Highly selective hydrogenation of aromatic chloronitro compounds to aromatic chloroamines with ionic-liquid-like copolymer stabilized platinum nanocatalysts in ionic liquids
Xiao Yuana, Ning Yanab, Chaoxian Xiaoa, Changning Lia, Zhaofu Feib, Zhipeng Caia, Yuan Kou*a and Paul J. Dyson*b
aPKU Green Chemistry Centre, Beijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, China. E-mail: yuankou@pku.edu.cn; Fax: +86 10 62751708; Tel: +86 10 62757792
bInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015, Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch; Fax: +41 21 693 98 85; Tel: +41 21 693 98 54
First published on 18th January 2010
Abstract
Platinum nanoparticles (PtNPs stabilized by an ionic-liquid-like-copolymer (IP) immobilized in various ionic liquids (ILs)) effectively catalyze the selective hydrogenation of aromatic chloronitro compounds to aromatic chloroamines, a reaction of considerable commercial significance. The preparation of 2,4-dichloro-3-aminophenol (DAP) has been primarily studied due to its important industrial applications. DAP is usually prepared from 2,4-dichloro-3-nitrophenol (DNP) by reduction with hydrogen using Ni- or Pt-based catalysts. Compared to reactions in molecular (organic) solvents, the ILs system provides superior selectivity with functionalized ILs containing an alcohol group demonstrating the best recyclability, and ultimately achieving a turnover number of 2025 which is 750 fold higher than Raney nickel catalyst. A universal catalyst–ionic liquid system for the conversion of aromatic chloronitro compounds to aromatic chloroamines was also established. TEM, XPS, IR spectroscopy were used to characterize the morphology of the nanocatalysts allowing their structure to be correlated to their activity.
Introduction
Hydrogenation reactions catalyzed by transition metal nanoparticles (NPs) immobilized in ionic liquids (ILs) have been extensively explored in recent years.1-6 Nanoparticles composed of first row transition metals including Co7 and Ni,8 and second and third row transition metals including Ru,9-16 Rh,7,12,17-23 Ir,7,12,17,21,24-30 Pd,18,31-34 and Pt,18,35-37 have been successfully immobilized in ILs and evaluated in different hydrogenation reactions. NPs suspended in ILs have also been used as catalysts38 and catalyst reservoirs39-41 in a range of other reactions. The advantage of these ‘soluble’ NP–ILs systems in catalysis corresponds to their high activity, selectivity and recyclability. In some cases the ILs themselves behave as effective NP stabilizers, as demonstrated by Dupont et al., nevertheless it has been shown that additional stabilizers can further enhance catalytic performance in certain reactions.3,19 Polyvinyl pyrrolidone (PVP) is the most commonly used NP stabilizer, but due to its poor solubility in ILs is only of limited use. Two strategies have previously been used to overcome this limitation. First, an ‘IL-like’ copolymer formed from the copolymerization of the PVP monomer and a vinylimidazolium unit is highly IL soluble,19 and second, hydroxyl-functionalized ILs effectively dissolve PVP.23,42-44 Both approaches were found to be highly effective, providing stable IL soluble NPs, which exhibit long lifetimes leading to high turnover numbers in hydrogenation reactions.NP-IL systems also exhibit excellent regioselectivity in the hydrogenation of complex substrates and also inhibit side reactions such as dehalogenation. For example, o-chloroaniline may be obtained in quantitative yield from o-chloronitrobenzene using a NP–IL system, whereas considerable dehalogenation is observed in molecular solvents.37 DFT calculations combined with IR spectroscopy showed that the exceptional selectivity arises from strong interactions between the IL cation and the nitro group of the substrate, thereby activating it selectively. Similarly, the selective hydrogenation of cinnamic aldehyde to cinnamyl alcohol has been demonstrated using a NP–IL system.45
The selective hydrogenation of aromatic chloronitro compounds to aromatic chloroamines is a reaction of considerable commercial significance. Prins et al. published an alternative method55 to reduce aromatic nitro compounds with hydrazine in the presence of an iron oxide catalyst and high selectivity was obtained. However, the disadvantage of this method is the danger of handling hydrazine. Notably, 2,4-dichloro-3-aminophenol (DAP) is a fine chemical with a high added-value usually prepared from 2,4-dichloro-3-nitrophenol (DNP) by reduction with hydrogen using Ni- or Pt-based catalysts (Scheme 1). The selective reduction of DNP to DAP is much more challenging than o-chloronitrobenzene, since it has four substituents attached to the aromatic ring, and there is a greater probability of dehalogenation side reactions taking place. Many patents and papers describe the preparation and use of DAP,46-48 nevertheless the catalytic efficiencies described tend to be poor. For example, the turnover number (TON) of Raney Ni in ethanol is around 3 with a selectivity of 70%. Based on the promise shown by NP–IL systems in selective hydrogenation reactions, especially for selectivity towards nitro-groups, we decided to evaluate their application in the reduction of DAP to DNP. Herein we describe the outcome of this study which shows that a PtNP–IL system exhibits excellent selectivity toward the desired product. Moreover, using functionalized ILs a TON of 2025 with a selectivity of 100% was achieved. The same catalyst system may also be used to convert a range of aromatic chloronitro compounds to aromatic chloroamines.
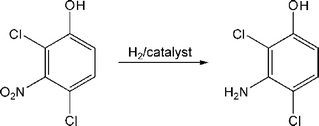 | ||
| Scheme 1 Catalytic synthesis of 2,4-dichloro-3-aminophenol via hydrogenation of 2,4-dichloro-3-nitrophenol. The synthesis is usually accompanied by significant dechlorination side-reactions. | ||
Experimental
Synthesis of the copolymer and ILs
The ionic-liquid-like-copolymer (IP)19 and ILs [Bmim][PF6], [Bmim][BF4], [C2OHmim][BF4], were prepared according to literature methods.49Nanoparticle preparation
PtNPs were prepared using a published method.50 In brief, NaOH (0.1 g, 2.5 × 10−3 mol) dispersed in ethylene glycol (5 mL) was added to H2PtCl6·6H2O (0.1 g, 1.9 × 10−4 mol) in ethylene glycol (5 mL). The mixture was heated to 433 K for 3 h under argon affording a dark-brown solution.Pt catalysts: The system was achieved by combining a 7.2 × 10−6 mol of the PtNP (0.38 mL ethylene glycol solution), IP or PVP (3.6 × 10−5 mol) with the appropriate solvent (5.0 mL).
Catalytic hydrogenation of aromatic chloronitro compounds
Hydrogenation of aromatic chloronitro compounds was performed at 363 K and 1.0 MPa of hydrogen in a stainless-steel autoclave (10 mL). Typically, the substrate (1.8 × 10−3 mol) and PtNP–IL solution (7.2 × 10−6 mol) were added in the autoclave, the autoclave charged with hydrogen, and then sealed and stirred for 2 h at 363 K. After reaction, diethyl ether (15 × 4 mL) was added to extract the products. The products were then analyzed on an Agilent 6820 gas chromatograph and Agilent 5975C/7890A GC-MS.Characterization of the PtNPs
Results and discussion
Hydrogenation of aromatic chloronitro compounds
PtNPs prepared according to a literature procedure were mixed with the IP or PVP in various solvents and evaluated as catalysts for the hydrogenation of DNP. For comparison purposes, Pt immobilized on activated carbon (Pt/AC) in methanol was evaluated in the same reaction. The results from these studies are summarized in Table 1.| Entry | Catalyst | Solvent | Stabilizer | Reaction time (h) | Reaction temperature (K) | Conversion (%) | Selectivity (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| DAP | MAP | other | |||||||
| a Reaction conditions: solvent (5 mL), Pt (7.2 × 10−3 mmol), DNP (1.8 × 10−3 mol), H2 (1.0 MPa), IP/PVP:Pt = 5:1.b From ref. 46. Reaction conditions: ethanol (900 mL,) Ni (9.0 g), DNP (85 g), H2 (2.0 MPa). | |||||||||
| 1 | Pt/AC(5wt.%) | CH3OH | - | 2 | 363 | 99.0 | 88.7 | 7.8 | - |
| 2 | Pt | CH3OH | PVP | 2 | 363 | 100 | 66.0 | 1.9 | 30.0 |
| 3b | Raney Ni | CH2CH3OH | - | - | 313 | - | 70.4 | - | - |
| 4 | Pt | [Bmim][PF6] | IP | 2 | 363 | 95.8 | 99.9 | <0.1 | - |
| 5 | Pt | [Bmim][BF4] | IP | 2 | 363 | 100 | 99.9 | <0.1 | - |
| 6 | Pt | [C2OHmim][BF4] | IP | 2 | 363 | 100 | 99.9 | <0.1 | - |
| 7 | Pt | [C2OHmim][BF4] | PVP | 2 | 363 | 100 | 99.9 | <0.1 | |
| 8 | Pt | [Bmim][BF4] | IP | 4 | 343 | 94.7 | 99.9 | <0.1 | - |
| 9 | Pt | [Bmim][BF4] | IP | 8 | 343 | 100 | 99.9 | <0.1 | - |
| 10 | Pt | [Bmim][BF4] | IP | 12 | 343 | 100 | 99.9 | <0.1 | - |
| 11 | Pt | [Bmim][BF4] | IP | 4 | 303 | 6.5 | 99.9 | <0.1 | - |
| 12 | Pt | [Bmim][BF4] | IP | 4 | 383 | 100 | 99.9 | <0.1 | - |
| 13 | Pt | [Bmim][BF4] | IP | 4 | 423 | 100 | 98.1 | 1.7 | - |
The selectivity for DAP catalyzed by 5wt% Pt/AC was 88.7%, with extensive dehalogenation observed, giving 2-chloro-3-aminophenol (MAP) in significant quantities. Using the PtNP–methanol solution, DNP underwent quantitative conversion, but the selectivity for DAP was even lower. These results are not too dissimilar to those reported previously, notably that Raney Ni in ethanol catalyzes the hydrogenation of DNP with a selectivity of only 70.4% for the desired product.46 Based on the low selectivity obtained in organic solvents the hydrogenation of DNP was assessed in ILs, including [Bmim][PF6], [Bmim][BF4] and [C2OHmim][BF4]. Under equivalent reaction conditions a dramatic improvement in selectivity was observed in all three ILs. Side (dehalogenation) reactions were suppressed and DAP was obtained in essentially quantitative yield.
It has previously been shown that dehalogenation is favored at high temperatures, usually occurring after reduction of the nitro-group,51,52 with the selectivity decreasing with reaction time and temperature. The reaction was therefore continued for 4, 8 and 12 hours (Table 1, entries 8, 9 and 10), albeit at a slightly reduced temperature, showing that selectively is essentially quantitative, merely with the conversion increasing. The effect of temperature was also explored, with the hydrogenation conducted at 303, 343, 383 and 423 K (Table 1, entries 11, 8, 12 and 13). The reaction was sluggish at 303 K (conversion < 10%), whereas at temperatures of 343 K and 383 K high conversions were observed with the selectivity for DAP being quantitative. The selectivity of DAP decreased slightly as the reaction temperature was increased further (to 423 K). Nevertheless, for the selective hydrogenation of DNP to DAP the PtNP–IL systems are superior, compared to catalysts operating in molecular solvent.
The IP protected PtNP in [C2OHmim][BF4] showed high conversion and perfect selectivity, so further substrates were evaluated using this system (see Table 2). Although the substrates contain different substituent groups, excellent selectivity to the corresponding aromatic chloroamines was achieved in all cases.
| Entry | Substrate | Cat./Sub. ratio | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|
| Reaction conditions: [C2OHmim][BF4] (5 mL), Pt (7.2 × 10−3 mmol), substrate (1.8 mmol), H2 (1.0 MPa) at 363 K, IP:Pt = 5:1. | ||||
| 1 | 2,6-dichloro-4-nitrophenol | 250 | 100 | 99.9 |
| 2 | 4-chloro-2-nitrophenol | 250 | 100 | 99.0 |
| 3 | 1,4-dichloro-2-nitrobenzene | 250 | 100 | 97.9 |
| 4 | 2,6-dichloro-3-nitrotoluene | 250 | 100 | 100 |
| 5 | 2,4-dichloro-6-nitroaniline | 250 | 100 | 98.0 |
The improved selectivity observed for the hydrogenation of nitro groups in ILs was previously attributed to weak non-covalent interactions formed between the ILs and the nitro group, as evidenced by IR spectroscopy and corroborated by DFT calculations.37 It is not unreasonable to assume that the reason for the exceptional selectivity observed here for the conversion of DNP to DAP (and other related substrates) is also due to hydrogen bonding between the ILs and nitro group thereby preferentially activating this group. To verify this hypothesis IR spectra of DNP dissolved in both ILs and molecular solvents were recorded, see Fig 1. The asymmetric stretching vibration of the nitro (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NO) group of DNP in methanol is observed at 1552 cm−1. In toluene the νNO vibration is almost unchanged (1551 cm−1), indicating that the change in polarity of the solvent has little influence on the strength of the NO bonds. However, there is a significant red shift to 1547 ± 1 cm−1 in the three ILs, implying that the NO bonds are slightly weakened (and activated) in the ILs. The stretching vibration of the C–Cl bonds in DNP (Fig 1b) are identical in all five solvents indicating that the nature of solvent does not influence the C–Cl bond strength. Thus, the weak interaction between the nitro group in DNP and the ILs probably contributes to the higher selectivity for the reaction in these solvents.
NO) group of DNP in methanol is observed at 1552 cm−1. In toluene the νNO vibration is almost unchanged (1551 cm−1), indicating that the change in polarity of the solvent has little influence on the strength of the NO bonds. However, there is a significant red shift to 1547 ± 1 cm−1 in the three ILs, implying that the NO bonds are slightly weakened (and activated) in the ILs. The stretching vibration of the C–Cl bonds in DNP (Fig 1b) are identical in all five solvents indicating that the nature of solvent does not influence the C–Cl bond strength. Thus, the weak interaction between the nitro group in DNP and the ILs probably contributes to the higher selectivity for the reaction in these solvents.
![IR spectra of (a) the asymmetric vibrations of the nitro groups and (b) the carbon–chlorine bond of DNP in (A) [Bmim][PF6], (B) [Bmim][BF4], (C) [C2OHmim][BF4], (D) methanol, and (E) toluene.](/image/article/2010/GC/b915299g/b915299g-f1.gif) | ||
| Fig. 1 IR spectra of (a) the asymmetric vibrations of the nitro groups and (b) the carbon–chlorine bond of DNP in (A) [Bmim][PF6], (B) [Bmim][BF4], (C) [C2OHmim][BF4], (D) methanol, and (E) toluene. | ||
Catalyst recycling
The ability to reuse the PtNP–IL systems was also investigated. After catalysis, the product(s) were extracted into diethyl ether, and the remaining PtNP–IL solution was charged with reactants for a subsequent hydrogenation. The recyclability of PtNP–[Bmim][BF4] and PtNP–[C2OHmim][BF4] solutions in the hydrogenation of DNP are shown in Fig 2.![Recycling of the Pt–[C2OHmim][BF4] (black) and Pt–[Bmim][BF4] (grey) systems in the hydrogenation of DNP. Reaction condition: temperature 343 K; hydrogen pressure 1.0 MPa; reaction time 2 h; DNP 0.9 mmol; 3.6 × 10−3 mmol of PtNP (IP:Pt = 5:1) in IL (5 mL).](/image/article/2010/GC/b915299g/b915299g-f2.gif) | ||
| Fig. 2 Recycling of the Pt–[C2OHmim][BF4] (black) and Pt–[Bmim][BF4] (grey) systems in the hydrogenation of DNP. Reaction condition: temperature 343 K; hydrogen pressure 1.0 MPa; reaction time 2 h; DNP 0.9 mmol; 3.6 × 10−3 mmol of PtNP (IP:Pt = 5:1) in IL (5 mL). | ||
The selectivity of the reaction was maintained at 100% throughout the recycling for both systems, demonstrating the excellent control of the PtNP–IL systems with respect to selectivity. However, in case of the PtNP–[Bmim][BF4] system the conversion steadily decreased during five runs, from ca. 95 to 60%. In comparison, the PtNP–[C2OHmim][BF4] system exhibits much better recycling characteristics and after 9 batches the conversion exceeds 80%, reaching a turnover number (TON) of 2025, which is 750 fold greater than the TON obtained using Raney Ni.46
Characterization of the PtNPs by TEM and XPS
One of the main problems associated with NP catalysis is that NPs are kinetically stable and therefore tend to aggregate during reaction, thereby becoming less active. ILs have been shown to help stabilize NPs, and in certain studies it has been shown that ILs with functional groups such as hydroxyl or nitrile groups, may further enhance the stability of the NPs as they can form additional interactions with the NP surface. In the case of the PtNP–[Bmim][BF4] system described herein, aggregation of the PtNP has been confirmed by TEM (Fig 3), presumably leading to the deterioration in catalytic activity during the recycling experiments. The PtNPs prepared in [Bmim][BF4] have an average particle size of 3.7 ± 0.2 nm, which after five catalytic cycles increases to 7.9 ± 0.4 nm. In [C2OHmim][BF4] aggregation of the PtNPs is scarcely observed; the average size of PtNPs prepared in [C2OHmim][BF4] is 3.7 ± 0.2 nm and after nine reaction cycles an average size of 3.8 ± 0.4 nm is observed.![TEM micrographs (scale bar = 20 nm) and size distribution of (a) PtNPs prepared in [Bmim][BF4], (b) PtNPs after five catalytic cycles in [Bmim][BF4] and (c) PtNPs after nine catalytic cycles in [C2OHmim][BF4].](/image/article/2010/GC/b915299g/b915299g-f3.gif) | ||
| Fig. 3 TEM micrographs (scale bar = 20 nm) and size distribution of (a) PtNPs prepared in [Bmim][BF4], (b) PtNPs after five catalytic cycles in [Bmim][BF4] and (c) PtNPs after nine catalytic cycles in [C2OHmim][BF4]. | ||
XPS measurements (Fig 4) were carried on the PtNPs in an attempt to establish why the PtNPs immobilized in [C2OHmim][BF4] are more stable than those in [Bmim][BF4]. The binding energy of the Pt 4f7/2 core-level in the PtNP–[Bmim][BF4] system has a value of 72.2 eV, 0.5 eV higher than in the PtNP–[C2OHmim][BF4] system (71.7 eV). Although binding energies are sensitive to the average valency and size of the NP, the difference observed here cannot be attributed to these factors. The lower binding energy of the PtNPs in [C2OHmim][BF4] suggests that additional electron transfer from the IL to the PtNPs is evident. After five catalytic cycles the 4 f7/2 core-level of PtNPs in [Bmim][BF4] changes from 72.2 eV to 70.7 eV (C), a dramatic shift towards the metallic state of Pt, and in accord with the TEM analysis that shows PtNP aggregation (see below). In contrast, after nine cycles the 4 f7/2 core-level value of the PtNPs in [C2OHmim][BF4] is 71.4 eV, almost the same value observed for the freshly prepared PtNPs. The constant binding energy for the PtNPs in [C2OHmim][BF4] provides additional evidence for the high stability of the PtNPs with respect to both their size and surface state. The XPS measurements, together with the TEM analysis, suggest that the hydroxyl groups in [C2OHmim][BF4] interact with the PtNPs helping to stabilize them. Indeed, alcohol functionalized ILs have been shown to be excellent media for the preparation of gold NPs.53
![Pt (4f) region of (a) Pt–[Bmim][BF4] before catalysis, (b) Pt–[C2OHmim][BF4] before catalysis, (c) Pt–[Bmim][BF4] after catalysis, (d) Pt–[C2OHmim][BF4] after catalysis.](/image/article/2010/GC/b915299g/b915299g-f4.gif) | ||
| Fig. 4 Pt (4f) region of (a) Pt–[Bmim][BF4] before catalysis, (b) Pt–[C2OHmim][BF4] before catalysis, (c) Pt–[Bmim][BF4] after catalysis, (d) Pt–[C2OHmim][BF4] after catalysis. | ||
Additional evidence for the interaction of OH groups with the surface of the PtNP is obtained from IR spectroscopy using H2S as a probe molecule54 (Fig 5).
![IR spectra of H2S absorbed on PtNPs (raw data, black line), fitting of symmetric vibrations of H2S (blue line), fitting for antisymmetric vibrations of H2S (green line) after the adsorption of H2S on (A) PtNP–[Bmim][BF4] and (B) Pt–[C2Omim][BF4] (difference spectra: after H2S adsorption minus spectrum without H2S – pink line). The red line corresponds to the sum of the blue and green lines.](/image/article/2010/GC/b915299g/b915299g-f5.gif) | ||
| Fig. 5 IR spectra of H2S absorbed on PtNPs (raw data, black line), fitting of symmetric vibrations of H2S (blue line), fitting for antisymmetric vibrations of H2S (green line) after the adsorption of H2S on (A) PtNP–[Bmim][BF4] and (B) Pt–[C2Omim][BF4] (difference spectra: after H2S adsorption minus spectrum without H2S – pink line). The red line corresponds to the sum of the blue and green lines. | ||
In [C2OHmim][BF4], the antisymmetric and symmetric vibrations of H2S were more overlapped than the case of Bmim[BF4], probably due to the formation of hydrogen bonding between H2S and OH groups. In Pt–Bmim[BF4], the γ(SH) wavenumber for the symmetric and antisymmetric vibrations are 2588 cm−1 and 2604 cm−1 (A) respectively. In Pt–[C2OHmim][BF4] (B), however, both peaks display a blue shift to 2592 cm−1 and 2607 cm−1, suggesting the OH groups in [C2OHmim][BF4] help to weaken the interaction of H2S on the surface of the PtNPs. The H2S-IR test gave further evidence for the presence of weak interactions between the PtNPs and OH groups in the IL. Moreover, the experiment indicates that the PtNPs in the hydroxyl group functionalized IL is poison resistant as it weakens the adsorption of poison (H2S) molecule to the metal surface.
Conclusions
A highly effective PtNP–IL system has been established for the selective synthesis of aromatic chloroamines from aromatic chloronitro compounds. Compared to other catalytic systems employing NPs or heterogeneous catalysts operating in molecular organic solvents, the PtNP–IL systems described herein exhibit excellent selectivity to the desired product, and the hydroxyl-functionalized IL system, display excellent stability allowing extensive recycling and high turnover numbers. This system provides a further example of an IL–catalyst regime which offers environmental benefits in terms of eliminating organic solvents from the catalytic process, although on this laboratory scale, organic solvents were used to extract the product. Moreover, and importantly, based on experimental evidence the role of the ILs can be directly ascribed to the high selectivity obtained and the high catalyst stability.Acknowledgements
This work was supported by the National Science Foundation of China (Project Nos 20773005, 20533010).References
- P. J. Dyson, Appl. Organomet. Chem., 2002, 16, 495 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS.
- P. Migowski and J. Dupont, Chem.–Eur. J., 2007, 13, 32 CrossRef.
- V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615 CrossRef CAS.
- L. D. PachÓn and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288 CrossRef.
- Y. L. Gua and G. X. Li, Adv. Synth. Catal., 2009, 351, 817 CrossRef.
- E. Redel, J. Krämer, R. Thomann and C. Janiak, J. Organomet. Chem., 2009, 694, 1069 CrossRef CAS.
- P. Migowski, G. Machado, S. R. Texeira, Maria. C. M. Alves, J. Morais, A. Traverse and J. Dupont, Phys. Chem. Chem. Phys., 2007, 9, 4814 RSC.
- G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner, S. R. Teixeira and J. Dupont, Chem. Eur. J., 2003, 9, 3263 CrossRef CAS.
- E. T. Silveira, A. P. Umpierre, L. M. Rossi, G. Machado, J. Morais, G. V. Soares, I. J. R. Baumvol, S. R. Teixeira, P. F. P. Fichtner and J. Dupont, Chem.–Eur. J., 2004, 10, 3734 CrossRef CAS.
- L. M. Rossi, J. Dupont, G. Machado, P. F. P. Fichtner, C. Radtke, I. J. R. Baumvol and S. R. Teixeira, J. Braz. Chem. Soc., 2004, 15, 904 CAS.
- G. S. Fonseca, E. T. Silveira, M. A. Gelesky and J. Dupont, Adv. Synth. Catal., 2005, 347, 847 CrossRef CAS.
- N. Yan, C. Zhao, C. Luo, P. J. Dyson, H. C. Liu and Y. Kou, J. Am. Chem. Soc., 2006, 128, 8714 CrossRef CAS.
- S. D. Miao, Z. M. Liu, B. X. Han, J. Huang, Z. Y. Sun, J. L. Zhang and T. Jiang, Angew. Chem., Int. Ed., 2006, 45, 266 CrossRef CAS.
- J. Wang, J. Feng, R. Qin, H. Fu, M. Yuan, H. Chen and X. Li, Tetrahedron: Asymmetry, 2007, 18, 1643 CrossRef CAS.
- M. H. G. Prechtl, M. Scariot, J. D. Scholten, G. Machado, S. R. Teixeira and J. Dupont, Inorg. Chem., 2008, 47, 8995 CrossRef CAS.
- G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner, S. R. Teixeira and J. Dupont, Chem. Eur. J, 2003, 9, 3263 CrossRef CAS.
- X. D. Mu, D. G. Evans and Y. A. Kou, Catal. Lett., 2004, 97, 151 CrossRef CAS.
- X. D. Mu, J. Q. Meng, Z. C. Li and Y. Kou, J. Am. Chem. Soc., 2005, 127, 9694 CrossRef CAS.
- C. Zhao, H. Z. Wang, N. Yan, C. X. Xiao, X. D. Mu, P. J. Dyson and Y. Kou, J. Catal., 2007, 250, 33 CrossRef CAS.
- I. S. Park, M. S. Kwon, K. Y. Kang, J. S. Lee and J. Parka, Adv. Synth. Catal., 2007, 349, 2039 CrossRef CAS.
- M. J. Jacinto, P. K. Kiyohara, S. H. Masunaga, R. F. Jardim and L. M. Rossi, Appl. Catal.,A, 2008, 38, 52 CrossRef.
- X. Yang, N. Yan, Z. Fei, R. M. C. Quesada, G. Laurenczy, L. K. Minsker, Y. Kou, Y. Li and P. J. Dyson, Inorg. Chem., 2008, 47, 7444 CrossRef CAS.
- J. Dupont, G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner and S. R. Teixeira, J. Am. Chem. Soc., 2002, 124, 4228 CrossRef CAS.
- G. S. Fonseca, J. D. Scholten and J. Dupont, Synlett., 2004, 1525 CAS.
- G. S. Fonseca, G. Machado, S. R. Teixeira, G. H. Fecher, J. Morais, M. C. M. Alves and J. Dupont, J. Colloid. Interface. Sci., 2006, 301, 193 CrossRef CAS.
- G. S. Fonseca, J. B. Domingos, F. Nome and J. Dupont, J. Mol. Catal., A, 2006, 248, 10 CrossRef CAS.
- J. D. Scholten, G. Ebeling and J. Dupont, Dalton Trans., 2007, 5554 RSC.
- L. S. Ott, S. Campbell, K. R. Seddon and R. G. Finke, Inorg. Chem., 2007, 46, 10335 CrossRef.
- Y. Zhu, C. Koh, A. T. Peng, A. Emi, W. Monalisa, L. K. J. Louis, N. S. Hosmane and J. A. Maguire, Inorg. Chem., 2008, 47, 5756 CrossRef CAS.
- J. Huang, T. Jiang, B. Han, H. Gao, Y. Chang, G. Zhao and W. Wu, Chem. Commun., 2003, 1654 RSC.
- J. Huang, T. Jiang, H. Gao, B. Han, Z. Liu, W. Wu, Y. Chang and G. Zhao, Angew. Chem. Int. Ed., 2004, 43, 1397 CrossRef CAS.
- J. Le Bras, D. K. Mukherjee, S. Gonzalez, M. Tristany, B. Ganchegui, M. Moreno-Manas, R. Pleixats, F. Henin and J. Muzart, New J. Chem., 2004, 28, 1550 RSC.
- A. P. Umpierre, G. Machado, G. H. Fecher, J. Morais and J. Dupont, Adv. Synth. Catal., 2005, 347, 1404 CrossRef CAS.
- C. W. Scheeren, G. Machado, J. Dupont, P. F. P. Fichtner and S. R. Texeira, Inorg. Chem., 2003, 42, 4738 CrossRef CAS.
- C. W. Scheeren, G. Machado, S. R. Teixeira, J. Morais, J. B. Domingos and J. Dupont, J. Phys. Chem. B, 2006, 110, 13011 CrossRef CAS.
- C. X. Xiao, H. Z. Wang, X. D. Mu and Y. Kou, J. Catal., 2007, 250, 25 CrossRef CAS.
- T. J. Geldbach, D. B. Zhao, N. C. Castillo, G. Laurenczy, B. Weyershausen and P. J. Dyson, J. Am. Chem. Soc., 2006, 128, 9773 CrossRef CAS.
- D. B. Zhao, Z. F. Fei, T. J. Geldbach, R. Scopelliti and P. J. Dyson, J. Am. Chem. Soc., 2004, 126, 15876 CrossRef CAS.
- F. Fei, D. B. Zhao, D. Pieraccini, W. H. Ang, T. J. Geldbach, R. Scopelliti, C. Chiappe and P. J. Dyson, Organometallics, 2007, 26, 1588 CrossRef CAS.
- Yang, Z. F. Fei, D. B. Zhao, W. H. Ang, Y. D. Li and P. J. Dyson, Inorg. Chem., 2008, 47, 3292 CrossRef CAS.
- M. Ruta, G. Laurenczy, P. J. Dyson and L. K. Minsker, J. Phys. Chem., C, 2008, 112, 17814 CrossRef CAS.
- D. Dorjnamjin, M. Ariunaa and Y. K. Shim, Int. J. Mol. Sci., 2008, 9, 807 Search PubMed.
- N. Yan, X. Yang, Z. Fei, Y. Li, Y. Kou and P. J. Dyson, Organometallics, 2009, 28, 937 CrossRef CAS.
- M. Zou, X. D. Mu, N. Yan and Y. Kou, Chin. J. Catal., 2007, 28, 5389 Search PubMed.
- US Pat., 4129414, 1978.
- DE Pat., 10160815, 2003.
- DE Pat., 102006020049, 2007.
- L. C. Branc o, J. N. Rosa, J. J. M. Ramos and C. A. M. Afonso, Chem. Eur. J, 2002, 8, 16.
- Y. Wang, J. W. Ren, K. Deng, L. L. Gui and Y. Q. Tang, Chem. Mater., 2000, 12, 1622 CrossRef CAS.
- X. D. Wang, M. H. Liang, J. L. Zhang and Y. Wang, Curr. Org. Chem., 2007, 11, 299 CrossRef CAS.
- B. Coq, A. Tijani and F. Figueras, J. Mol. Catal., 1991, 68, 331 CrossRef CAS.
- L. Z. Ren, L. J. Meng and Q. H. Lu, Chem. Lett., 2008, 37, 106 CrossRef CAS.
- F. Maugé, A. Sahibed-Dine, M. Gaillard and M. Ziolek, J. Catal., 2002, 207, 353 CrossRef CAS.
- M. Benza, A. M. van der Kraanb and R. Prins, Appl. Catal. A: General, 1998, 172, 149 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |