Fate and removal of estrogens in municipal wastewater†
LeeAnn
Racz
and
Ramesh K.
Goel
*
Department of Civil and Environmental Engineering, University of Utah, Ste 104, 122 S. Central Campus Drive, Salt Lake City, UT 84112-0610, USA. E-mail: rgoel@eng.utah.edu; Fax: +1 315 801 585 5477; Tel: +1 315 801 581-6110
First published on 25th November 2009
Abstract
Natural and synthetic estrogens are some of the most potent endocrine disrupting compounds found in municipal wastewater. Much research has been conducted on the source and fate of estrogens in wastewater treatment plants. Sorption and biodegradation are the primary removal mechanisms for estrogens in activated sludge systems, which are widely used biological treatment techniques for municipal wastewater treatment. However, when removal of estrogens in a wastewater treatment plant is incomplete, these compounds enter the environment through wastewater discharges or waste activated sludge at concentrations that can cause endocrine-reproductive system alterations in birds, reptiles and mammals. Therefore, studies have also focused on potential advanced treatment technologies with the aim of removing the compounds before discharging wastewater effluent or disposing waste sludge. This review discusses the physiological effects of these estrogens and the degree of problems estrogens pose as they enter the wastewater stream. Thereafter, this review also analyzes their fate in wastewater treatment systems and how they may reach drinking water sources. Furthermore, this review includes a discussion on various treatment technologies being investigated and future research trends for this pressing environmental issue.
 LeeAnn Racz | LeeAnn Racz is currently an Environmental Engineering PhD candidate at the University of Utah working under Dr. Ramesh Goel. She is also an active duty Air Force officer with a position as a Bioenvironmental Engineer, and has been stationed in New Mexico, South Korea, Colorado, Texas and Utah. She holds a professional engineer license and is currently studying nitrifying biomass and estrogens in wastewater. |
Environmental impactMicropollutants in water, though in trace concentrations, can exist in sufficient quantities in water sources to cause detrimental effects in animals and humans. Endocrine disrupting compounds, particularly natural and synthetic hormones, are a class of micropollutants that have gained increasing attention as researchers focus their attention on contaminants of emerging concern. This review provides current and in-depth knowledge of the fate, transport, effects and treatment of natural (17β-estradiol, estrone and estriol) and synthetic (17α-ethinylestradiol) estrogens to the readers. We discuss the issues associated with estrogens in municipal wastewater and offer a critical analysis of areas requiring further study. |
1. Introduction
As early as the 1930s, scientists have reported that certain synthetic and natural compounds can mimic the endogenous hormones in the endocrine systems of animals.1,2 Following casual observation by anglers of hermaphrodite fish in wastewater treatment plant lagoons, surveys confirmed that fish exhibited these unusual characteristics.3–6 Another survey revealed that male trout downstream of a wastewater treatment plant effluent produce vitellogenin, an egg yolk protein normally associated with sexually mature females, when stimulated by estradiol, a natural estrogen.7 Now referred to as endocrine-disrupting compounds (EDCs), these natural and synthetic compounds have gained attention as concerns have increased for micropollutants in municipal wastewater.6,8 In particular, the estrogenic class of EDCs, comprised of compounds that mimic or block natural estrogens by binding to the estrogen receptor,9 include some of the most potent chemicals linked to reproductive effects in aquatic species.10 Although controversial, some researchers have also suggested associations between EDCs in the environment and human sperm quality11–13 as well as breast, testicular and prostate cancers.9,13–17Estrogens are some of the most potent EDCs found in wastewater. Four estrogens most commonly found in wastewater include three natural steroids (17β-estradiol (E2), estrone (E1) and estriol (E3)) and one synthetic compound (17α-ethinyl estradiol (EE2), used in contraceptives and hormone replacement therapy). Past reviews have focused on estrogen contributions by wastewater and livestock manure and removal mechanisms in engineered systems.18,19 This critical review, however, discusses the physiological effects of these estrogens, the degree of problems estrogens pose as they enter the wastewater stream, their fate in wastewater treatment systems, how they may reach drinking water sources, and various treatment technologies being investigated.
2. Estrogenicity
The estrogenicity of a chemical is determined by its ability to bind to the estrogen receptor, thereby mimicking or blocking the activity of natural estrogens.20–22 The human estrogen receptor contains a gap in the ligand-binding domain which is nearly twice the size required for E2, thus providing space for a variety of other molecules to bind with the estrogen receptor.23 Although the human estrogen receptor and the estrogen receptors of aquatic species do not have identical functional activities,24 all vertebrate species are likely similar in their nonspecific ligand-binding domains.25 Specifically, estrogenicity is dependent on the size and degree of the branching alkyl group and its position on the phenol ring.20 In fact, this feature of estrogen receptor proteins might be responsible for some minor loss of estrogenic activity in wastewater as non-estrogenic compounds may bind to estrogen receptors,26–31 and chemical matrix effects can limit estrogen uptake.31 Hu and Aizawa21 developed a quantitative structure-activity relationship (QSAR) model describing the binding affinity of phenolic compounds, including estrogens, to the estrogen receptor. This model incorporates expressions for bulk effect, polarity effects, and hydrogen-bonding effects.Total estrogenicity of a sample is commonly measured in order to avoid exhaustive chemical analysis in chemically complex samples, including wastewaters.31 For example, bioassays such as the yeast estrogen screen (YES)6 and the ER-CALUX,32 use reporter genes to quantify relative overall estrogen activity of a sample. Table 1 lists the relative estrogenicities of the three natural and one synthetic estrogen according to the YES bioassay.31 Similar proportions have been reported by others using bioassays with human breast cancer cells.33,34 E1 and E2 generally represent greater than 98% of the total estrogenicity of a wastewater effluent.35,36 Natural and synthetic estrogens have three to seven orders of magnitude greater estrogenic potencies than other EDCs identified in wastewater.8,35,37 Of these, E2 and EE2 are the most potent.8,38–41 Although E1 has an estrogenic potency one-fifth of E2, the quantity of E1 in wastewater effluents can be greater than ten times that of E2, making E1 a very important EDC in the aquatic environment.31,42,43 There is another natural estrogen, 17α-estradiol, but it is rarely studied as it is about one-tenth as potent as 17β-estradiol (E2) and seldom detected.40,44
| Compound | Structure | Molecular Weight | Log Kowa | Henry's Law constant (atm m3 mol−1)b | Relative estrogenic activity (YES bioassay)c |
|---|---|---|---|---|---|
| a Lai et al.157 b Bodzek and Dudziak240 c Conroy et al.31 d A, B, C, and D refer to the ring nomenclature. The C-atom on which the functional group is placed is referred to as 3 or 17. | |||||
| Estrone (E1) d |
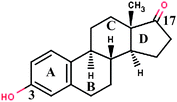
|
270.37 | 3.43 | 3.80 × 10−10 | 0.20 |
| 17β-Estradiol (E2) |
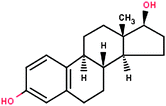
|
272.39 | 3.94 | 3.64 × 10−11 | 1.0 |
| Estriol (E3) |
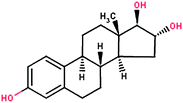
|
288.39 | 2.81 | 3.80 × 10−10 | 0.01 |
| 17α-Ethinylestradiol (EE2) |
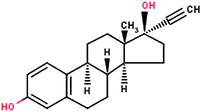
|
296.40 | 4.15 | 7.94 × 10−12 | 1.32 |
The predicted-no-effect-concentration (PNEC) is 1 ng L−1 for E2 and 3–5 ng L−1 for E1,45,46 while the lowest observable effect concentration (LOEC) affecting production of vitellogenin in juvenile female rainbow trout is reported as 14 ng L−1 for E2 and 3.3 ng L−1 for E1. Purdom et al.7 reported less than 1 ng L−1 of EE2 can stimulate male rainbow trout to produce vitellogenin, while Länge et al.47 found a concentration of 4 ng L−1 EE2 can cause failure in the male fathead minnow to develop normal secondary sexual characteristics. Kiyoaki et al.48 reported vitellogenin production in male Japanese medaka at LOECs of 32 ng L−1 for E1 and 5.0 ng L−1 for E2. Other studies claim exposure to estrogen levels as low as 1 ng L−1 can cause the development of intersex characteristists in roach and fish.8,49–53 Measurable changes in fish reproduction can result at E2 and EE2 concentrations as low as 2 ng L−1.4–6,54–58 Over a three-year study in a Canadian lake, estrogens at a concentration of 5–6 ng L−1 feminized male fish and killed the entire fish population.59 Birds, reptiles and mammals in polluted areas can experience endocrine-reproductive system alterations as well.60 Furthermore, freshwater worms can bioaccumulate EE2, making possible a transfer to benthivores and subsequent secondary poisoning of predators.61
3. Extent of estrogen contamination
In humans and other mammals, estrogens are primarily metabolized in the liver. These transformations include oxidation, hydroxylation, deoxidation, and methylation prior to the final conjugation with glucuronic acid or sulfate.62,63 These natural and synthetic estrogens are then excreted in urine and feces largely as inactive polar conjugates,64–69 examples of which are illustrated in figure 1. These estrogen conjugates have potencies of more than four orders of magnitude less than that of E2.70 While approximately 85–90% of fecal estrogens are in the unconjugated form, only 5–10% of the total estrogens are eliminated with the feces, making fecal free estrogens account for only a small fraction of the total estrogens.42,71 The remaining excreted estrogens are primarily in the conjugated forms. Biological processes then deconjugate the excreted conjugates to the active unconjugated forms72,73 (table 1).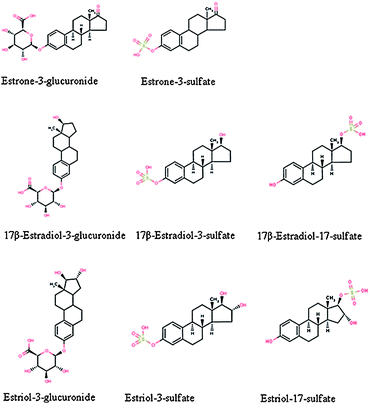 | ||
| Fig. 1 Structures of selected conjugated natural estrogens. | ||
Women typically excrete 0.5 to 5 µg day−1 of E2 (up to approximately 400 µg day−1 for pregnant women), but it is also excreted by men.62,65,66,74,75 E1 is an important pregnancy estrogen for women as well as a metabolite of E2 and is excreted at a rate of 3 to 20 µg day−1. The excretion rate by women for E3 is up to about 64 µg day−1.65,66,75 As wastewater treatment plants are not designed to remove estrogens or other micropollutants, estrogens that are not degraded during the wastewater treatment process are then released to the environment with the effluent. In densely-populated areas, wastewater treatment effluents can make up a large portion of flow in the receiving waters, especially during dry periods when low flow rates mean little dilution.76 Therefore, treated municipal wastewater is one of the most likely sources of estrogenic compounds in the environment.6,8,53,77–94
While humans produce natural estrogens in the microgram range daily, concentrations of these compounds in municipal wastewater and surface water receiving effluent is reported in the nanogram per liter level.95–102 Estrogen concentrations of 1–500 ng L−1 have been found in municipal wastewater (influents), with the distribution of concentrations generally following the pattern E1 > E2 > E3 > EE2.68,74,103,104 Concentrations of 1–500 ng L−1 are also commonly found in wastewater effluents.8,22,46,95,96,98,101,102,105–111 With dilution, receiving surface waters have been found to contain E1 and E3 to a few ng L−1,69,112 E2 up to 27 ng L−1,113 and EE2 up to 20 ng L−1.114 However, even these trace concentrations are environmentally relevant.
4. Fate of estrogens in wastewater treatment
Estrogen removal during wastewater treatment is well-documented.72,96,98,107,109,115–123 Following deconjugation, excreted estrogens are primarily removed from wastewater in an activated sludge system either by sorption or biodegradation as illustrated in figure 2.124–130 Although sorption occurs quickly,126,131–133 biodegradation is the primary removal means for estrogens in wastewater.68,100,134–136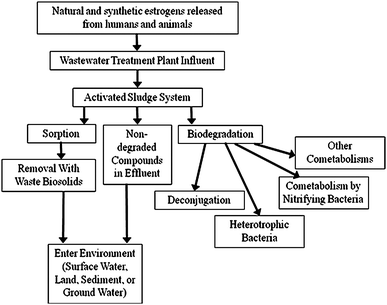 | ||
| Fig. 2 Fate of estrogens in a wastewater treatment plant and the environment. | ||
In activated sludge, E2 can be removed 44 to 99.9%,42,68,96,102,135,137–140 E3 can be removed 18 to almost 100%,42,97,101,141 E1 can be removed up to 98%,68,96,102,137,140 and EE2 removal efficiencies vary from 34% to almost 100%.68,69,96,99 There are considerable day-to-day variations in effluent concentrations with no clear temporal trend despite consistency in wastewater flow.120 Johnson and Williams142 developed a model for the relationship between influent and effluent estrogen concentrations, but application of this model gives mixed results.143 Variations in wastewater treatment processes and operational conditions are generally regarded as the reason for fluctuations in removal efficiencies and effluent concentrations.19 In particular, natural estrogens are poorly removed in highly loaded plants.101,144,145 Furthermore, plants with solids retention times (SRTs) longer than 10 days tend to achieve better estrogen removal.101,137,144,146,147 Plants with good nitrification, which requires long SRTs, also demonstrate better estrogen removal.129,146
4.1 Sorption
Estrogen compounds have low Henry's Law coefficients (less than 1.0 × 10−3 atm m3 mol−1), making any volatilization negligible.148 Also, since they have high octanol–water partition coefficients (Kow) and are hydrophobic, theoretically they can readily sorb onto solids in the wastewater activated sludge process.126 Free unconjugated estrogens are more hydrophobic than conjugated estrogens, and therefore are more likely to sorb onto sludge.40,103 Clara et al.149 and De Gusseme et al.150 demonstrated that sorption can account for up to about 30% removal of EE2. However, other researchers report that sorption typically accounts for less than 10% of the estrogen removal from wastewater.68,103,117,135,151 Sorption variability among different studies can be attributed to sorbent-specific characteristics such as organic carbon content, particle size, pH, salinity, and ion content that varies from plant to plant.112,129,149 Xu et al.152 suggest that larger flocs, such as those in suspended growth reactors, are more porous allowing for more intraparticle mass transfer, and therefore result in greater sorption than in membrane bioreactors. On the other hand, EE2 may sorb better to membrane bioreactor sludge because of the sludge hydrophobic properties.153 Yi et al.131 explained that sorption was more important in nitrifying sludge when the initial ammonium concentration was 37.3 mg NH4+-N L−1, but biodegradation became more relevant at higher ammonium concentrations. Since ammonia concentrations are known to regulate ammonia monooxygenase (AMO) activity during nitrification,154 there is less of the cometabolic activity likely responsible for EE2 degradation at lower initial ammonia concentrations. As the initial ammonia concentration increases, respiration and cometabolism become more important, although the overall amount of sorbed estrogens may remain unchanged.While sorption can generally be described as following Freundlich isotherms, sorption behavior is often quantified as the specific sorption coefficient KD (L kg−1 total suspended solids (TSS)).126,129,131,149,151 This coefficient is the ratio of equilibrium concentrations of a dissolved compound in a system containing a sorbent (sludge) and an aqueous phase (eq. 1)
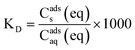 | (1) |
It is interesting to note that estrogens can desorb from sludge as well. Thermodynamic studies reveal that the low binding energies of E1, E2, E3, and EE2 make physical sorption dominant over chemical sorption. Therefore, estrogen sorption onto biomass is reversible to some extent, albeit with a slower rate than the initial sorption.126,152 This can have important implications as sorbed estrogens are known to be released into the water phase during sludge dewatering, leaving the dewatering filtrate with estrogen concentrations greater than those in influent or effluent.70 However, when biomass undergoes repeated sorption and desorption cycles, hysteresis can occur in which desorption can fail to restore the full sorption capacity of the biomass. When this happens, the relative effluent concentrations of estrogens can increase by at least a factor of nine.132
4.2 Biodegradation
Following initial sorption, additional losses are attributable to biodegradation.126,133,147,158,159 Biodegradation is the primary removal means for estrogens in wastewater.68,100,134–136 These mechanisms include (1) deconjugation, (2) degradation as a carbon source for heterotrophic bacteria, (3) cometabolism with nitrifying biomass, or (4) other cometabolisms. Generally, the natural estrogens, E2 in particular, are quite readily biodegradable.72 However, EE2 is not nearly as easily biologically removed.160 The ethinyl group of EE2 is assumed to sterically hinder enzyme expression, substrate-receptor binding, and EE2 metabolism.161 In addition, it is important to note that the bacterial communities in municipal wastewater treatment sludge have a much greater capacity to biodegrade estrogens than industrial wastewater treatment sludge, which mineralized only 4% of E2 in one study.156Wastewater treatment plants with long SRTs, especially those performing nitrification, are generally effective at removing estrogens.101,137,144,146,147 Long SRTs, namely greater than 10 days, allow the enrichment of slowly growing bacteria, such as nitrifying bacteria, and the establishment of a more diverse ecological community. For this reason, membrane bioreactors with long SRTs have been shown to effectively degrade estrogens. De Gusseme et al.150 demonstrated greater than 99% removal of EE2 (83 ng L−1 initial concentration, loading rates up to 208 ng L−1 d−1, membrane flux rate 6.9 L m−2 h−1) in a membrane bioreactor applied with a nitrifier enrichment culture. Conversely, estrogens are poorly removed in mere primary settling8,68 as well as trickling filters.96,134,137,162 Poor trickling filter removal of estrogens has been attributed to lower hydraulic retention times than those found in suspended sludge systems, as well as estrogen saturation on the biofilm.98 Nevertheless, products of estrogen biodegradation are reported as non-estrogenic.64,163
The major metabolite of E2 is E1.64,72,171 Subsequent E2 degradation products include E3, 16α-hydroxyestrone, 2-methoxyestradiol, and 2-methoxyestrone.171 E3 is also transiently converted to 16α-hydroxyestrone under aerobic conditions.172,173 However, one study could not degrade E3 below a threshold initial concentration of 80 µg L−1 in an isolated culture.172 It has been suggested that E1 biodegradation might be the rate-limiting step for converting E2 and E1 to non-estrogenic products64 as several studies observed better removal of E2 than E1 in wastewater treatment plants.96,97,106,156,165 In fact, E2 is transformed to E1 even under anaerobic conditions, although to not as great an extent as under aerobic conditions.147,171,174–176
Yu et al.64 suggest that E2 degradation patterns might be explained by two possible degradation mechanisms: cometabolic (non-growth) and metabolic (growth). Cometabolism, where enzymes whose primary function is not for estrogen degradation, could be responsible for rapid and stoichiometric degradation of E2 to E1 since enzymes, such as mono- and di-oxygenase, are already present.132 Under these conditions, E1 degradation is not likely. Metabolic activity, on the other hand, where bacteria use estrogens as a carbon source, might be credited with complete removal of E2 and E1. This mechanism explains degradation patterns observed in other studies in which E2 was rapidly oxidized to E1 (within 3–7 hours with activated sludge) which is more slowly further converted to E3 (within 72 hours), the major excretion product.167,171
Heterotrophic degradation pathways appear to begin by oxidizing the secondary alcohol on the C17 position (D-ring) of E2 to ketone.64 EE2 degradation, however, has been suggested by Haiyan et al.176 as direct oxidation of EE2 to E1 with a subsequent cleavage of the A- and B-rings to unsaturated acids which are further mineralized to carbon dioxide and water.
It appears that degradation rates of estrogens increase with increasing initial concentrations,172 presumably as greater initial concentrations provide greater substrate for bacteria that metabolize these compounds. Furthermore, the greater the concentration of total organic compounds, the less easily estrogens are degraded, possibly because bacteria preferentially utilize other organic compounds before utilizing estrogens.158
| NH3 + O2 + 2e− + 2H+ → NH2OH + H2O | (2) |
| NH2OH + H2O + 1/2 O2 → NO2− + 2 H2O + H+ | (3) |
Nitrite oxidizing bacteria (NOB), such as Nitrospira and Nitrobacter, then convert the nitrite to NO3− as given in equation (4). This reaction is rate-limiting in the overall nitrification process.
| NO2− + 1/2 O2 → NO3− | (4) |
AOB oxidize ammonia to hydroxylamine via the ammonia monooxygenase (AMO) enzyme. AMO inserts oxygen into C–H bonds, catalyzing the hydroxylation of alkanes to produce primary and secondary alcohols.178,179 This hydroxylation is attributed as converting estrogens into hydrophilic products essentially devoid of estrogenic activity.146,180 Therefore, estrogen degradation in nitrifying biomass is typically attributed to cometabolism via AMO.125,146,181 The AMO enzyme is particularly important for EE2, the most recalcitrant estrogen. EE2 has been successfully degraded in nitrifying sludge where it could not be removed by heterotrophs alone.132,146,150,181 De Gusseme et al.150 suggested a possible competition between ammonia and estrogens, specifically EE2, for removal by the AMO enzyme as maximum EE2 removal rates occur when ammonia concentration is minimal. This same study, along with Yi and Harper132 and Shi et al.181 suggested that AOB are likely responsible for the first degradation step of EE2, while heterotrophic bacteria in a mixed culture might remove the subsequent metabolites. However, nitrification could be less important for degrading E2 as it is suggested that heterotrophic bacteria are responsible for its degradation instead.181 Interestingly, lab-scale nitrification bioreactors continued to degrade estrogens, specifically EE2, months after being starved of ammonium indicating that even ammonia-starved AOB maintain population density and activity.182 Also, nitrifying activated sludge does not require an adaptation period before degrading EE2.146
While the evidence of estrogen degradation by AMO cometabolism is compelling, our discussion of nitrifying bacterial degradation must also include the work by Gaulke et al.183 This research reported that in a co-culture of ammonia oxidizing bacteria, EE2 was transformed via abiotic nitration rather than being removed via cometabolism. These researchers suggested that EE2 removal in wastewater treatment plants is more likely achieved by heterotrophic activity rather than cometabolic degradation. Therefore, these conflicting results present some controversy as to the true means by which estrogens, particularly EE2, are removed in a wastewater treatment plant.
5. Pathway of estrogens to water through wastewater
Under some circumstances, wastewater effluent is discharged into a body of water that at some point downstream could be used as a water source.247–249 This scenario explains how estrogens, particularly the recalcitrant EE2, could potentially be found in drinking water. In addition, estrogens can accumulate in wastewater sludge72,184 and can accumulate even more on soluble organic compounds found in soils.185 In fact, wastewater irrigation increases the sorptive properties of soluble organic compounds in soil.184,186 When wastewater is used to irrigate soils or digested wastewater sludge is applied to soils, the mineralization of E2 was found to be particularly slow, with complete degradation estimated to occur in 12 to 18 months.185 Incidentally, the application of manure to structured soils has also been found to contribute estrogens to the aquatic environment,243 particularly when manure is applied to areas where streamwater derives from drainage water.187 Lucas and Jones188 likewise found that urine enhances the leaching and persistence of estrogens in soils, a condition likely to be found where livestock are raised. Estrogens can also leach from dairy farm waste lagoons and impact groundwater.189,190 A similar scenario exists for landfill disposal sites through which leachate with estrogenic activity reaches groundwater or surface water.191–193 Therefore, estrogens can reach ground or surface water since colloidal and dissolved organic matter could act as soluble sorbents.185,190,194,244–246 In deeper water-saturated soil depths, microbial activity depletes oxygen, leading to anaerobic conditions.195 Since biodegradation is much slower in the absence of oxygen than in aerobic conditions,147,171,174–176 accumulation and transport of estrogens in the groundwater might be possible.6. Additional treatment technologies
While activated sludge wastewater treatment systems can be effective in removing significant amounts of estrogens from wastewater, estrogens still exist in the effluent at environmentally relevant concentrations. Therefore, other treatment technologies have been explored with the intention of finding suitable polishing techniques to further reduce effluent concentrations. These technologies include chemical removal, activated carbon, chlorination, ozonation, ultraviolet irradiation, membrane separation, and other novel approaches.6.1 Chemical removal
Wastewater effluents and surface waters contain organic colloidal material ranging in size from 0.001 to one micron in diameter. The benzene A-ring in estrogen compounds is thought to be responsible for estrogen sorption on organic matter.196 Yamamoto and Liljestrand122 found high correlations between aromatic properties of organic colloids and E2 sorption. Holbrook et al.197 and Zhou et al.198 estimated that between 1 and 60% of E2 and EE2 can be associated with colloidal material. Despite this wide range of colloidal sorption, colloids possess a greater sorption affinity for E2 and EE2 compared to either sediment or suspended particles.197,199–201 In fact, colloidal organic carbon at a concentration of 10 mg L−1 produced a 20% decrease in the YES response to E2.202 Consequently, reduction in the colloidal organic content concentration of effluent wastewater may reduce the effluent concentrations of E2 and EE2.Coagulation and flocculation are methods commonly used to remove these particles quickly. Coagulation is first employed by rapidly mixing metal salts such as aluminium sulfate and ferric chloride with colloids. The positive charge on the coagulants neutralizes the negative charge on the colloids enabling the colloids to agglomerate. Flocculation, the process by which particle collisions are brought about hydrodynamically by slow mixing, then allows the coagulated colloids to agglomerate into larger flocs that can settle quickly. Although it is possible to remove effluent colloids with coagulation and flocculation, there is limited data on the ability of colloidal material produced during biological wastewater treatment processes to sorb estrogenic compounds. The data that does exist indicates a wide range of sorption capacities, although one study demonstrated only 18% removal with no biological unit involved.134 Therefore, while this approach may have some value, it is unclear to what extent this value may be as non-sorbed estrogens are not affected by this approach.
6.2 Activated carbon
Activated carbon efficiently removes nonpolar organic compounds, specifically those compounds with log Kow greater than 2, primarily via hydrophobic interactions.81 Snyder et al.203 found that both powdered activated carbon (PAC) (5 mg L−1) and granular activated carbon (GAC) removed greater than 90% of estrogens (100–200 ng L−1 initial concentrations). However, dissolved organic compounds, surfactants and humic acids compete with binding sites and can block pores within the activated carbon structure.203,204 Fukuhara et al.205 also found substantial reduction in the ability of GAC to remove estrogens in secondary effluent when compared to pure water. Therefore, the use of activated carbon in treating activated carbon wastewater effluent with considerable amounts of dissolved organic compounds can be problematic.PAC is added to a treatment system as a feed “chemical”, and continuously provides fresh carbon. Furthermore, it can be used when required, such as during low-flow events when the risk of micropollutants may be greater.203 PAC performance can be improved by increasing the retention time.206,207 However, PAC use has the disadvantage of requiring proper disposal of spent PAC. GAC, on the other hand, requires regular replacement or regeneration in order to be effective in removing estrogens, particularly in wastewater effluent with relatively high levels of organic carbon.203 Thermal regeneration of GAC requires significant amounts of energy, which can add considerable expense and environmental risk to the treatment process.
6.3 Chlorination
Free chlorine, specifically hypochlorous acid (HOCl) and the hypochlorite ion (OCl−), is commonly used for disinfection of wastewater effluent. HOCl readily reacts with phenolic compounds by stepwise substitution of chlorine to the aromatic ring at the 2, 4, and 6 positions. The more highly chlorinated the phenolic reactant, the more likely the aromatic ring is to undergo oxidative rupture of the benzene ring.208 Using estrone (E1) as an example, figure 3 illustrates the proposed pathway for the degradation of estrogens by chlorination.209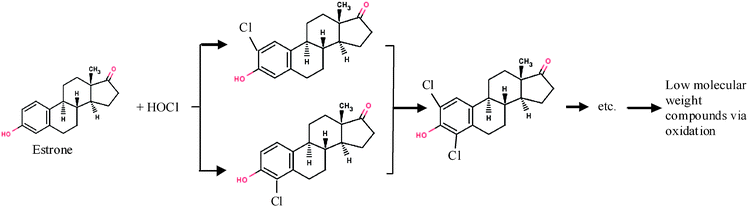 | ||
| Fig. 3 Proposed degradation pathway for estrone by chlorination. | ||
Laboratory studies have demonstrated success in transforming estrogens by chlorination.210 Lee and Von Gunten209 achieved 90% conversion of EE2 with chlorine, but increased the rate of EE2 transformation by a factor of 3 with the addition of 0.25 µM Br−. Furthermore, the brominated products of EE2 are estrogenic, albeit orders of magnitude less than the parent compound and decreasing in estrogenicity with increasing bromination.211 However, the accelerating effect of Br− diminishes in the presence of dissolved organic matter as it consumes bromine faster than estrogens. Schiliro et al.212 observed that chlorination of wastewater treatment plant effluents seemed to have reduced the overall estrogenicity in the effluent. In contrast, Matsui and Takigami170 found conflicting results and reported chlorine as ineffective in degrading estrogens. Hu et al.213,214 also observed that E2 degradation by chlorination was incomplete. One explanation for these observations may be that ammonia is chlorinated to form chloramines about one thousand times faster than phenol is chlorinated. Therefore, small amounts of chlorophenols or much slower rates of formation of chlorophenols would be expected in the presence of ammonia,208 which could be the case where a wastewater treatment plant does not include nitrification. Furthermore, there can be the potential to form chlorinated organic intermediates, which can also pose an environmental risk.
6.4 Ozonation
Ozone (O3) is often used as a disinfectant and oxidizer. O3 reacts with organic compounds either directly as molecular O3 or through the formation of hydroxyl radicals (·OH). The concentration of ·OH is generally quite low in comparison to O3 in most treatment applications. Molecular O3 is a selective electrophile that quickly reacts with amines, phenols, and double bonds in aliphatic compounds. The ·OH, however, is less selective and about nine orders of magnitude faster in reacting with organic compounds.215–218 At neutral or basic pH, ozone reacts quickly with phenols.217 The reaction of O3 with the phenolate anion is the predominant reaction, leading to a modification of the phenol ring.219 Estrogens have been removed by 90 to greater than 99% (20 to 350 ng L−1 initial concentration) with an applied O3 dose of 1 to 1.25 mg L−1.220,221 Huber et al.219 reduced estrogenicity of water containing EE2 by a factor of over 200 (up to 3 mg L−1 initial concentration). However, there was a slow reappearance of 0.1 to 0.2% of the initial EE2 concentration after ozonation. While estrogens are easily removed with O3, the concentration of ·OH must be increased in order to remove other more recalcitrant compounds. For this reason, treatment facilities may use advanced oxidation processes in which ozonation is combined with hydrogen peroxide (O3/H2O2) or ultraviolet irradiation (UV/O3).222,241,2426.5 Ultraviolet irradiation
Ultraviolet (UV) irradiation is widely used to disinfect water and wastewater effluent. Light absorption of estrogens is attributed to the photoactive phenolic group.223,224 Direct photolysis occurs via direct absorption of light. Indirect photolysis is caused when photosensitizers, such as nitrate and dissolved organic matter, absorb light and generate reactive oxygenated radicals that subsequently degrade other compounds.225 Indeed, faster photodegradation rates of EE2 have been observed in waters with greater concentrations of dissolved organic matter.226 Both monochromatic (λ = 254 nm) and polychromatic (λ > 290 nm) UV irradiation gave similar results.223 However, typical doses used for disinfection (less than 5 to 30 mJ cm−2) are several orders of magnitude lower than those used for treating micropollutants.81 As such, UV treatment alone is not an economically reasonable option for removing estrogens from wastewater. Alternatively, UV irradiation (250 W lamp) coupled with Fe(III) and H2O2 or TiO2 can remove over 98% of E1, E2 and EE2.227,228Besides artificial light, sunlight has been known to degrade estrogens via photolysis to some degree. For instance, EE2 has light absorption bands that extend into the 290–350 nm region, overlapping the solar spectrum and making the compound subject to direct photolysis.229 Laboratory studies demonstrated that both E2 and EE2 could be degraded in river waters with half-lives of at least 10 days under 12 hours of bright sunshine each day,174 and estuarine water yielded a half-life of less than 1.5 day.229 Photodegradation of E1 is expected to provide a half-life of 12 days at best.174 Surface waters contain dissolved organic compounds that absorb light to generate radicals that degrade compounds as noted above. However, in surface waters photodegradation is limited by water turbidity and light attenuation in the upper depths from the surface.226
6.6 Membrane separation
Because estrogens have a relatively small molecular size, only estrogens associated with particles or colloidal organic matter can be removed via microfiltration (MF) and ultrafiltration (UF) (0.1 to 0.4 µm). For this reason, MF and UF are of little value for steroid removal unless used as membrane bioreactors.150,203 On the other hand, Snyder et al.203 demonstrated that reverse osmosis membranes and tight nanofiltration removed greater than 99% of estrogens (100 ng L−1 initial concentration). Besides size exclusion, charge repulsion and adsorption are also responsible for estrogen rejection in membrane processes.19 In cases where the membrane pore size is larger than the compound of interest, rejection is attributed to sorption rather than sieving and is reversible.230,231 However, these removed compounds become concentrated in brine streams. Since this brine would have much greater toxicity than the wastewater influent, it cannot be disposed of in natural waters.6.7 Other techniques
Other novel treatment techniques have been explored, though many of these experiments used estrogen concentrations orders of magnitude greater than those found in typical municipal wastewater. Kimura et al.43 demonstrated that at a dose of 200 Gy (J kg−1), ionizing60 Co γ-rays generated hydroxyl radicals which eliminated the estrogen activity from E2 in wastewater. Manganese oxide (MnO2), a strong oxidizing agent and sorbent, removed 93% of EE2 (loading rate of 5 µg L−1 d−1).232,233 Electrolysis shows promise as it has removed EE2 between 85% (10 µg L−1 initial concentration in drinking water) and 98% (1 mg L−1 initial concentration in treated wastewater effluent).234 Even ultrasound (1.1 to 2.1 W mL−1 sonication units) could degrade about 80% of estrogens (initial concentrations 1 to 10 µg L−1).235 As an applied treatment technique, organo-montmorillonites have also been demonstrated to remove 68 to 85% E2 (0.1 to 1.5 mg L−1 initial concentration) via sorption.236 In addition, an unsaturated vertical-flow constructed wetland with a depth of 7.5 cm was able to remove 68, 84 and 75% of E1, E2 and EE2, respectively (0.39 to 10.49 ng L−1 initial concentrations).237 Cajthaml et al.163 also found two species of fungi that could degrade greater than 90% of EE2.7. Conclusions and future trends
Natural and synthetic estrogens are a growing environmental concern with significant ecological risks. Since wastewater treatment plant effluents and sludges are the likely primary source of these compounds in the environment, much attention has been focused on their behavior in wastewater treatment systems. There have been several mechanisms, such as heterotrophic activity, nitrifying cometabolism, other cometabolism, and abiotic transformations, offered as explanations for estrogen degradation in wastewater treatment plants. However, some of these explanations conflict with each other and prevent a clear understanding of the exact degradation paths. It is feasible that in a mixed culture such as activated sludge there could be a combination of removal mechanisms at work from both heterotrophs and nitrifiers. Nevertheless, future studies will likely research the relative importance of nitrifiers and heterotrophs.132Much attention has also been placed on studying methods of removing estrogens prior to discharging effluent or disposing waste sludge. While advanced treatment systems such as chemical removal, activated carbon, chlorination, ozonation, ultraviolet irradiation, membrane separation, and other novel approaches may be effective, their current capital and operation costs may make them not viable options.238
Past attempts to model estrogen removal in wastewater treatment plants have been confounded by the considerable removal variability observed in real systems. Even among plants of the same type and consistency in wastewater flow, it has been difficult to generate models that accurately predict the inflow, sorption, biodegradation, and outflow of estrogens. There is a keen need to obtain a reliable comprehensive estrogen mass balance model in order to optimize wastewater plant operations and improve estrogen removal.
Research to date has primarily focused on studying the behavior of a single or select few compounds. However, there could be other significant effects from the interaction of other compounds in real systems. For instance, antibiotics are known to also exist in municipal wastewater. Antibiotics inhibit the ability of bacteria to degrade estrogens and compete as the primary carbon source with the estrogens. Thus, additional research is needed to understand the interactions and competitive biodegradation pathways between these two kinds of compounds.18
Currently, available analytical techniques limit research to study estrogens in concentrations at greater than may actually exist in wastewater and natural systems. The concentration at which estrogens are studied is an important parameter as many observations and conclusions can differ between low (ng L−1 range) and high (mg L−1 range) concentrations. The case with other organic compounds is often that mineralization can occur at low concentrations, but cometabolism dominates at high concentrations.239 Biodegradation studies at concentrations greater than those found in real systems may lead to erroneous conclusions about the occurrence, rates, and products of microbial transformation. Furthermore, it is often true that there exists a threshold concentration below which a substrate cannot support bacterial growth. This observation may explain the persistence of some low levels of otherwise biodegradable organic compounds in natural environments. Therefore, it may not be clear whether these compounds are intrinsically resistant or they remain because of their low concentrations.239
The concentrations of estrogens in studies can affect the degradation kinetics as well. High concentrations of substrate can saturate the initial enzymatic reactions, leading to zero-order (linear) kinetics. This is particularly true in cases where cells cannot appreciably reproduce either because there is a threshold or because the cell number is too large. On the other hand, lower concentrations of substrate become rate-limiting, and the cell numbers increase at a rate proportional to the falling concentration of substrate, resulting in first-order kinetics.239 This phenomenon is evidenced in biodegradation studies that applied mg L−1 ranges of estrogens to activated sludge resulting in zero-order kinetics.167,168,172 Other studies that used estrogen concentrations closer to those found in wastewater (ng L−1 to µg L−1) observed first-order kinetics instead.117,149,181 The issue of analytical thresholds ought to be mitigated in the future with advances in instrumentation and detection limits. The development of robust standard methods that allow the measurement of estrogens at environmentally relevant concentrations will also go far to aid our understanding of estrogen removal mechanisms.
Estrogen sorption to biomass, colloids and other particles may remove the compounds from the effluent. However, it shifts the problem to the solid phase as these particles are removed in waste sludge. There is potential for estrogens to be removed during sludge digestion as this process requires a long SRT. However, given the potential for estrogens to desorb as well as colloidal and dissolved organic matter to act as soluble sorbents, estrogens that remain in digested sludge can still reach ground or surface water with leachate from biomass applied to soil or disposed of in landfills. Furthermore, there is a lack of research on the estrogen degradation profile at different soil depths. Another important consideration is the sorption of estrogens in wastewater treatment effluent to sediments and soils, particularly where effluents are discharged to anoxic areas such as mangroves, given the stability of estrogens where oxygen is lacking. Therefore, the preferred option is to degrade these compounds altogether rather than rely on sorption processes.
References
- J. W. Cook, E. C. Dodds, C. L. Hewett and W. Lawson, Proc. R. Soc. London, Ser. B, 1934, 114, 272–286 CrossRef CAS.
- B. S. Walker and J. C. Janney, Endocrinology, 1930, 14, 389–392 CrossRef CAS.
- S. Gimeno, H. Komen, P. W. M. Venderbosch and T. Bowmer, Environ. Sci. Technol., 1997, 31, 2884–2890 CrossRef CAS.
- L. D. Arcand-Hoy, A. C. Nimrod and W. H. Benson, Int. J. Toxicol., 1998, 17, 139–158 CrossRef CAS.
- V. J. Kramer, S. Miles-Richardson, S. L. Piernes and J. P. Giesy, Aquat. Toxicol., 1998, 40, 335–360 CrossRef CAS.
- E. J. Routledge, D. Sheahan, C. Desbrow, G. C. Brighty, M. Waldock and J. P. Sumpter, Environ. Sci. Technol., 1998, 32, 1559–1565 CrossRef CAS.
- C. E. Purdom, P. A. Hardiman, V. J. Bye, N. C. Eno, C. R. Tyler and J. P. Sumpter, Chem. Ecol., 1994, 8(4), 275–285 Search PubMed.
- C. Desbrow, E. J. Routledge, G. C. Brighty, J. P. Sumpter and M. Waldock, Environ. Sci. Technol., 1998, 32, 1549–1558 CrossRef CAS.
- EPA, Special report on environmental endocrine disruption: An effects assessment and analysis, EPA/630/R-96/012, Office of Research and Development, Washington, DC, 1997 Search PubMed.
- L. T. Chang, N. W. Yu, C. Y. Hsu and H. W. Liu, Gen. Comp. Endocrinol., 1996, 102(3), 299–306 CrossRef CAS.
- R. M. Sharpe and N. E. Skakkebaek, Lancet, 1993, 341, 1392–1395 CrossRef CAS.
- R. Stone, Science, 1994, 265, 308–310 CAS.
- E. Carlsen, A. Giwercman, N. Keiding and N. E. Skakkebaek, Environ. Health Perspect., 1995, 103, 137–139 CrossRef.
- V. Krishnan and S. Safe, Toxicol. Appl. Pharmacol., 1993, 120, 55–61 CrossRef CAS.
- U. G. Ahlborg, L. Lipworth, L. Titus-Ernustoff, C.-C. Hsieh, A. Hanberg, J. Baron, D. Trichopoulos and H. O. Adami, Crit. Rev. Toxicol., 1995, 25, 463–531 CrossRef CAS.
- J. Ashby, Environ. Toxicol. Pharmacol., 1997, 3, 87–90 CrossRef CAS.
- B. E. Gillesby and T. R. Zacharewski, Environ. Toxicol. Chem., 1998, 17, 3–14 CrossRef CAS.
- S. K. Khanal, B. Xie, M. L. Thompson, S. Sung, S.-K. Ong and J. van Leeuwen, Environ. Sci. Technol., 2006, 40(21), 6537–6546 CrossRef CAS.
- Z. Liu, Y. Kanjo and S. Mizutani, Sci. Total Environ., 2009, 407, 731–748 CrossRef CAS.
- E. J. Routledge and J. P. Sumpter, J. Biol. Chem., 1997, 272(6), 3280–3288 CrossRef CAS.
- J.-Y. Hu and T. Aizawa, Water Res., 2003, 37, 1213–1222 CrossRef CAS.
- C. Miège, S. Karolak, V. Gabet, M.-L. Jugan, L. Oziol, M. Chevreuil, Y. Levi and M. Coquery, TrAC, Trends Anal. Chem., 2009, 28(2), 186–195 CrossRef CAS.
- A. M. Brzozowski, A. C. W. Pike, Z. Dauter, R. E. Hubbard, T. Bonn, O. Engtröm, L. Öhman, G. L. Greene, J.-A. Gustafsson and M. Carlquist, Nature, 1997, 389, 753–758 CrossRef CAS.
- F. Petit, Y. Valotaire and F. Pakdel, Eur. J. Biochem., 1995, 233, 584–592 CrossRef CAS.
- W. Körner, P. Spengler, Y. Bolz, W. Schuller, V. Hanf and J. W. Metzger, Environ. Toxicol. Chem., 2001, 20(10), 2142–2151 CrossRef CAS.
- B. S. Katzenellenbogen, M. A. Miller, R. L. Eckert and K. Sudo, J. Steroid Biochem., 1983, 19, 59–68 CAS.
- H. Kohno, O. Gandini, S. W. Curtis and K. S. Korach, Steroids, 1994, 59, 572–578 CrossRef CAS.
- E. McInerney and B. S. Katzenellenbogen, J. Biol. Chem., 1996, 271, 24172–24178 CrossRef CAS.
- M. Nichols, J. M. Rientjes and A. F. Stewartm, EMBO J., 1998, 17, 765–773 CrossRef CAS.
- A. C. W. Pike, A. M. Brzozowski, J. Walton, R. E. Hubbard, A. G. Thorsell, Y. L. Li, J.-Å. Gustafsson and M. Carlquist, Structure, 2001, 9(2), 145–153 CrossRef CAS.
- O. Conroy, A. E. Sáez, D. Quanrud, W. Ela and R. G. Arnold, Sci. Total Environ., 2007, 382, 311–323 CrossRef CAS.
- J. Legler, C. E. Van den Brink, A. Brouwer, A. J. Murk, P. T. van der Saag, A. D. Vethaak and B. van der Burg, Toxicol. Sci., 1999, 48, 55–66 CrossRef CAS.
- T. Furuichi and S. Masunaga, J. Water Waste, 2002, 44, 46–51 Search PubMed.
- J. P. Giesy, V. J. Kramer, W. G. Helferich, A. Bergman and E. Klasson-Wehler, Toxicol. Appl. Pharmacol., 1997, 144, 363–376 CrossRef.
- N. Nakada, H. Nyunoya, M. Nakamura, A. Hara, T. Iguchi and H. Takada, Environ. Toxicol. Chem., 2004, 23, 2807–2815 CrossRef.
- J. M. Nelson, F. Bishay, A. V. Roodselaar, M. Ikonomou and F. C. Law, Sci. Total Environ., 2007, 374, 80–90 CrossRef CAS.
- H. Tanaka, Y. Yakou, A. Takahashki, T. Higashitani and K. Komori, Water Sci. Technol., 2001, 43(2), 125–132 Search PubMed.
- S. A. Snyder, T. L. Keith, D. A. Verbrugge, E. M. Snyder, T. S. Gross, K. Kannan and J. P. Giesy, Environ. Sci. Technol., 1999, 33, 2814–2820 CrossRef CAS.
- S. A. Snyder, D. L. Villeneuve, E. M. Snyder and J. P. Giesy, Environ. Sci. Technol., 2001, 35, 3620–3625 CrossRef CAS.
- H. Takigami, N. Taniguchi, T. Matsuda, M. Yamada, Y. Shimizu and S. Matsui, Water Sci. Technol., 2000, 42(7–8), 45–51 Search PubMed.
- L. C. Folmar, M. J. Hemner, D. Denslow, K. Kroll, J. Chen, A. Cheek, H. Richman, H. Meredith and E. G. Grau, Aquat. Toxicol., 2002, 60(1–2), 101–110 CrossRef CAS.
- G. D'Ascenzo, A. Di Corcia, A. Gentili, R. Mancini, R. Mastropasqua, M. Nazzari and R. Samperi, Sci. Total Environ., 2003, 302, 199–209 CrossRef CAS.
- A. Kimura, M. Taguchi, Y. Ohtani, Y. Shimada, H. Hiratsuka and T. Kojima, Radiat. Phys. Chem., 2007, 76, 699–706 CrossRef CAS.
- D. P. Edwards and W. L. McGuire, Endocrinology, 1980, 107(4), 884–891 CrossRef CAS.
- K. L. Thorpe, R. I. Cummings, T. H. Hutchinson, M. Schloze, G. Brighty, J. P. Sumpter and C. R. Tyler, Environ. Sci. Technol., 2003, 37(6), 1142–1149 CrossRef CAS.
- T. P. Rodgers-Gray, S. Jobling, S. Morris, C. Kelly, S. Kirby, A. Janbakhsh, J. E. Harries, M. J. Waldock, J. P. Sumpter and C. R. Tyler, Environ. Sci. Technol., 2000, 34(8), 1521–1528 CrossRef CAS.
- R. Länge, T. H. Hutchinson, C. P. Croudace and F. Siegmund, Environ. Toxicol. Chem., 2001, 20, 1216–1227 CrossRef CAS.
- K. Kiyoaki, K. Mijahima, T. Higashitani, N. Nakada, K. Komori and Y. Suzuki, J. Am. Water Resour. Assoc., 2009, 45(1), 22–34 Search PubMed.
- S. Jobling, M. Nolan, C. R. Tyler, G. Brighty and J. P. Sumpter, Environ. Sci. Technol., 1998, 32, 2498–2506 CrossRef CAS.
- J. Harries, A. Janbakhsh, S. Jobling, P. Matthiessen, J. Sumpter and C. Tyler, Environ. Toxicol. Chem., 1999, 18, 932–937 CAS.
- T. Colborn, F. S. Vom Saal and A. M. Soto, Environ. Health Perspect., 1993, 101, 378–383 CrossRef CAS.
- C. R. Tyler, S. Jobling and J. P. Sumpter, Crit. Rev. Toxicol., 1998, 28, 319–361 CrossRef CAS.
- C. R. Tyler and C. R. Routledge, Pure Appl. Chem., 1998, 70(9), 1795–1804 CAS.
- G. H. Panter, R. S. Thompson and J. P. Sumpter, Aquat. Toxicol., 1998, 42, 243–253 CrossRef CAS.
- A. Lister and G. Van Der Kraak, Water Qual. Res. J. Can., 2001, 36, 175–190 Search PubMed.
- C. D. Metcalfe, T. L. Metcalfe, Y. Kiparisis, B. G. Koenig, C. Khan, R. Hughes, T. R. Croley, R. E. March and T. Potter, Environ. Toxicol. Chem., 2002, 21, 16–23 CrossRef.
- A. D. Pickering and J. P. Sumpter, Environ. Sci. Technol., 2003, 37, 331A–336A CrossRef CAS.
- P. D. Hansen, H. Dizer, B. Hock, A. Marx, J. Sherry, M. McMaster and C. H. Blaise, TrAC, Trends Anal. Chem., 1998, 17, 448–451 CrossRef CAS.
- J. Pelley, Environ. Sci. Technol., 2003, 37, 313A–314A CrossRef CAS.
- P. Preziosi, Pure Appl. Chem., 1998, 70, 1617–1631 CrossRef CAS.
- M. Liebig, P. Egeler, J. Oehlmann and T. Knacker, Chemosphere, 2005, 59, 271–280 CrossRef CAS.
- C. Lauritzen, in Grundlagen und Klinik der menschlichen Fortpflanzung, ed. H. P. G. Schneider, C. Lauritzen and E. C. Nieschlag, Walter de Gruyter, Berlin, 1988 Search PubMed.
- E. Mutschler, in Arzneimittelwirkungen, Lehrbuch der Pharmakologie und Toxikologie. Textbook of pharmacology and toxicology, Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart, 1996 Search PubMed.
- C.-P. Yu, H. Roh and K.-H. Chu, Environ. Sci. Technol., 2007, 41, 486–492 CrossRef CAS.
- T. Fotsis, P. Järvenpää and H. Alderkreutz, J. Steroid Biochem., 1980, 12, 503–508 CrossRef CAS.
- T. Fotsis and H. Adlercreutz, J. Steroid Biochem., 1987, 28, 203–213 CrossRef CAS.
- A. Turan, in Endocrinically Active Chemicals in the Environment, Federal Environmental Agency of Germany, Berlin, pp. 15–20 Search PubMed.
- M. Muller, F. Rabenoelina, P. Balaguer, D. Patureau, K. Lemenach, H. Budzinski, D. Barceló, M. López de Alda, M. Kuster, J.-P. Delgenès and G. Hernandez-Raquet, Environ. Toxicol. Chem., 2008, 27(8), 1649–1658 CrossRef CAS.
- M. Cargouët, D. Perdiz, A. Moutassim-Souali, S. Tamisier-Karolak and Y. Lévi, Sci. Total Environ., 2004, 324(1–3), 55–66 CrossRef CAS.
- S. Matsui, H. Takigami, T. Matsuda, N. Taniguchi, J. Adachi, H. Kawami and Y. Shimizu, Water Sci. Technol., 2000, 42(12), 173–179 Search PubMed.
- H. Adlercreutz and P. Jarvenpaa, J. Steroid Biochem., 1982, 17, 639–645 CrossRef CAS.
- T. A. Ternes, P. Kreckel and J. Mueller, Sci. Total Environ., 1999, 225, 91–99 CrossRef CAS.
- J. Dray, F. Dray, F. Tiller and A. Ulman, Ann. Inst. Pasteur, 1972, 123, 853 Search PubMed.
- A. C. Johnson, A. Belfroid and A. C. Di Corcia, Sci. Total Environ., 2000, 256, 163–173 CrossRef CAS.
- J. Römbke, T. Knacker and P. Stahlschmidt-Allner, Studie über Umweltprobleme im Zusammenhang mit Arzneimitteln. Study about environmental problems in context with drugs, F + E Vorhaben Nr. 106 04 121, Umweltbundesamt, Berlin, 1996 Search PubMed.
- R. J. Williams, M. D. Jürgens and A. C. Johnson, Water Res., 1999, 33, 1663–1671 CrossRef CAS.
- M. K. Sellin, D. D. Snow, D. L. Akerly and A. S. Kolok, J. Am. Water Resour. Assoc., 2009, 45(1), 14–21 Search PubMed.
- C. Björkblom, E. Högfors, L. Salste, E. Bergelin, P.-E. Olsson, I. Katsiadaki and T. Wiklund, Environ. Toxicol. Chem., 2009, 28(5), 1063–1071 CrossRef CAS.
- D. W. Kolpin, E. T. Furlong, M. T. Meyer, E. M. Thurman, S. D. Zaugg, L. B. Barber and H. T. Buxton, Environ. Sci. Technol., 2002, 36, 1202–1211 CrossRef CAS.
- J. Legler, L. M. Zeinstra, F. Schuitemaker, P. H. Lanser, J. Bogerd, J. Brouwer, A. D. Vethaak, P. de Voogt, J. M. Albertinka and B. van der Burg, Environ. Sci. Technol., 2002, 36, 4410–4415 CrossRef CAS.
- S. A. Snyder, P. Westerhoff, Y. Yoon and D. L. Sedlak, Environ. Eng. Sci., 2003, 20(5), 449–469 CrossRef CAS.
- N. Nakada, T. Tanishima, H. Shinohara, K. Kiri and S. Tanada, Water Res., 2006, 40, 3297–3303 CrossRef CAS.
- B. L. Tan, D. W. Harker, J. F. Muller, F. D. Leusch, L. A. Tremblay and H. F. Chapman, Chemosphere, 2007, 69, 644–654 CrossRef CAS.
- M. Hewitt and M. Servos, Water Qual. Res. J. Can., 2001, 36, 319–330 Search PubMed.
- P. Chambers, M. Allard, S. Walker, J. Marsalek, J. Lawrence and M. Servos, Water Qual. Res. J. Can., 1997, 32, 659–713 Search PubMed.
- M. Servos, P. Chambers, R. Macdonald and G. Van Der Kraak, Threats to Sources of Drinking Water and Aquatic Ecosystem Health in Canada, NWRI Scientific Report Series No. 1. ISSN 1499-5905: No. 1, 2001 Search PubMed.
- D. Bennie, C. Sullivan, H.-B. Lee, T. Peart and R. Maguire, Sci. Total Environ., 1997, 193, 263–275 CrossRef CAS.
- K. Onda, S. Y. Yang, A. Miya and T. Tanaka, Water Sci. Technol., 2002, 46, 367–373 Search PubMed.
- P. Spengler, W. Körner and J. W. Metzger, Environ. Toxicol. Chem., 2001, 20, 2133–2141 CrossRef CAS.
- S. Castiglioni, R. Bagnati, R. Fanelli, F. Pomati, D. Calamari and E. Zuccato, Environ. Sci. Technol., 2006, 40(1), 357–363 CrossRef CAS.
- J. P. Sumpter and S. Jobling, Environ. Health Perspect., 1995, 103(Suppl. 7), 173–178 CAS.
- C. H. Huang and D. L. Sedlak, Environ. Toxicol. Chem., 2001, 20, 133–139 CAS.
- T. Hashimoto, K. Onda, Y. Nakamura, K. Tada, A. Miya and T. Murakami, Water Res., 2007, 41, 2117–2126 CAS.
- K. L. Thorpe, G. Maack, R. Benstead and C. R. Tyler, Environ. Sci. Technol., 2009, 43, 2976–2982 CrossRef CAS.
- A. C. Belfroid, A. Van der Horst, A. D. Vethaak, A. J. Schafer, G. B. J. Rijs, J. Wegener and W. P. Cofino, Sci. Total Environ., 1999, 225, 101–108 CrossRef CAS.
- T. A. Ternes, M. Stumpf, J. Mueller, K. Haberer, R. D. Wilken and M. Servos, Sci. Total Environ., 1999, 225, 81–90 CrossRef CAS.
- C. Baronti, R. Curini, G. Ascenzo, A. Corcia and R. Samperi, Environ. Sci. Technol., 2000, 34, 5059–5066 CrossRef CAS.
- R. F. Chimchirian, R. P. S. Suri and H. Fu, Water Environ. Res., 2007, 79(9), 969–974 CrossRef CAS.
- A. D. Vethaak, J. Lahr and R. V. Kuiper, Toxicology, 2002, 181–182, 147–150.
- T. Yoshimoto, F. Nagai, J. Fujimoto, K. Watanabe, H. Mizukoshi, T. Makino, K. Kimura, H. Saino, H. Sawada and H. Omura, Appl. Environ. Microbiol., 2004, 70(9), 5283–5289 CrossRef CAS.
- M. Clara, N. Kreuzinger, B. Strenn, O. Gans and H. Kroiss, Water Res., 2005, 39(1), 97–106 CrossRef CAS.
- S. Zorita, L. Mårtensson and L. Mathaisson, Sci. Total Environ., 2009, 407(8), 2760–2770 CrossRef CAS.
- M.-L. Janex-Habibi, A. Huyard, M. Esperanza and A. Bruchet, Water Res., 2009, 43, 1565–1576 CrossRef CAS.
- T. de Mes, G. Zeeman and G. Lettinga, Rev. Environ. Sci. Bio/Technol., 2005, 4, 275–311 Search PubMed.
- D. G. J. Larsson, M. Adolfsson-Erici, J. Parkkonen, M. Pettersson, A. H. Berg, P. E. Olsson and L. Forlin, Aquat. Toxicol., 1999, 45, 91–97 CrossRef.
- H. B. Lee and T. E. Peart, J. AOAC Intl., 1998, 81, 1209–1216 Search PubMed.
- W. Körner, U. Bolz, W. Süßmuh, G. Hiller, W. Schuller and H. Hagenmaier, Chemosphere, 2000, 40, 1131–1142 CrossRef.
- M. J. Lopez de Alda and D. Barcelo, J. Chromatogr., A, 2001, 911, 203–210 CrossRef CAS.
- K. Komori, H. Tanaka, Y. Okayasu, M. Yasojima and C. Sato, Water Sci. Technol., 2004, 38, 3047–3055 Search PubMed.
- H. M. Kuch and K. Ballschmiter, Environ. Sci. Technol., 2001, 35, 3201–3206 CrossRef CAS.
- G. W. Aherne and R. Briggs, J. Pharm. Pharmacol., 1989, 41, 735–736 CAS.
- B. Lei, S. Huang, Y. Zhou, D. Wang and Z. Wang, Chemosphere, 2009, 76, 36–42 CrossRef CAS.
- G. G. Ying, R. S. Kookana and Y. J. Ru, Environ. Int., 2002, 28(6), 545–551 CrossRef CAS.
- K. Barel-Cohen, L. S. Shore, M. Shemesh, A. Wenzel, J. Mueller and N. Kronfeld-Schor, J. Environ. Manage., 2006, 78, 16–23 Search PubMed.
- M. Esperanza, M. T. Suidan, F. Nishimura, Z. Wang, G. Sorial, A. Zalfiro, P. McCaule, R. Brenner and G. Sayles, Environ. Sci. Technol., 2004, 38, 3028–3035 CrossRef CAS.
- R. D. Holbrook, J. T. Novak, T. J. Grizzard and N. G. Love, Environ. Sci. Technol., 2002, 36, 4533–4539 CrossRef CAS.
- A. Joss, H. Andersen, T. Ternes, P. Richle and H. Siegrist, Environ. Sci. Technol., 2004, 38, 3047–3055 CrossRef CAS.
- M. Petrovic, M. Sole, M. J. L. de Alda and D. Barcelo, Environ. Toxicol. Chem., 2002, 21, 2146–2156 CrossRef CAS.
- T. A. Ternes, H. Anderson, D. Gilberg and M. Bonerz, Anal. Chem., 2002, 74(14), 3498–3504 CrossRef CAS.
- R. J. Williams, A. C. Johnson, J. J. L. Smith and R. Kanda, Environ. Sci. Technol., 2003, 37, 1744–1750 CrossRef CAS.
- R. S. S. Wu, B. S. Zhou, D. J. Randall, N. Y. S. Woo and P. K. S. Lam, Environ. Sci. Technol., 2003, 37(6), 1137–1141 CrossRef CAS.
- H. Yamamoto and H. M. Liljestrand, Water Sci. Technol., 2003, 47(9), 77–84 Search PubMed.
- G.-G. Ying and R. S. Kookana, Environ. Sci. Technol., 2003, 37(7), 1256–1260 CrossRef CAS.
- I. Flemming and H. S. Bent, in Evaluation of analytical chemical methods for detection of estrogens in the environment. Denmark, 2003, vol. No. 44, pp. 1–69 Search PubMed.
- Y.-X. Ren, K. Nakano, M. Nomura, N. Chiba and O. Nishimura, Water Res., 2007, 41, 3089–3096 CrossRef CAS.
- Y.-X. Ren, K. Nakano, M. Nomura, N. Chiba and O. Nishimura, Water Res., 2007, 41, 2341–2348 CrossRef CAS.
- T. A. Ternes and A. Joss, in Human Pharmaceuticals, Hormones and Fragrances – The Challenge of Micropollutants in Urban Water Management. IWA Publishing, London, 2006 Search PubMed.
- G. H. Panter, R. S. Thompson, N. Beresford and J. P. Sumpter, Chemosphere, 1999, 38, 3579–3596 CrossRef CAS.
- M. Carballa, F. Omil and J. M. Lema, Environ. Sci. Technol., 2007, 41(3), 884–890 CrossRef CAS.
- K. Langford and J. N. Lester, in Endocrine Disruptors in Wastewater and Sludge Treatment Processes, ed. J. W. Birkett and J. N. Lester, CRC Press, Boca Raton, FL, 2003, pp. 103–144 Search PubMed.
- T. W. Yi, W. F. Harper, R. D. Holbrook and N. G. Love, J. Environ. Eng., 2006, 132(11), 1527–1529 CrossRef CAS.
- T. Yi and W. F. Harper, Water Res., 2007, 41, 1543–1553 CrossRef CAS.
- Y. Suzuki and T. Maruyama, Water Res., 2006, 40, 1061–1069 CrossRef CAS.
- A. Svenson, A. S. Allard and M. Ek, Water Res., 2003, 37, 4433–4443 CrossRef CAS.
- H. Andersen, H. Siegrist, B. Sørensen and T. A. Ternes, Environ. Sci. Technol., 2003, 37(18), 4021–4026 CrossRef.
- O. Braga, G. A. Smythe and A. J. Schafer Feitz, Environ. Sci. Technol., 2005, 39, 3351–3359 CrossRef CAS.
- M. R. Servos, D. T. Bennie, B. K. Burnison, A. Jurkovic, R. McInnis, T. Neheli, A. Schnell, P. Seto, S. A. Smyth and T. A. Ternes, Sci. Total Environ., 2005, 336, 155–170 CrossRef CAS.
- P. Adler, T. Steger-Hartmann and W. Kalbfus, Acta Hydrochim. Hydrobiol., 2001, 29, 227–241 CrossRef CAS.
- F. C. Robert, P. S. S. Rominder and H. X. Fu, Sci. Total Environ., 2009, 407(2), 731–748 CrossRef CAS.
- L. Lishman, S. A. Smyth, K. Sarafin, S. Kleywegt, J. Toito, T. Peart, B. Lee, M. Servos, M. Beland and P. Seto, Sci. Total Environ., 2006, 367(2–3), 544–558 CrossRef CAS.
- A. Lagana, A. Bacaloni, I. D. Leva, A. Faberi, G. Fago and A. Marino, Anal. Chim. Acta, 2004, 501, 79–88 CrossRef CAS.
- A. C. Johnson and R. J. Williams, Environ. Sci. Technol., 2004, 38, 3649–3658 CrossRef CAS.
- A. Bertin, P. A. Inostroza and R. A. Quinones, Sci. Total Environ., 2009, 407, 4965–4971 CrossRef CAS.
- N. Kreuzinger, M. Clara, B. Strenn and H. Kroiss, Water Sci. Technol., 2004, 50(5), 149–156 Search PubMed.
- T. A. Ternes, in Pharmaceuticals and Personal Care Products in the Environment, ed. C. G. Daughton and T. L. Jones-Lepp, American Chemical Society, Washington, DC, 2001, pp 1–16 Search PubMed.
- J. S. Vader, C. G. van Ginkel, F. M. G. M. Sperling, J. de Jong, W. de Boer, J. S. de Graaf, M. van der Most and P. G. W. Stokman, Chemosphere, 2000, 41(8), 1239–1243 CrossRef CAS.
- T. Hashimoto and T. Murakami, Water Res., 2009, 43, 573–582 CrossRef CAS.
- E. Namkung and B. E. Rittmann, J. Water Pollut. Control Fed., 1987, 59(7), 670–678 Search PubMed.
- M. Clara, B. Strenn, E. Saracevic and N. Kreuzingber, Chemosphere, 2004, 56, 843–851 CrossRef CAS.
- B. De Gusseme, B. Pycke, T. Hennebel, A. Marcoen, S. E. Vlaeminck, H. Noppe, N. Boon and W. Verstraete, Water Res., 2009, 43, 2493–2503 CrossRef CAS.
- T. A. Ternes, N. Herrmann, M. Bonerz, T. Knacker, H. Siegrist and A. Joss, Water Res., 2004, 38, 4075–4084 CrossRef CAS.
- K. Xu, W. F. Harper and D. Zhao, Water Res., 2008, 42(3146), 3152.
- T. Yi and W. F. Harper, Proceedings of the 78th Annual Water Environment Federation Technical Exposition and Conference, Washington, D.C., 2005 Search PubMed.
- L. Sayavedra-soto, N. Hommes, S. Russell and D. Arp, Mol. Microbiol., 1996, 20(3), 541–548 CrossRef CAS.
- M. Carballa, G. Fink, F. Omil, J. M. Lema and T. Ternes, Water Res., 2008, 42, 287–295 CrossRef CAS.
- A. C. Layton, B. W. Gregory, J. R. Seward, T. W. Schultz and G. S. Sayler, Environ. Sci. Technol., 2000, 34, 3925–3931 CrossRef CAS.
- K. M. Lai, K. L. Johnson, M. D. Scrimshaw and J. N. Lester, Environ. Sci. Technol., 2000, 34, 3890–3894 CrossRef CAS.
- T. Urase and T. Kikuta, Water Res., 2005, 39, 1289–1300 CrossRef CAS.
- T. Kikuta and T. Urase, Environ. Eng. Res. JSCE, 2002, 39, 87–98 Search PubMed.
- B. Stumpe and B. Marschner, Chemosphere, 2009, 74, 556–562 CrossRef CAS.
- B. Pauwels, K. Wille, H. Noppe, H. De Brabander, T. Van de Wiele, W. Verstraete and N. Boon, Biodegradation, 2008, 19, 683–693 CrossRef CAS.
- A. C. Johnson, R. J. Williams, P. Simpson and R. Kanda, Environ. Pollut., 2007, 147, 194–202 CrossRef CAS.
- T. Cajthaml, Z. Křesinová, K. Svodobová and M. Möder, Chemosphere, 2009, 75, 745–750 CrossRef CAS.
- L. S. Shore, M. Gurevitz and M. Shemesh, Bull. Environ. Contam. Toxicol., 1993, 51, 361–366 CrossRef CAS.
- A. C. Johnson and J. P. Sumpter, Environ. Sci. Technol., 2001, 35, 4697–4703 CrossRef CAS.
- R. L. Gomes, M. D. Scrimshaw and J. N. Lester, Environ. Sci. Technol., 2009, 43, 3612–3618 CrossRef CAS.
- S. Weber, P. Leuschner, P. Kämpfer, W. Dott and J. Hollender, Appl. Microbiol. Biotechnol., 2005, 67, 106–112 CrossRef CAS.
- K. Fujii, S. Kikuchi, M. Satomi, N. Ushio-Sata and N. Morita, Appl. Environ. Microbiol., 2002, 68(4), 2057–2060 CrossRef CAS.
- J. H. Shi, Y. Suzuki, B. C. Lee, S. Nakai and M. Hosomi, Water Sci. Technol., 2002, 45(12), 175–179 Search PubMed.
- S. Matsui and H. Takigami, Awwa 2000 Annual Conference, Sunday Workshop, Denver, CO, 2000 Search PubMed.
- H. B. Lee and D. Liu, Water, Air, Soil Pollut., 2002, 134, 351 CrossRef.
- J. Ke, W. Zhuang, K. Y.-H. Gin, M. Reinhard, L. T. Hoon and J.-H. Tay, Appl. Microbiol. Biotechnol., 2007, 75, 1163–1171 CrossRef CAS.
- P. Järvenpää, T. Kosunen, T. Fotsis and H. Adlercreuta, J. Steroid Biochem., 1980, 13, 345–349 CrossRef CAS.
- M. D. Jürgens, K. I. Holthaus, A. C. Johnson, J. L. Smith, M. Hetheridge and R. J. Williams, Environ. Toxicol. Chem., 2002, 21, 480–488 CrossRef CAS.
- M. A. Dytczak, K. L. Londry and J. A. Oleszkiewicz, Water Environ. Res., 2008, 80(1), 47–52 CAS.
- R. Haiyan, J. Shulan, N. ud din Ahman, W. Dao and C. Chengwu, Chemosphere, 2007, 66(2), 340–346 CrossRef.
- B. E. Rittmann and P. L. McCarty, in Environmental Biotechnology: Principles and Applications, McGraw-Hill Higher Education, New York, 2001 Search PubMed.
- M. R. Hyman and P. M. Wood, Biochem. J., 1983, 212, 31–37 CAS.
- M. R. Hyman, I. B. Murton and D. J. Arp, Appl. Environ. Microbiol., 1988, 54, 3187–3190 CAS.
- E. W. Bergink, H. J. Koosterboer, W. H. M. van der Velden, J. van der Viles and M. S. Winter, in Steroids and Endometrial Cancer, ed. V. M. Jasonni, Raven Press, New York, 1983 Search PubMed.
- J. Shi, S. Fujisawa, S. Nakai and M. Hosomi, Water Res., 2004, 38, 2323–2330 CrossRef CAS.
- I. Forrez, M. Carballa, N. Boon and W. Verstraete, J. Chem. Technol. Biotechnol., 2009, 84, 119–125 CrossRef CAS.
- L. S. Gaulke, S. E. Strand, T. F. Kalhorn and H. D. Stensel, Environ. Sci. Technol., 2008, 42, 7622–7627 CrossRef CAS.
- H. Dizer, B. Fischer, I. Sepulveda, E. Loffredo, N. Senesi, F. Santana and P.-D. Hanse, Environ. Toxicol., 2002, 17(2), 105–112 CrossRef CAS.
- B. Stumpe and B. Marschner, Sci. Total Environ., 2007, 374, 282–291 CrossRef CAS.
- E. W. Peterson, R. K. Davis and H. A. Orndorff, J. Environ. Qual., 2000, 29, 826–834 CAS.
- J. Kjær, P. Olsen, K. Bach, H. C. Barlebo, F. Ingerslev, M. Hansen and B. H. Sørensen, Environ. Sci. Technol., 2007, 41, 3911–3917 CrossRef.
- S. D. Lucas and D. L. Jones, Soil Biol. Biochem., 2009, 41, 236–242 CrossRef CAS.
- S. Arnon, O. Dahan, S. Elhanany, K. Cohen, I. Pankratov, A. Gross, Z. Ronen, S. Baram and L. S. Shore, Environ. Sci. Technol., 2008, 42, 5521–5526 CrossRef CAS.
- M. Lægdsmand, H. Andersen, O. H. Jacobsen and B. Halling-Sørensen, J. Environ. Qual., 2009, 38(3), 955–964 CrossRef CAS.
- T. A. Ternes, A. Joss and H. Siegrist, Environ. Sci. Technol., 2004, 38, 392A–398A CrossRef CAS.
- H. Zhang, C.-H. Chang, F. Lü, D.-J. Lee, P.-J. He, L.-M. Shao and A. Su, Sci. Total Environ., 2009, 407, 879–886 CrossRef CAS.
- E. Noaksson, M. Linderoth, A. T. C. Bosveld, L. Norrgren, Y. Zebühr and L. Balk, Sci. Total Environ., 2003, 305, 87–103 CrossRef CAS.
- R. L. Gomes and J. N. Lester, in Endocrine Disruptors in Wastewater and Sludge Treatment Processes, ed. J. W. Birkett and J. N. Lester, CRC Press, Boca Raton, FL, 2003, pp. 177–217 Search PubMed.
- T. E. Ziegler and D. J. Wittwer, Am. J. Primatol., 2005, 67, 159–174 CrossRef CAS.
- K. P. C. Vollhardtand N. E. Shore, in Organische Chemie, Wiley-VCH Verlag, Weinheim, 2000 Search PubMed.
- R. D. Holbrook, N. G. Love and J. T. Novak, Environ. Sci. Technol., 2004, 38, 3322–3329 CrossRef CAS.
- J. L. Zhou, R. Liu, A. Wilding and A. Hibberd, Environ. Sci. Technol., 2007, 41, 206–213 CrossRef CAS.
- K. I. E. Holthaus, A. C. Johnson, M. D. Jurgens, R. J. Williams, J. J. L. Smith and J. E. Carter, Environ. Toxicol. Chem., 2002, 21(12), 2526–2535 CrossRef CAS.
- J. C. Bowman, J. L. Zhou and J. W. Readman, Mar. Chem., 2002, 77, 263–276 CrossRef.
- C. N. Duong, D. Schlenk, N. I. Chang and S. D. Kim, Chemosphere, 2009, 76, 395–401 CrossRef CAS.
- R. D. Holbrook, J. T. Novak and N. G. Love, Environ. Toxicol. Chem., 2005, 24, 2717–2724 CrossRef CAS.
- S. A. Snyder, S. Adham, A. M. Redding, F. S. Cannon, J. DeCarolis, J. Oppenheimer, E. C. Wert and Y. Yoon, Desalination, 2007, 202, 156–181 CrossRef CAS.
- Y. P. Zhang and J. L. Zhou, Water Res., 2005, 39, 3991–4003 CrossRef CAS.
- T. Fukuhara, S. Iwasaki, M. Kawashima, O. Shinohara and I. Abe, Water Res., 2006, 40(2), 241–248 CrossRef CAS.
- Y. Yoon, P. Westerhoff and S. A. Snyder, Water, Air, Soil Pollut., 2005, 166, 343–351 CrossRef CAS.
- P. Westerhoff, Y. Yoon, S. Snyder and E. Wert, Environ. Sci. Technol., 2005, 39, 6649–6663 CrossRef CAS.
- S. D. Faust and J. V. Hunter, in Principles and applications of water chemistry, John Wiley and Sons, New York, 1967 Search PubMed.
- Y. Lee and U. von Gunten, Environ. Sci. Technol., 2009, 43, 480–487 CrossRef CAS.
- P. West, Awwa 2000 Annual Conference, Sunday Workshop, Denver, CO, 2000 Search PubMed.
- A. Flores and E. M. Hill, Chemosphere, 2008, 73(7), 1115–1120 CrossRef CAS.
- T. Schiliro, C. Pignata, R. Rovere, E. Fea and G. Gilli, Chemosphere, 2009, 75, 335–340 CrossRef CAS.
- J.-Y. Hu, T. Aizawa and S. Kubo, Environ. Sci. Technol., 2002, 36, 1980–1987 CrossRef CAS.
- J.-Y. Hu, S. J. Cheng, T. Aizawa, Y. Terao and S. Kunikane, Environ. Sci. Technol., 2003, 37, 5665–5670 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Ref. Data, 1988, 17, 513–851 Search PubMed.
- W. R. Haag and C. C. D. Yao, Environ. Sci. Technol., 1992, 26, 1005–1013.
- J. Hoigné and H. Bader, Water Res., 1983, 17(2), 173–183 CrossRef CAS.
- J. Hoigné and H. Bader, Water Res., 1983, 17(2), 185–194 CrossRef CAS.
- M. M. Huber, T. A. Ternes and U. Von Gunten, Environ. Sci. Technol., 2004, 38, 5177–5186 CrossRef CAS.
- S. A. Snyder, E. C. Wert, D. J. Rexing, R. E. Zegers and D. D. Drury, Ozone: Sci. Eng., 2006, 28, 445–460 CrossRef CAS.
- T. Hashimoto, K. Takahashi and T. Murakami, Water Sci. Technol., 2006, 54(10), 87–93 Search PubMed.
- E. J. Rosenfeldt and K. G. Linden, Environ. Sci. Technol., 2004, 38, 5476–5483 CrossRef CAS.
- P. Mazellier, L. Méité and J. De Laat, Chemosphere, 2008, 73, 1216–1223 CrossRef CAS.
- H. M. Coleman, E. J. Routledge, J. P. Sumpter, B. R. Eggins and J. A. Byrne, Water Res., 2004, 38, 3233–3240 CrossRef CAS.
- R. G. Zeep and D. M. Cline, Environ. Sci. Technol., 1977, 11, 359–366 CAS.
- V. Matamoros, A. Duhec, J. Albaigés and J. M. Bayona, Water, Air, Soil Pollut., 2009, 196, 161–168 CrossRef CAS.
- M. J. Benotti, B. D. Stanford, E. C. Wert and S. A. Snyder, Water Res., 2009, 43, 1513–1522 CrossRef CAS.
- X. Feng, S. Ding, J. Tu, F. Wu and N. Deng, Sci. Total Environ., 2005, 345, 229–237 CrossRef CAS.
- Y. Zuo, K. Zhang and Y. Deng, Chemosphere, 2006, 63, 1583–1590 CrossRef CAS.
- S. Chang, D. T. Wait, A. I. Schafer and A. G. Fane, Desalination, 2002, 146, 381–386 CrossRef CAS.
- S. Chang, D. T. Wait, A. I. Schafer and A. G. Fane, Environ. Sci. Technol., 2003, 37, 3158–3163 CrossRef CAS.
- I. Forrez, M. Carballa, H. Noppe, H. De Brabander, N. Boon and W. Verstraete, Water Res., 2009, 43, 77–86 CrossRef CAS.
- J. De Rudder, T. van de Wiele, W. Dhooge, F. Comhaire and W. Verstraete, Water Res., 2004, 38, 184–192 CrossRef CAS.
- B. Pauwels, S. Deconink and W. Verstraete, J. Chem. Technol. Biotechnol., 2006, 81, 1338–1343 CrossRef CAS.
- H. Fu, R. P. S. Suri, R. F. Chimchirian, E. Helmig and R. Constable, Environ. Sci. Technol., 2007, 41(16), 5869–5874 CrossRef CAS.
- J. Zhou, Y. Wan and L. Liao, Ninth International Congress for Applied Mineralogy, Brisbane, Australia, 2008 Search PubMed.
- H.-L. Song, K. Nakano, T. Taniguchi, M. Nomura and O. Nishimura, Bioresour. Technol., 2009, 100, 2945–2951 CrossRef CAS.
- O. A. H. Jones, P. G. Green, N. Voulvoulis and J. N. Lester, Environ. Sci. Technol., 2007, 41, 5085–5089 CrossRef CAS.
- M. Alexander, Environ. Sci. Technol., 1985, 19(2), 106–111.
- M. Bodzek and M. Dudziak, Desalination, 2006, 198, 24–32 CrossRef CAS.
- P.-J. Chen, K. G. Linden, D. E. Hinton, S. Kashiwada, E. J. Rosenfeldt and S. W. Kullman, Chemosphere, 2006, 65(7), 1094–1102 CrossRef CAS.
- P.-J. Chen, E. J. Rosenfeldt, S. W. Kullman, D. E. Hinton and K. G. Linden, Sci. Total Environ., 2007, 376(1–3), 18–26 CrossRef CAS.
- Z. Zhao, K. F. Knowlton and N. G. Love, in Fate of Pharmaceuticals in the Environment and in Water Treatment Systems, ed. D. S. Aga, CRC Press, Boca Raton, FL, 2008, ch. 13, pp. 291–329 Search PubMed.
- F. X. M. Casey, G. L. Larsen, H. Hakk and J. Simunek, Environ. Sci. Technol., 2003, 37, 2400–2409 CrossRef CAS.
- F. X. M. Casey, J. Simunek, J. Lee, G. L. Larsen and H. Hakk, J. Environ. Qual., 2005, 34, 1372–1379 CrossRef CAS.
- B. S. Das, L. S. Lee, P. S. C. Rao and R. P. Hultgren, Environ. Sci. Technol., 2004, 38, 1460–1470 CrossRef CAS.
- P. E. Stackelberg, E. T. Furlong, M. T. Meyer, S. D. Zaugg, A. K. Henderson and D. B. Reissman, Sci. Total Environ., 2004, 329(1–3), 99–113 CrossRef CAS.
- T. Heberer, Toxicol. Lett., 2002, 131(1–2), 5–17 CrossRef CAS.
- M. J. Focazio, D. W. Kolpin, K. K. Barnes, E. T. Furlong, M. T. Meyer, S. D. Zaugg, L. B. Barber and M. E. Thurman, Sci. Total Environ., 2008, 402(2–3), 201–216 CrossRef CAS.
Footnote |
| † Part of a themed issue dealing with water and water related issues. |
| This journal is © The Royal Society of Chemistry 2010 |