thermal reaction of calcium levulinate, and (4) regeneration of calcium oxide in a lime kiln. Steps 1 and 2 are mildly exothermic, and step 3 is endothermic. Step 4 can be either exothermic or endothermic depending on the relative amount of char formed.
The pyrolytic nature of TDO promises a tolerance of varying composition and purity of a levulinic acid feedstock produced from municipal solid wastes. Furthermore, the economics of production of biofuels from calcium levulinate would be much improved when integrated within an existing industrial forest products complex such as a kraft pulp mill. Such a complex already operates a lime kiln, and has facilities for processing lignocellulosic biomass, treatment of waste water, and generation of steam and electricity from biomass.
Conclusions
We have presented a novel process for the thermal deoxygenation (TDO) of biomass. Functionalized organic acids such as levulinic acid can be neutralized with calcium hydroxide and then subjected to high temperatures to form significantly deoxygenated and energy densified products. We have determined kinetic parameters for the process when applied to levulinic acid and proposed a mechanism for the formation of the identified products.To be candidates for hydrocarbon transportation fuels, the TDO products will require further upgrading. Hydrodeoxygenation of the aqueous fraction of pyrolysis oils has been accomplished by hydrogenation and dehydration using Ru and Pt catalysts.22 Such hydrodeoxygenation of the TDO products would likely yield a feedstock similar in properties to the naptha fraction of crude oil which could then be further refined using existing petroleum processing technologies. Ring opening and isomerization could be accomplished with noble metal catalysts on acidic supports,23 and resulting C7–C12 paraffins would be suitable for use as a jet fuel blending agent.
Using waste lignocellulosic biomass, it may be possible to fuel a sizable fraction of the existing US transportation fleet without vehicle modification using a bio-based hydrocarbon source. While this may seem like an optimistic claim, the US Departments of Energy and Agriculture have proposed that it is possible to replace nearly 30% of the US petroleum consumption with bio-based fuels by 2030, using approximately 1 billion dry tons of biomass feedstock per year. This capacity exists with only modest improvements to land use and no disruption to food supplies.4 The use of domestically available biomass as a feedstock would not only improve the energy security of a country, it would provide a sustainable and carbon-neutral fuel which has the potential to help mitigate the effects of global climate change. Optimally such a process would be co-located at a kraft pulp mill.
Experimental
The TDO reaction was characterized using thermogravimetric analysis (TGA). Total carbon analysis of the char formed in the TGA was performed by the University of Maine Analytical Lab and Soil Testing Service. Full experimental details can be found in the ESI.† Products of TDO were characterized using several methods. Nuclear magnetic resonance spectroscopy was used to determine functionality present in the product mixture. GC/MS was used to identify individual products, and oxygen bomb calorimetry was used to obtain higher heating value.Acknowledgements
Funding for this work was provided by DOE grant #DE-FG02-07ER46373. The authors acknowledge valuable discussions with Peter van Walsum which contributed to development of the TDO approach. We also thank Paige Case for invaluable assistance with preparation and characterization of calcium levulinate, Bruce Jensen for help with developing the presented mechanism, Nick Hill and Amos Cline for their assistance in assembling experimental apparatus, Jincy Joseph, Cirila Baker, and Brian Frederick for their assistance in performing and interpreting NMR spectra, Mohit Bhatia for assistance with experiment design, and Elizabeth Stemmler for her assistance with GC/MS.Notes and references
- Ibrahim Sami Nashawi, Adel Malallah and Mohammed Al-Bisharah, Energy Fuels, 2010, 24, 1788–1800 CrossRef.
- Y. H. Percival Zhang, J. Ind. Microbiol. Biotechnol., 2008, 35, 367–375 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106(9), 4044–4096 CrossRef CAS.
- R. D. Perlack, L. L. Wright, A. F. Turhollow, R. L. Graham, B. J. Stokes and D. C. Erbach, Biomass as Feedstock for a Bioenergy and Bioproducts Industry: The Technical Feasibility of a Billion Ton Annual Supply, U.S. Department of Energy, 2005 Search PubMed.
- T. Werpy, G. Petersen, A. Aden, J. Bozell, J. Holladay, J. White, Amy Manheim, D. Eliot, L. Lasure and S. Jones, Top Value Added Chemicals From Biomass. Volume 1 – Results of Screening for Potential Candidates From Sugars and Synthesis Gas, U.S. Department of Energy, Washington, DC, 2004 Search PubMed.
- S. Fitzpatrick, ACS Symp. Ser., 2006, 921, 271–287 CAS.
- Bob McCormack, Presented at 8th Diesel Engine Emissions Reduction Conference, August 25–29, Coronado, CA, 2003.
- PNNL Hydrogen Analysis Resource Center. http://hydrogen.pnl.gov/cocoon/morf/hydrogen/site_specific/fuel_heating_calculator?canprint=false (accessed 3 March 2010).
- Jesse Q. Bond, David Martin Alonso, Dong Wang, Ryan M. West and James A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS.
- Juan Carlos Serrano-Ruiz, Dong Wang and James A. Dumesic, Green Chem., 2010, 12, 574 RSC.
- E. G. R. Ardagh, A. D. Barbour, G. E. McClellan and E. W. McBride, Ind. Eng. Chem., 1924, 16(11), 1133–1139 CrossRef CAS.
- Societe LeFranc et Cie, British Patent 216
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 120, February 26, 1925.
120, February 26, 1925. - S. K. Adelson, “Pyrolytic Decomposition of Volatile Fatty Acid Salts,” Master's Thesis, Texas A&M University, 1994.
- Mark T. Holtzapple, Richard R. Davison, Kyle M. Ross, Salvador Aldrett-Lee, Murlidhar Nagwani, Chang-Ming Lee, Champion Lee, Seth Adelson, William Kaar, David Gaskin, Hiroshi Shirage, Nan-Sheng Chang, Vincent S. Chang and Mitchell E. Loescher, Appl. Biochem. Biotechnol., 1999, 77–79, 609–631 CrossRef.
- M. T. Holtzapple, N. S. Chang, S. Aldrett and R. R. Davison, Chem. Eng. Sci., 2000, 55(23), 5721–5732 CrossRef.
- M. T. Holtzapple, P. Thanakoses and N. A. A. Mostafa, Appl. Biochem. Biotechnol., 2003, 105–108, 523–546.
- F. K. Agbogbo and M. T. Holtzapple, Appl. Biochem. Biotechnol., 2009, 132(1–3), 997–1014.
- Michael Renz, Eur. J. Org. Chem., 2005, 979–988 CrossRef CAS.
- Leon Rand, Walter Wagner, Peter O. Warner and L. Robert Kovac, J. Org. Chem., 1962, 27, 1034–1035 CrossRef CAS.
- S. Beis, S. Mukkamala, N. Hill, J. Joseph, C. Baker, B. Jensen, E. Stemmler, C. Wheeler, B. Frederick, A. van Heiningen, A. Berg and W. DeSisto, Bioresources, 2010, 5(3), 1408–1424 Search PubMed.
- Oscar Chiantore, Massimo Lazzari, Francesco Ciardelli and Silvia De Vito, Macromolecules, 1997, 30(9), 2589–2597 CrossRef CAS.
- Tushar P. Vispute and George W. Huber, Green Chem., 2009, 11, 1433–1445 RSC.
- G. B. McVicker, M. Daage, M. S. Touvelle, C. W. Hudson, D. P. Klein, W. C. Baird, Jr., B. R. Cook, J. G. Chen, S. Hantzer, D. E. W. Vaughan, E. S. Ellis and O. C. Feeley, J. Catal., 2002, 210, 137–148 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, kinetic analysis, and mass spectra tables. See DOI: 10.1039/c005067a |
| This journal is © The Royal Society of Chemistry 2010 |