Production of liquid hydrocarbon transportation fuels by oligomerization of biomass-derived C9 alkenes
David Martin Alonso, Jesse Q. Bond, Juan Carlos Serrano-Ruiz and James A. Dumesic*
University of Wisconsin-Madison, Department of Chemical and Biological Engineering, Madison, 53706, WI, USA. E-mail: dumesic@engr.wisc.edu; Fax: +1 608 262 5434
First published on 26th April 2010
Abstract
A process is described to produce renewable liquid fuels, similar to existing petroleum-derived transportation fuels, through the oligomerization over solid acid catalysts of C9-alkenes derived from γ-valerolactone (GVL). Larger, non-terminal alkenes are shown to be less reactive than short chain α-alkenes for oligomerization over solid acid sites, and Amberlyst-70 has been identified to be an active and stable catalyst with sufficient acidity to couple C9-alkenes. The inhibiting effect of water on alkene oligomerization can be minimized, because C9 alkenes derived from aqueous solutions of GVL separate spontaneously from water. The effect of other impurities arising from the cascade process for production of C9 alkenes from GVL, such as 5-nonanone and 5-nonanol, has been studied. Ketones are shown to be inert, while alcohols readily dehydrate on acid sites, producing an equivalent of water, which inhibits the rate of oligomerization. Small amounts of 5-nonanol present with C9-alkenes (< 1%) have a promotional effect, due to swelling of the catalyst by polar molecules; however, large amounts of 5-nonanol lead to inhibition of oligomerization. Other more reactive alkenes present in C9-alkenes produced from GVL, such as hexene and heptene isomers, compete for acid sites with the nonene feed. These smaller, more reactive alkenes are readily coupled at high conversion. Accordingly, with this process approximately 50 kg of liquid hydrocarbons can be produced from 100 kg of GVL retaining more than 90% of its energy content.
Introduction
The energy demands of the transportation sector are becoming increasingly difficult to fulfil in this era of diminishing fossil fuel resources1,2 and concerns about growing levels of CO2 in the atmosphere.3,4 For example, the transportation sector presently accounts for 28% of total annual energy consumption in the United States, with more than 95% of this energy supplied by petroleum, such that the transportation sector accounts for more than 70% of the petroleum consumed in the U.S. annually.5 This strong dependence on petroleum-derived fuels requires the development of new approaches for producing transportation fuels from renewable resources. Currently there are attractive, although long-term, alternatives to the internal combustion engine proposed for transportation applications, such as solar cells and hydrogen fuel cells. Nevertheless these technologies require further developments for widespread use in the transportation market, and successful implementation of such energy sources would require replacement of current combustion engines and construction of facilities to supply and distribute the appropriate fuels. Accordingly, the most immediate option to provide renewable energy for the transportation sector is direct substitution of petroleum by liquid bio-fuels that can be used with the established infrastructure.First-generation bio-fuels, such as bio-ethanol and bio-diesel, have shown that it is technically possible to replace petroleum (at least partially) by biomass-derived fuels. However, the demand for transportation fuels is sufficiently high that these bio-fuels can satisfy only a small portion of the energy needs by the transportation sector.6-8 Furthermore, first-generation bio-fuels utilize edible biomass as a feedstock, contributing to competition for food sources and increases in the cost of food production. Second-generation bio-fuels derived from lignocellulosic biomass have been proposed as alternatives to supply transportation energy, while not negatively impacting global food production.7 In this respect, levulinic acid, which can be obtained by acid hydrolysis of biomass, has been proposed as a renewable building block that can be transformed into various bio-fuels. Methyl and ethyl esters of levulinic acid have been proposed as blending agents for petroleum diesel.9 Levulinic acid can be hydrogenated to γ-valerolactone (GVL), an important intermediate which has been proposed as a substitute for blending of ethanol in gasoline at levels of 10% v/v,10 or converted into methyltetrahydrofuran (MeTHF), which can be blended up to 70% in gasoline.11 One of the drawbacks of these alternatives is that these compounds are limited to serving as blending agents, with petroleum-derived alkanes still being required as the main component of the fuels. Additionally, all of these alternative fuels suffer from low energy density and have limited applicability for use as jet or diesel fuels. Furthermore, they require the widespread introduction of oxygenated molecules in the transportation fuel infrastructure.
A new process has recently been proposed to upgrade GVL to liquid hydrocarbon fuels.12 In this process, GVL undergoes ring-opening and hydrogenation to produce pentanoic acid over a Pd/niobia catalyst, which is subsequently converted via ketonization to 5-nonanone over niobia and/or a ceria–zirconia catalyst.13 This C9 ketone can then undergo successive hydrogenation and dehydration to produce a mixture of linear C9 alkenes (nonenes), which can be hydrogenated to produce n-nonane for use in diesel fuel. One of the main advantages of the aforementioned processes for conversion of GVL to pentanoic acid and various C9 species is that in each step involving aqueous feeds or water by-products, the organic products separate spontaneously, thereby reducing the difficulty of purification steps. In the present paper, we explore another alternative to the above listed processes, which is oligomerization of C9 alkenes over an acid catalyst to produce C18 alkenes that can, upon hydrogenation, be used as jet fuel (Fig. 1).
 | ||
| Fig. 1 Reaction pathway for conversion of GVL to liquid transportation fuels. | ||
Oligomerization of small, terminal alkenes has been studied in the literature, using both homogeneous and heterogeneous catalysts;14-16 however, studies of the oligomerization of large, non-terminal alkenes (such C9 species) are limited, and generally utilize homogeneous catalysts.17 The coupling of non-terminal alkenes over solid acids is reported to be more difficult than analogous reactions of α-alkenes.18 Accordingly, we have investigated the effect of different reaction parameters, such as temperature, pressure, and weight-hourly space velocity (WHSV) on the oligomerization of C9 alkenes. With the goal of establishing an efficient process with minimal separation/purification requirements, we have paid attention to possible effects of feed impurities, caused by the cascade nature of this process, on the oligomerization of nonenes. In this respect, we have demonstrated that a stream of 5-nonanone produced from GVL can be successfully converted to C18 alkenes, without the need for complex separation steps. Overall mass balances for the production of jet fuel range hydrocarbons from both cellulose and GVL have been included.
2. Experimental
2.1. Materials
1-Nonanol (Sigma-Aldrich >98%), 5-nonanone (Alfa-Aesar, 98%), 1-hexene (Acros Organics 97%), 1-octene (Sigma-Aldrich 98%), trans-2-octene (Sigma-Aldrich 97%), trans-3-octene (Sigma-Aldrich 98%), trans-4-octene (Sigma-Aldrich 98%), 1-nonene (Fluka >99.5%), 1-decene (Fluka >99.5%), and γ-valerolactone (Sigma-Aldrich >98%) were used as received without further purification. Feeds of 5-nonanol were prepared by hydrogenation of 5-nonanone in a sealed stainless steel batch reactor (Parr Instruments). The reactor was charged with 2 g of Ru/C (Sigma-Aldrich) and 100 g of 5-nonanone in a 300 mL Teflon liner. The reactor was purged with He and heated to 423 K, followed by addition of H2 to a pressure of 40 bar, and stirring the solution at 1000 rpm. The reactor pressure was maintained by providing subsequent hydrogen doses as the reaction proceeded toward completion. Typically, nearly complete conversion of 5-nonanone with 100% selectivity to 5-nonanol was achieved in 24–30 h of reaction.Non-terminal nonenes were prepared by dehydration of 5-nonanol in a packed bed reactor operating in a down flow configuration. Initially, 2 g of crushed Amberlyst-70 (Rohm and Haas) was mixed with 12 g of carbon support (CABOT black pearls 1300), an inert packing material used to fill the reactor volume, and this mixture was loaded in a 0.5 inch stainless steel tubular reactor between two end-plugs of fused silica and quartz wool. The reactor was surrounded with aluminium blocks externally heated by an oven to ensure uniform bed temperature. The temperature was set at 413 K, and a continuous sweep of helium (60 cm3(STP) min−1) was passed through the reactor operating at atmospheric pressure to vaporize the nonene and prevent further oligomerization. A mass-flow controller (5850 Brooks Instruments) was used to control the flow rate of He, and an HPLC pump (Lab Alliance Series 1) was used to control the liquid flow of 5-nonanol to the reactor.
5-Nonanone was prepared from an aqueous solution of GVL in a dual-catalyst bed flow reactor, similar to the flow reactor described above. The feed to the reactor consisted of a 60 wt% solution of GVL in water at a flow rate of 0.05 mL min−1, along with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v He:H2 gas mixture at a rate of 50 cm3(STP) min−1. The total pressure was controlled with a back-pressure regulator (GO BP-60). In the first catalyst bed in the reactor, GVL was ring opened and hydrogenated to pentanoic acid over 2.5 g of Pd/Nb2O5 at 598 K and a pressure of 14 bar. In the same reactor but in a second catalyst bed, pentanoic acid was ketonized over 3 g of Ce0.5Zr0.5O2 at 698 K and a pressure of 14 bar. Mass-flow controllers (5850 Brooks Instruments) were used to control the flow rates of He and H2, and an HPLC pump (Lab Alliance Series 1) was used to control the liquid flow of GVL.
:1 v/v He:H2 gas mixture at a rate of 50 cm3(STP) min−1. The total pressure was controlled with a back-pressure regulator (GO BP-60). In the first catalyst bed in the reactor, GVL was ring opened and hydrogenated to pentanoic acid over 2.5 g of Pd/Nb2O5 at 598 K and a pressure of 14 bar. In the same reactor but in a second catalyst bed, pentanoic acid was ketonized over 3 g of Ce0.5Zr0.5O2 at 698 K and a pressure of 14 bar. Mass-flow controllers (5850 Brooks Instruments) were used to control the flow rates of He and H2, and an HPLC pump (Lab Alliance Series 1) was used to control the liquid flow of GVL.
Amberlyst-70 (Rohm & Haas Company) was rinsed prior to use with distilled water until the pH of the effluent solution stabilized and showed no indication of residual acidity. The Amberlyst-70 catalyst was then dried overnight at 393 K and stored in a glass vial at ambient conditions. A catalyst of sulfated zirconia, SO42−/ZrO2, was prepared by calcination in air at 773 K for 3 h of SO42−/Zr(OH)4 (Mel Chemicals). Catalysts consisting of H-ZSM-5 (Engelhard), SiO2/Al2O3 (Grace Davidson), and Nafion SAC 13 (Sigma-Aldrich) were used as received.
2.2. Reaction kinetics studies
Initial reaction kinetics studies of all catalysts and studies of the effect of chain length and double bond position in the oligomerization reaction were carried out in batch mode using 10 mL sealed glass vials (Alltech). In these studies, 2 g of feed and 0.3 g of catalyst were loaded into a vial, followed by sealing the vial and heating to the reaction temperature inside an oil bath. The solution in the vial was stirred vigorously using a magnetic stirrer. The vials were cooled after 4 h to room temperature and a liquid sample was filtered and analyzed.Studies of dehydration and oligomerization reactions were carried out in a packed bed flow reactor operating in a down flow configuration, similar to the flow reactor described above for dehydration of 5-nonanol, containing 2 g of crushed Amberlyst-70 mixed with 12 g of carbon support to fill the reactor volume. Helium was used as a sweep gas and to pressurize the reactor as necessary. The total pressure was controlled with a back-pressure regulator (GO BP-60). A syringe pump (Teldyne Isco Model 290D) was used to control the liquid flow of nonene to the reactor. The products were collected at room temperature in a gas–liquid separator (Jerguson Gage and Valve).
2.3. Product analyses
The liquid effluents from the reactors were analyzed using a gas-chromatograph (Shimadzu GC2010) with an FID detector and RTx-5 column. A Shimadzu GC-2010 with a mass spectrometer and DB-5ms column was used for product identification. Gas phase alkenes were analyzed using a gas-chromatograph (Agilent) with an FID detector and a RTc-5 column. Total carbon balances typically closed to within 10%. The extent of cracking for all the experiments was low, and less than 1% of the carbon appears in the gas phase. When alcohols are present in the feed, two liquid phases are collected, one organic and one aqueous phase. No carbon was identified in the aqueous phase. Based on experimental replicates, the errors associated with alkene conversion and oligomer yield are within 3% for batch experiments and within 2% for flow experiments.3. Results and discussion
3.1 Initial reactivity and stability studies
The potential for oligomerization of long-chain, non-terminal alkenes (such as 2,3, and 4-nonenes, referred to as nonene in the remainder of the text) obtained from the hydrogenation and subsequent dehydration of commercial 5-nonanone, was investigated by screening the reactivity of various solid acid catalysts in a batch reactor. Table 1 shows the results obtained with different solid acid catalysts. Silica-alumina was not active for the oligomerization of nonenes, while zeolite H-ZSM5 and sulfated zirconia showed measurable but very low activity. Nafion SAC 13 and Amberlyst-70 showed higher catalytic activities, achieving nonene conversions of 38% and 43%, respectively, after a holding time of 4 h at 433 K, and these catalysts were selected for further stability experiments. The product distributions indicate that nonene is mainly converted into dimers, although small amounts of trimers and larger oligomers were also present.| Entry | Catalyst | T (K) | Nonene conversion (%) | Alkenes product carbon distribution (% by mass) | |||||
|---|---|---|---|---|---|---|---|---|---|
| C9− | C9 | C10-C17 | C18 | C18+ | Others* | ||||
| *Ketones and alcohols (mainly unreacted 5-nonanol and 5-nonanone from previous reactions).a Feed with < 0.2% of 5-nonanol.b Feed with 4.9% of 5-nonanol.c Feed with 97% of 5-nonanol.d 8 h batch. | |||||||||
| 1 | Silicaa | 433 | 0 | 0 | 99 | 1 | 0 | 0 | 0 |
| 2 | HZSM5a | 433 | 1 | 0 | 98 | 1 | 0 | 0 | 1 |
| 3 | SZa | 433 | 1 | 0 | 98 | 1 | 0 | 0 | 1 |
| 4 | Nafion SAC 13a | 433 | 38 | 0 | 61 | 2 | 33 | 3 | 1 |
| 5 | Amberlyst-70a | 433 | 44 | 1 | 56 | 3 | 36 | 4 | 1 |
| 6 | Amberlyst-70 1st batchb | 433 | 45 | 0 | 53 | 4 | 37 | 4 | 2 |
| 7 | Amberlyst-70 2nd batchb | 433 | 46 | 0 | 52 | 3 | 39 | 4 | 2 |
| 8 | Amberlyst-70 3rd batchb | 433 | 10 | 0 | 86 | 1 | 9 | 1 | 2 |
| 9 | Amberlyst-70 4th batchb | 433 | 3 | 1 | 93 | 1 | 3 | 0 | 2 |
| 10 | Amberlyst-70 driedb | 433 | 55 | 0 | 43 | 2 | 48 | 5 | 1 |
| 11 | Amberlyst + H2Ob | 433 | 1 | 1 | 92 | 1 | 0 | 0 | 4 |
| 12 | Amberlyst-70 1st batcha | 433 | 42 | 1 | 56 | 2 | 35 | 4 | 1 |
| 13 | Amberlyst-70 4th batcha | 433 | 52 | 0 | 47 | 2 | 45 | 4 | 2 |
| 14 | Amberlyst-70c | 433 | 5 | 0 | 1 | 1 | 0 | 0 | 98 |
| 15d | Amberlyst-70a | 433 | 90 | 0 | 10 | 6 | 71 | 11 | 1 |
Entries 6 to 9 show the nonene conversion in successive runs at 433 K reusing the same Amberlyst-70 catalyst without any regeneration or wash between runs. The conversion is similar in the first two runs, but it decreases in the 3rd and 4th runs. This deactivation of the catalyst can be attributed either to the leaching of active species or to inhibition by water, the latter of which has been reported previously.19 In the present study, nonene was prepared by dehydration of 5-nonanol, resulting in a stoichiometric water by-product. Even though nonene and water separate spontaneously and it is not necessary to purify the nonene product to remove water, trace quantities of nonanol present in the nonene feed undergo dehydration over the acid catalysts and thus produce water at the reaction conditions tested for oligomerization. Given its pronounced effect on oligomerization activity, the subsequent accumulation of this small amount of water during successive batch experiments could be responsible for the observed decrease in catalytic activity. It can be seen in entry 10 that the catalytic activity can be recovered by drying the Amberlyst-70 at 443 K for 4 h, indicating that deactivation is not caused by leaching of active species, but rather it is associated with inhibition by water. After in situ drying, an enhancement in the activity can be observed, which can potentially be attributed to physical changes in the catalyst (Amberlyst) upon exposure to water in the reaction medium. In agreement with this conclusion, entry 11 shows that the addition of an equimolar amount of water (i.e., water:nonene=1:1) to the catalyst is sufficient to completely suppress the reaction. To minimize the quantity of water present, nonene with less than 0.2 wt% of nonanol was subsequently used as the feed. In this case the catalyst retains its initial activity for 4 subsequent cycles without regeneration (entries 12,13). Entry 14 shows that coupling the dehydration and oligomerization reactions by using 5-nonanol as the feed to the batch reactor is not a viable processing option, because of extensive inhibition of the reaction caused by the equimolar amount of water. A longer batch reaction (8 h) was carried out (Entry 15) to show that high conversions (90%) can be achieved with minimal degradation products. However, increased residence times cause a shift toward heavier oligomeric products. At these conditions, we observe that 11% wt% of the product is present as C18+ alkenes, predominately C27.
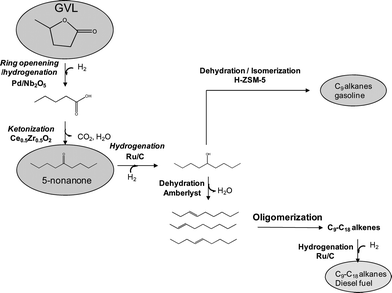
A potential alternative to minimize effects of water inhibition and to allow the direct use of nonanol as the feed to the oligomerization process is to use a continuous flow system with a gas sweep through the reactor. This approach would (i) remove the water generated during nonanol dehydration and (ii) decrease the partial pressure of the water in the reactor. Commercial 1-nonanol was selected to study oligomerization activity and catalyst stability using a flow reactor with a sweep gas through the reactor. The use of this commercial feed eliminates the possible influence of residual impurities from the preparation of nonene from 5-nonanone and 5-nonanol. Fig. 2a shows that it is possible to dehydrate 1-nonanol (>96% conversion) to form nonene using a continuous flow system with 40 cm3(STP) min−1 of helium as sweep gas, and also to achieve oligomerization to heavier alkenes (>80% conversion) in a single step using Amberlyst-70 as catalyst. It can be seen in Fig. 2b that if a flowing gas sweep is not used, then the rates of dehydration and oligomerization decrease with time and stabilize after 40 h at levels that are 3 times lower than those achieved using a gas sweep. Without the inclusion of a sweep gas for water removal, the conversion of 1-nonanol by dehydration decreases to 30%. Due the high reactivity of terminal alkenes and the decrease in the amounts of nonene and water produced from dehydration, the conversion of nonene by oligomerization increases to 95%, indicating that the rate of nonene oligomerization is limited by the rate of nonanol dehydration. Similar experiments were carried out using Nafion SAC 13 as catalyst. It was observed that the activity decreases with time on stream, and it was not possible to recover the activity after drying the catalyst. This results suggests that the deactivation is probably associated with leaching of active species, and further experiments were not carried out using this catalyst.
 | ||
| Fig. 2 Reaction rate for 1-nonanol dehydration (squares) and subsequent oligomerization (circles) versus time-on-stream. Catalyst: Amberlyst-70, 1 bar, 433 K, and 1.25 h−1 (feed, 145 μmol gcat−1 min−1). (a) Flowing 40 cm3(STP) min−1 of He. (b) No sweep gas. | ||
3.2. Effect of the length and double bond position
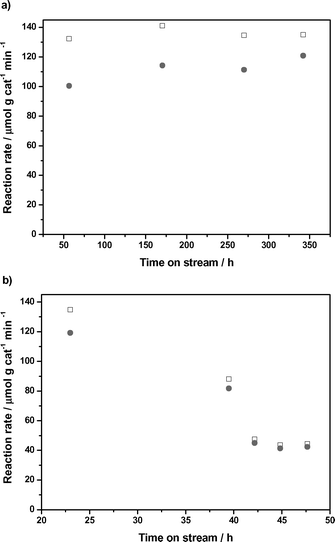
The difficulty in achieving dehydration and oligomerization of 1-nonanol is exacerbated when using 5-nonanol as the feed. At the same conditions used above for conversion of 1-nonanol, we observe that while 100% of the 5-nonanol is converted to nonenes, the rate of oligomerization is 50 times lower than that observed during 1-nonanol conversion. Even when we prepare a feed of nonene from 5-nonanol at conditions leading to less than 0.2 wt% of 5-nonanol, we find that the oligomerization reaction rate in the absence of a gas sweep and in the absence of water is still two times lower than that achieved using 1-nonanol as the feed. It has been reported for butene oligomerization that the rate of oligomerization of non-terminal alkenes is lower than that of α-alkenes.20As biomass-derived nonene is produced through dehydration of 5-nonanol, the oligomerization feed will necessarily be a mixture of (largely non-alpha) nine carbon alkenes. To study the effect of double bond position and chain length, batch experiments were conducted using a variety of alkene feeds. Fig. 3 shows conversion observed for a series of C8 alkenes (trans-1,2,3, and 4 octenes). The rate of oligomerization is higher for 1-octene (38%) than for octene isomers having an internal double bond, e.g., almost double the rate observed for 4-octene (22%). The differences are less significant among the isomers, 2-octene (27%) and 3-octene (25%). It is evident that the position of the double bond affects the rate of alkene oligomerization, and it is more difficult to oligomerize non-terminal alkenes.
 | ||
| Fig. 3 Effect of the position of the double bond in a C8 alkene in oligomerization conversion. Catalyst: Amberlyst-70, 423 K, 4 h batch reaction. | ||
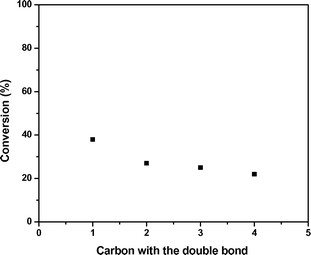
Fig. 4 shows the conversions observed for oligomerization of 1-hexene, 1-octene, 1-nonene, and 1-decene. It can be seen that chain length also has a pronounced effect, and that the rate of oligomerization decreases when the length of the alkene is increased. Similar effects have been previously reported for other reactions in the presence of methanol, where the decrease in rate has been attributed to steric factors21 and changes in molecular polarity.22 Batch experiments conducted using a mixed feed of 50 wt% 1-octene and 1-decene demonstrated that feed composition has an interesting effect on alkene reactivity and product distribution through cross coupling of alkenes of various molecular weight. In the case of 1-octene and 1-decene, the main products observed are C18 (42% of products) and C16 (40%). This cross-coupling affects the conversions observed for individual monomers, such that 1-octene conversion decreases from 38% to 32% while the conversion of 1-decene increases from 5% to 10% as decene monomers preferentially couple with more reactive octene monomers. From this study, it can thus be concluded that alkene oligomerization becomes increasingly difficult for large, non-terminal alkenes, highlighting the importance of documenting an optimized strategy for coupling nonene via acid-catalyzed oligomerization.
 | ||
| Fig. 4 Effect of the length of α-alkenes in oligomerization conversion. Catalyst: Amberlyst-70, 423 K, 4 h batch reaction. | ||
3.3. Effects of reaction conditions
Table 2 shows the conversion and product distribution achieved using a continuous flow reactor at different reaction conditions. To eliminate possible effects of inhibition by water, we have used a nonene feed containing less than 0.2 wt% of 5-nonanol. Entries 1–3 show that it is possible to increase the conversion of nonene by reducing the WHSV, which has an important effect on the final product composition. When the WHSV is reduced from 2.19 to 0.44 h−1, the rate of nonene conversion is decreased from 4.35 to 2.53 mmoles of nonene per gram of catalyst per hour. The concentration of C18 dimers inside the reactor increases with residence time, leading to competitive adsorption of C9 and C18 species on the acid sites, giving rise to further reaction to produce C27 trimers. In particular, the mass ratio of C18 dimers to C27 trimers decreases from 8 to 5 at the lower space velocity. A decrease in the reaction temperature by 20 K leads to a decrease in conversion by a factor of 3, and a decrease in the percentage of trimers in the final product by a factor of four (Entry 4). At these low temperatures, cracking reactions are negligible, and in all the cases the amounts of C9- and C10-C17 alkenes in the final product are lower than 2% and 5%, respectively.| Entry | WHSV (h−1) | P (bar) | T (K) | Nonene conversion (%) | Alkenes product carbon distribution (% by mass) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| C9− | C9 | C10–C17 | C18 | C18+ | Other* | |||||
| Feed | 1 | 94 | 1 | 2 | 0 | 2 | ||||
| *Ketones and alcohols (mainly unreacted 5-nonanol and 5-nonanone from previous reactions).a Flow of 40 cm3(STP) min−1 of He | ||||||||||
| 1 | 2.19 | 1 | 433 | 25 | 1 | 70 | 3 | 22 | 3 | 2 |
| 2 | 1.08 | 1 | 433 | 37 | 2 | 59 | 4 | 28 | 5 | 2 |
| 3 | 0.44 | 1 | 433 | 73 | 2 | 25 | 5 | 57 | 11 | 1 |
| 4 | 0.44 | 1 | 413 | 27 | 1 | 68 | 3 | 24 | 2 | 2 |
| 5 | 0.44 | 1 | 423 | 41 | 1 | 56 | 3 | 35 | 4 | 1 |
| 6 | 0.44 | 14 | 423 | 61 | 1 | 37 | 2 | 47 | 13 | 1 |
| 7a | 0.44 | 1 | 423 | 7 | 1 | 88 | 1 | 8 | 0 | 2 |
When the pressure of the reactor was increased to 14 bar under helium (entry 6), the conversion was observed to increase from 41% to 61%. It is known that the rate of alkene oligomerization is higher in the liquid phase than in gas phase,15 and an increase in pressure to 14 bar is sufficient to retain nonene (b.p. ≈ 423 K) in the liquid phase at the reaction temperature. The increase in the partial pressures of the alkenes in the reactor favors adsorption of nonene and products, increasing the surface concentration of dimers and thus resulting in the formation of larger amounts of trimers. If the nonene partial pressure is decreased by introducing a helium sweep through the reactor, then the conversion of nonene decreases to 7% (Entry 7). At these conditions the surface concentration of dimers is very low and no trimers are observed.
3.4. Effect of the impurities from GVL conversion
An important challenge facing the seamless integration of biomass upgrading strategies is the documentation and control of potential negative effects that feed impurities can have on the process. In the pathway proposed in Fig. 1, the main impurities expected to be present in the nonene feed will be 5-nonanone and 5-nonanol. To isolate the effect of 5-nonanone on oligomerization activity, a commercial source of 1-nonanol was used as the feed for the oligomerization process. Fig. 5 shows that addition of 5-nonanone (to levels of 5 wt%) has no effect on either the rate of nonanol dehydration or the rate of oligomerization of the resulting nonene product. Analysis of the final product stream shows the presence of 5-nonanone at the same concentration as in the feed, confirming that 5- nonanone can be considered to be inert under the conditions studied. The effect of nonanol impurities in nonene is more complex, because nonanol undergoes dehydration over acid sites, producing water which we have demonstrated to inhibit nonene oligomerization. Fig. 6 shows that the presence of small amounts of nonanol (e.g., 1 wt%) in the feed cause a slight increase in the conversion of nonene. This effect can be related to the small amount of water produced by nonanol dehydration. Amberlyst-70 is a polymeric resin that swells in polar solvents,23 which changes its physical properties and causes an increase in its catalytic activity. It has been reported that the BET surface area of Amberlyst-70 increases by 4 orders of magnitude when it swells in a polar solvent like water.24 This effect of swelling could compensate the inhibiting effect caused by trace quantities of water. A further increase in the amount of 5-nonanol to 6 wt% causes a decrease in nonene conversion, although the conversion is still higher than that for conversion of pure nonene. The inhibiting effect of water is evident when pure 5-nonanol is used as the feed, where the conversion is 3 times lower than that observed for pure nonene. This result indicates that in the presence of equimolar quantities of water, nonene oligomerization is severely inhibited despite any potential benefit upon swelling to the physical structure of the catalyst.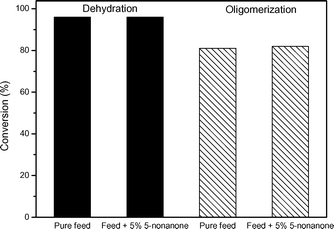 | ||
| Fig. 5 Effect of the presence of 5-nonanone in the feed in the dehydration of 1-nonanol and subsequent oligomerization. Catalyst: Amberlyst-70, 1 bar, 433 K, and 1.25 h−1. | ||
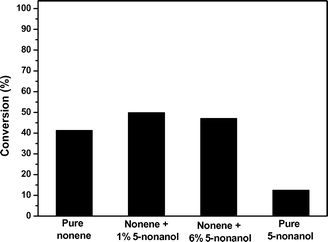 | ||
| Fig. 6 Effect of the presence of 5-nonanol in the feed in the oligomerization of C9 alkenes. Catalyst: Amberlyst-70, 1 bar, 423 K, and 0.44 h−1. | ||
3.5. Use of GVL derived from 5-nonanone
As indicated in Fig. 1, an organic stream enriched in 5-nonanone (that spontaneously separates from water) can be produced from aqueous solutions of GVL. The reaction takes place with the intermediate formation of pentanoic acid (over a bifunctional Pd/Nb2O5) and subsequent ketonization of the carboxylic acid over ceria-zirconia. Although the C9-ketone can be produced with high yields (around 85%) at optimized conditions,12 several side reactions can take place in the combined process. Thus, the 5-nonanone-enriched organic layer contains impurities that could affect the subsequent nonene oligomerization step. The main impurities detected in the organic phase were lower n-alkanes (in the C4–C5 range, produced by decarbonylation and hydrogenation of pentanoic acid) and lower linear ketones (2-hexanone and 3-heptanone produced by scission of 5-nonanone in the α and β positions). The formation of these unwanted products can be controlled by adjusting reaction conditions and/or catalyst formulation. For example, by decreasing the Pd loading over niobia (to the level of 0.1 wt%) the formation of the decarbonylation/hydrogenation products can be minimized, and only 5% of the carbon fed as GVL is converted to butane and pentane. As a consequence of the working pressure in the system, these gaseous butane and pentane by-products appeared in the organic liquid effluent phase. Importantly, due to the unfunctionalized nature of these by-products, we do not anticipate any detrimental effect on the nonene oligomerization step, other than a possible minimal dilution effect. On the other hand, the challenge in the pentanoic acid ketonization process is to control the degree of scission of the resulting C9-ketone which leads to the onset of lower ketones. Unlike the formation of pentanoic acid from GVL, we found that complete ketonization of pentanoic acid to 5-nonanone required high temperatures (698 K). At these conditions, the control of the space velocity is important to avoid scission of the C9 ketone. We observed, for example, that 5–10% of the carbon fed as GVL formed hexanone and heptanone in the organic layer.To demonstrate the feasibility of an integrated process, GVL-derived ketones (primarily 5-nonanone), prepared as described in the preceding section, were processed via hydrogenation and dehydration as described previously for commercial 5-nonanone feeds. The resulting product was rich in nonene isomers (83%, molar basis) and contained small quantities of the anticipated impurities such as hexenes (6%), heptenes (7%), 2-hexanone (<1%), 3-heptanone (<1%), 2-hexanol (<1%), 3-hexanol (<1%), 5-nonanone (3%), and 5-nonanol (<1%). This GVL-derived alkene stream was then fed to a downflow oligomerization reactor packed with Amberlyst-70. The reactor was operated at a 423 K and 14 bar with a WHSV of 0.44 h−1, which were the optimal conditions for oligomerization reported in section 3.3. We observed a lower conversion of the nonene fraction (48%) compared to that achieved using a pure 5-nonanone derived feed (61%), and higher conversions of the shorter, more reactive C6 and C7 alkenes (80% and 70% respectively). The predominant feed impurities do not adversely affect the reaction; rather, they are coupled alongside the nonene to form larger, C15–C16 alkenes. In Fig. 7 it can be observed that after 50 h on stream the decrease in activity for all of the alkenes is more significant, and we observed 25% conversion for C9, 55% for C6, and 45% for C7. This decrease in conversion can be directly associated with the accumulation of water produced by the dehydration of the different alcohols, as previously reported; however, further experiments must be carried out to determine possible effect of traces of other minor impurities present in the sample.
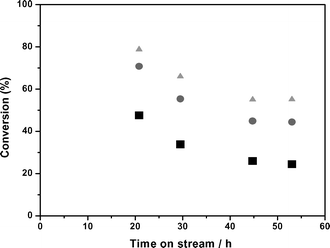 | ||
| Fig. 7 Conversion for GVL-derived alkenes versus time. Catalyst: Amberlyst-70. 14 bar, 423 K, and 0.44 h−1. Nonenes (squares); heptenes (circles); hexenes (triangles). | ||
3.6 Overview of final yields for the process
It has been demonstrated that the reactions shown in Fig. 1 can be carried out with good yields. Furthermore, the aqueous and organic products separate spontaneously with no carbon detected in the aqueous phase, thus eliminating the need for expensive separation/purification steps. At optimal reaction conditions, in the first bed of a dual-bed system, pentanoic acid can be produced from GVL with 92% total carbon yield. This organic phase can be upgraded to ketones over a second bed with a carbon yield of 90%.12 Experimentally, we have observed that 5-nonanone can be converted to 5-nonanol with yields over 99%. Comparably high yields of nonene can be obtained from 5-nonanol dehydration. Considering the aforementioned yields and that 1 mole of CO2 is produced in the ketonization of 2 moles of pentenoic acid, using a basis of 100 kg of GVL, it is possible to produce 53 kg of C9 alkenes that can be hydrogenated to produce alkane fuels or upgraded via oligomerization. In the production of 5-nonanone, the predominate side reaction is C–C scission of the C9 product to C6 and C7 ketones. However, these ketones are easily processed together with the C9 product to produce a more diverse oligomerization feedstock, which broadens the molecular weight range of alkenes in the final oligomer distribution. These side products increase the total quantity of upgradeable alkenes available to 57 kg per 100 kg of GVL. Depending on the space velocity used in the oligomerization reaction, over 90% conversion of the alkene feed can be achieved. Experimental data (Entry 15 Table 1) indicate that at high conversions (90%), the final oligomerization product contains 11% by mass of C18+ alkenes, which would require additional processing to be appropriate for transportation fuel blends. These heavier compounds would reduce the direct yield of alkenes in the transportation fuel range to 50 kg per 100 kg of GVL, although larger alkenes can still be processed through conventional refining methods. However, because its higher energy density, these 50 kg of alkenes would retain more than 90% of the energy content in 100 kg of GVL.The Biofine process25,26 can be used to produce GVL-derived alkenes from cellulosic feedstocks, according to which 50% of the total mass of C6 sugars can be converted to levulinic acid, that can be hydrogenated quantitatively (yields >97%27-29) to GVL. On this basis, approximately 24.5 kg of liquid fuels could be produced from 100 kg of cellulose. This amount is smaller than the amount of ethanol (30 kg)30 or MTHF (31 kg)31 that can be produced from 100 kg of cellulose. However, this slightly lower yield is compensated by the higher heat of combustion per kg of the alkenes produced, 1.3 and 1.6 times higher than MTHF and ethanol, respectively. In comparison to triglyceride-based pathways for alkane production, high yields of jet fuel range products have been reported through decarboxylation/hydrotreating of vegetable oils (71% by mass32). However these processes typically occur at high pressures (50 bar), and it has been estimated by Li, et al.33 that approximately 10–13.5 moles of H2 are required per mole of triglyceride for the decarboxylation/hydrotreating of triglyceride-based feedstocks. Similar numbers are reported for aqueous phase processing options based on aldol condensation and dehydration–hydrogenation schemes. For example, the production of C15 alkanes through condensation of HMF–acetone–HMF would consume 14 moles of H2.34 The process reported here requires of 7 moles of external H2 for the preparation of one mole of octadecane from cellulose, thus reducing the hydrogen demand considerably from other alternative pathways. Thus, GVL upgrading to alkenes offers a potentially useful strategy for processing plant waste/lignocellulosic residues from the non-edible fraction of the biomass, which is less expensive and more abundant than triglyceride feedstocks.
Conclusions
We have demonstrated that it is possible to achieve good yields of alkenes in the range of C18–C27 from oligomerization of C9 alkenes produced from biomass-derived GVL, even though the rate of oligomerization of non-terminal C9 alkenes is slower than that of shorter, terminal alkenes. These C18–C27 compounds are in the molecular weight range suitable for jet and diesel fuel applications. The extent of oligomerization can be modified by varying the reaction conditions to yield product distributions tailored to various applications. For example, if a jet fuel range product is desired, a product rich in linear C9 species and branched C18 species can be produced by operating at lower temperatures and elevated space velocities.We have observed that oxygenated compounds like 5-nonanone (a potential impurity present in streams of C9 alkenes produced from biomass-derived GVL) have no effect on the rate of oligomerization. Alcohols in the feed, such as 5-nonanol, can present a problem for oligomerization, because they undergo dehydration under reaction conditions to produce water, which has a strongly inhibiting effect on the rate of alkene oligomerization. Interestingly, because Amberlyst-70 swells in polar media to produce a higher surface area, the presence of small amounts of nonanol leads to an increase in the rate of oligomerization. This effect and the fact that nonene separates spontaneously from water, allow the production of diesel and jet fuel range alkenes from 5-nonanone without the need for complicated purification strategies. Overall, we estimate that it is possible to produce approximately 50 kg of C9–C18 alkenes from 100 kg of GVL retaining more than 90% of its energy content.
Acknowledgements
This work was supported through funding from the Defense Advanced Research Projects Agency (Surf-cat: Catalysts for Production of JP-8 range molecules from Lignocellulosic Biomass). The views, opinions, and/or findings contained in this article/presentation are those of the author/presenter and should not be interpreted as representing the official views or policies, either expressed or implied, of the Defense Advanced Research Projects Agency or the Department of Defense. In addition, this work was supported in part by the U.S. Department of Energy Office of Basic Energy Sciences.Notes and references
- D. Goswami and F. Kreith, in, Handbook of energy efficiency and renewable energy, ed. D. Goswami and F. Kreith, CRC Press, Boca Raton, FL, 2007, pp. 1–24 Search PubMed.
- S. Shafiee and E. Topal, Energy Policy, 2009, 37, 181–189 CrossRef.
- European Enviromental Agency. European Community and Member States Greenhouse Gas Emission Trends 1990–1998. Luxembourg, 2000 Search PubMed.
- C. Rosenzweig, D. Karoly, M. Vicarelli, P. Neofotis, Q. Wu, G. Casassa, A. Menzel, T. L. Root, N. Estrella, B. Seguin, P. Tryjanowski, C. Liu, S. Rawlins and A. Imeson, Nature, 2008, 453, 353–357 CrossRef CAS.
- Energy Information Administration, Annual Energy Review 2008, U.S. Department of Energy, Washington DC 2008. http://www.eia.doe.gov/emeu/aer/pdf/aer.pdf Search PubMed.
- D. J. Hayes, Catal. Today, 2009, 145, 138–151 CrossRef CAS.
- G. W. Huber, Breaking the Chemical and Engineering Barriers to Lignocellulosic Biofuels: Next Generation Hydrocarbon Biorefineries, U. Massachusetts Amherst, 2007. http://www.ecs.umass.edu/biofuels/Images/Roadmap2-08.pdf Search PubMed.
- J. R. Bartle and A. Abadi, Energy Fuels, 2010, 24, 2–9 CrossRef CAS.
- Texaco/NYSERDA/Biofine, Ethyl Levulinate D-975 Diesel Additive Test Program. Glenham, NY, 2000 Search PubMed.
- I. T. Horvàth, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–498 CrossRef CAS.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574–577 RSC.
- M. Renz, Eur. J. Org. Chem., 2005, 979–988 CrossRef CAS.
- A. de Klerk, Ind. Eng. Chem. Res., 2005, 44, 3887–3893 CrossRef CAS.
- J. P. G. Pater, P. A. Jacobs and J. A. Martens, J. Catal., 1998, 179, 477–482 CrossRef CAS.
- R. J. Quann, L. A. Green, S. A. Tabak and F. J. Krambeck, Ind. Eng. Chem. Res., 1988, 27, 565–570 CrossRef CAS.
- J. Skupinska, Chem. Rev., 1991, 91, 613–648 CrossRef CAS.
- J. F. Knifton, J. R. Sanderson and P. E. Dai, Catal. Lett., 1994, 28, 223–230 CrossRef CAS.
- J. Q. Bond, D. Martin Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS.
- W. Keim, B. Hoffmann, R. Lodewick, M. Peuckert, G. Schmitt, J. Fleischhauer and U. Meier, J. Mol. Catal., 1979, 6, 79–97 CrossRef CAS.
- Y. Liu, E. Lotero and J. G. Goodwin Jr, J. Catal., 2006, 243, 221–228 CrossRef CAS.
- D. Martin Alonso, M. L. Granados, R. Mariscal and A. Douhal, J. Catal., 2009, 262, 18–26 CrossRef.
- P. R. Siril, H. E. Cross and D. R. Brown, J. Mol. Catal. A: Chem., 2008, 279, 63–68 CrossRef CAS.
- R. Bringue, M. Iborra, J. Tejero, J. F. Izquierdo, F. Cunill, C. Fite and V. J. Cruz, J. Catal., 2006, 244, 33–42 CrossRef CAS.
- US Pat., 4897497S, 1990.
- US Pat., 5608105, 1997.
- H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49–54 CrossRef CAS.
- H. Heeres, R. Handana, D. Chunai, C. B. Rasrendra, B Girisuta and H. J. Heeres, Green Chem., 2009, 11, 1247–1255 RSC.
- L. Deng, J. Li, D. M. Lai, Y. Fu and Q. X. Guo, Angew. Chem., Int. Ed., 2009, 48, 6529–6532 CrossRef CAS.
- L. R. Lynd, J. H. Cushman, R. J. Nichols and C. E. Wyman, Science, 1991, 251, 1318–1323 CrossRef CAS.
- D. J. Hayes, S. Fitzpatrick and M. H. B. Hayes, in Biorefineries-Industrial Processes and Products. Status Quo and Future Directions, ed. B. Kamm, P. R. Gruber and M. Kamm, WILEY-VCH, Weinheim, 2006, vol. 1 Search PubMed.
- G. W. Huber, P. O'Connor and A. Corma, Appl. Catal., A, 2007, 329, 120–129 CrossRef CAS.
- L. X. Li, E. Coppola, J. Rine, J. L. Miller and D. Walker, Energy Fuels, 2010, 24, 1305–1315 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |