Towards the conversion of carbohydrate biomass feedstocks to biofuels via hydroxylmethylfurfural
Olusola O.
James
*ac,
Sudip
Maity
c,
Lamidi Ajao
Usman
b,
Kolawole O.
Ajanaku
a,
Olayinka O.
Ajani
a,
Tolu O.
Siyanbola
a,
Satanand
Sahu
c and
Rashmi
Chaubey
c
aDepartment of Chemistry, Covenant University, Canaan Land, Ota, Nigeria. E-mail: jamesoladele2003@yahoo.com; Tel: + 00 91 9031127204
bDepartment of Chemistry, University of Ilorin, Ilorin, Nigeria
cLiquid Fuels Section, Central Institute of Mining and Fuel Research, Digwadih Campus, Dhanbad 828108, JH, India
First published on 3rd September 2010
Abstract
This review appraises the chemical conversion processes recently reported for the production of hydroxylmethylfurfural (HMF), a key biorefining intermediate, from carbohydrate biomass feedstocks. Catalytic sites or groups required for the efficient and selective conversion of hexose substrates to HMF are examined. The principle of concerted catalysis was used to rationalise the dehydration of fructose and glucose to HMF in non-aqueous media. A survey of reported reaction routes to diesel-range biofuel intermediates from HMF or furfural is presented and self-condensation reaction routes for linking two or more HMF and furfural units together toward obtaining kerosene and diesel-range biofuel intermediates are highlighted. The reaction routes include: benzoin condensation, condensation of furfuryl alcohols, hetero Diels–Alder reaction and ketonisation reaction. These reaction routes are yet to be exploited despite their potential for obtaining kerosene and diesel-range biofuel intermediates exclusively from furfural or hydroxylmethylfurfural.
 Olusola O. James | Olusola O. James received his BSc and MSc (Chemistry) degrees from the University of Ilorin, Ilorin, Nigeria. He is presently a doctoral researcher at Central Institute of Mining and Fuel Research (CIMFR), Dhanbad, India. His research interests include heterogeneous catalysis with special focus on acid–base reactions, Fischer–Tropsch synthesis, biomass conversions to biofuels and chemicals. |
 Sudip Maity | Sudip Maity is working as a Scientist at the Central Institute of Mining and Fuel Research, FRI, Dhanbad, India for the last 12 years. He did his PhD in High Temperature & Pressure phase transitions studies. He is working in the field of catalyst research for Fischer–Tropsch synthesis for coal-to-oil and gas-to-oil technologies. He is also working on the molecular structure of coal using XRD and the production of hydrogen from renewable sources by non-thermal plasma techniques. He has several patents and publications. |
Lamidi Ajao Usman has been an organic chemistry lecturer in the department of Chemistry University of Ilorin, Ilorin Nigeria. He obtained his PhD from Ladoke Akintola University of Technology Ogbomoso, Nigeria, under the supervision of Prof. N. O. Olawore. His research interests are Food chemistry, oleo- & biochemicals and more recently essential oils. He is co-author of over eighteen publications. |
Kolawole O. Ajanaku received his PhD degree in Industrial Chemistry from the University of Ibadan, Nigeria. He is currently a lecturer at department of Industrial Chemistry, Covenant University, Ota, Nigeria. His research interests include material science, corrosion studies, solid waste management, biofuels and biomass conversions. He has co-authored over 25 peer-reviewed publications. |
Broader contextShort-carbon-chain alcohols (ethanol and butanol) are the most prominent biofuels from carbohydrate biomass. Production of these biofuels is based on relatively slow fermentation and energy-demanding distillation processes. In recent times, the range of biofuels obtainable from carbohydrate biomass has increased considerably with the use of faster and energy efficient chemical catalytic methods. One of the products of chemical catalytic methods is dimethylfuran (DMF). DMF is considered to be a better fuel than ethanol in terms of production efficiency, energy density, handling and storage. This has lead to greater interest in chemical catalytic methods for producing a wide range of fuels from carbohydrate substrates. To this end, hydroxylmethylfurfural (HMF) has been identified as a key intermediate. To date kerosene and diesel-range fuel intermediates have not been produced from HMF without the need for complementary compounds or feedstocks (obtained using a biological method or derived from petroleum). Here, we point out that kerosene and diesel-fuel intermediates can be obtained with HMF alone as the feedstock via self-coupling (C–C bond formation). The resulting intermediates can then be converted to their respective fuels in a single step via hydrogenolysis as in the case of HMF to DMF. |
1. Introduction
Our global society is rapidly evolving and the demand for energy in increasing at a high rate. More than eighty percent of worldwide energy needs are presently being met from fossil fuels and other non-renewable energy resources. In particular, the transportation sector, which is witnessing the fastest growth rate in energy consumption, derives its fuels almost entirely from petroleum crude. Correspondingly, this sector also has associated with it the highest growth rate in the emission of green house gases (GHGs) over the last decade. However, increasing anthropogenic emission of GHGs had been linked to global warming; and global warming is a major concern worldwide. This accounts for the global advocacy for a paradigm shift from fossil fuels energy resources to renewable energy systems. Another motivation for the search for alternatives to fossil fuels is the quest for some measure of independence and security in terms of energy supply. Extensively investigations are being carried on development of efficient and economic systems for harnessing energy from carbon-free renewable sources (such as solar, hydroelectric, wind, geothermal, tidal and nuclear) in order to expand the spectrum of their applications at competitive prices compared to the existing fossil fuels based systems.1,2However, the application of carbon-free renewable energy resources in the transportation sector appears not to be feasible in the near future. Hence, the production of transportation fuels from carbon-neutral biomass is widely accepted as a sustainable alternative to petroleum-derived fuels. In this respect, the biofuel market has increased greatly in recent times and a competitive biofuels sector has become or assumed both national and global strategic goals towards sustainable development. In this sector, bioethanol and biodiesel (from carbohydrate and lipid biomass respectively) are the main global biofuel commodities. Although bioethanol and biodiesel are classified as first generation biofuels, their production has been associated with concerns about adverse effect on global food supply, loss of forest resources and threats to biodiversity; the rate of consumption of biodiesel and its incorporation rate in the energy mixed target of the European Commission for the transportation sector significantly outpaced that of bioethanol (see Fig. 1).2,3
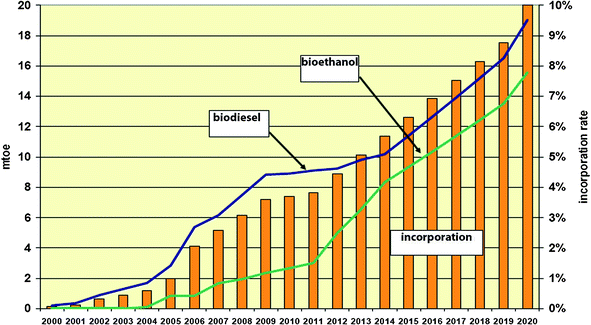 | ||
| Fig. 1 Biodiesel and bioethanol demand and the incorporation rate until 2020 in the . | ||
This is due to the properties of biodiesel which make it a closer substitute for its petroleum counterpart than bioethanol. Biodiesel can be used directly in diesel engines without prior modification. Conversely, bioethanol is not a close substitute for petrol and it cannot be used directly in petrol engines. It is generally employed as a blend with petrol up to 85%, E85, in flexible fuel vehicles. In addition, ethanol is highly hygroscopic and slightly corrosive. Biodiesel is typically produced through chemical catalytic methods and the net energy balance of the process is generally adjudged to be significantly positive. Most commercial processes for conversion of carbohydrate biomass to biofuels are via biological methods which are characterised by slower reaction rates than chemical catalytic methods. Also, the net energy balance of bioethanol production via fermentation of sugars followed by distillation had been indicated to be low and in some cases negative.4
Carbohydrate biomass resources are abundant, widely distributed and cheaper than lipid biomass, thus, production of biofuels from carbohydrate biomass is expected to be more sustainable than lipid biomass. The above, in addition to the aforementioned limitations of bioethanol as a direct substitute for petrol, has inspired the development of chemical catalytic methods for producing promising biofuels from carbohydrate biomass. In this regard, hydroxylmethylfurfural (HMF) had been identified as a key biorefining intermediate for chemicals and biofuels. The potential of HMF as a biofuel intermediate had been demonstrated by Dumesic and associates.5,6
A biofuel that will qualify as a transportation fuel needs to meet criteria that are similar to their counterparts obtained from petroleum. The molecular formula of HMF, C6H6O3, reveals that it is oxygen rich and hydrogen deficient compared to other petroleum-derived fuels. Its conversion to a compound that will be useful as a transportation fuel requires processes that will deplete and enrich its oxygen and hydrogen contents respectively. It is also obvious from the molecular formula that HMF is not of sufficiently long carbon chain to produce a compound that will be suitable as a diesel-range fuel. One of the strategies used for increasing the carbon chain length of HMF involves carbon–carbon (C–C) coupling through condensation with a suitable compound of appropriate carbon length. This yields another intermediate compound which can be subsequently transformed to diesel-range biofuel by decreasing its oxygen and increasing its hydrogen content. It is observed that the need to couple HMF (through C–C bond formation) with another compound (which implies another feedstock) in order to form a diesel-range intermediate from HMF was due to the limitation imposed by the reaction route adopted. HMF is a multifunctional molecule that can undergo a variety of reactions including self-condensation through C–C bond formation which can afford intermediates to kerosene and diesel-range fuels. These self-condensation reactions of HMF are yet to be explored toward biofuel production. The highlights of these reactions constitutes the main focus of the paper. However, for a better appreciation of the conversion processes from carbohydrate substrates to biofuels, we appraise recent developments in the production of HMF from different substrates.
2. Production of hydroxylmethylfurfural
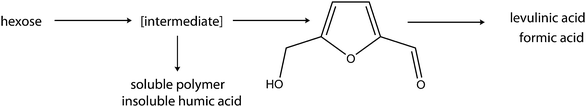
HMF is expected to play a central role as a precursor to chemicals and biofuels from carbohydrate biomass. Hence a cheap, readily available starting material and efficient process for the commercial production of HMF is one of the major challenges that needs to be solved in order to make biofuels competitive with fossil fuels. Although the investigation of the chemistry of HMF dates back to the 19th century, research on this important chemical is still ongoing. Past research findings in this area are summarised in review reports, notable among which are those of Faury et al.,7 Kustar,8 Cottieras and Desertes9 and lately Lewkowski.10 The report of Lewkowski on this subject to the best of our knowledge appears to be the most recent and exhaustive. It discusses the synthesis, chemistry and applications of HMF and its derivatives. It also highlights the important aspects of previous reviews by several researchers.It is generally accepted that the synthesis of HMF is based on the triple dehydration of hexose (Scheme 1). Apart from hexoses, other substrates that can be used are oligo- and polysaccharides. The dehydration reaction can occur in either aqueous or non-aqueous media, usually in the presence of acid catalysts. The dehydration of hexoses to HMF, when carried out in non-aqueous media, is found to be more efficient in terms of HMF yield and reaction temperature than those in aqueous media. The higher HMF yields in non-aqueous solvents have been attributed wholly or in part to the inhibition of the degradation of HMF to levulinic acids. Non-aqueous solvents such as dimethylsulfoxide (DMSO), n-butanol, acetone, dioxane, polyglycol ether and dimethylformamide (DMF) have been used as the reaction medium in the dehydration of hexose to HMF. Among the non-aqueous solvents, reports on the use of DMSO constitute the greatest number in the literature. The dehydration of fructose in DMSO was also found to be possible in the absence of a catalyst.11 However, the use of these solvents also suffers from the drawback of the poor solubility of hexose and the problem of separation of the HMF from the reaction medium. In addition, there are concerns about the economic feasibility and ecological impact of the use of the non-aqueous solvents in the large -scale production of HMF.12,13 This may account for why there is not yet a process based on a non-aqueous medium system for the commercial production of HMF. However, the solubility problems can be addressed by the application of mixed-solvent (water–organic) systems. Here non-aqueous solvents are usually miscible with water and thus act as phase modifiers of the aqueous medium.
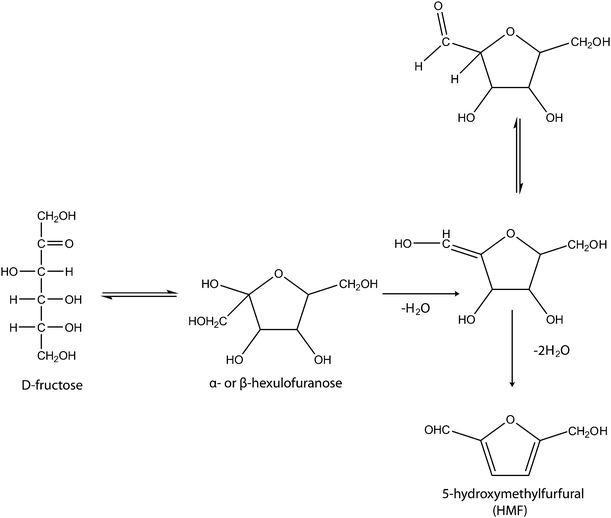
The common hexose substrates normally used for the production of HMF are fructose and glucose. It has been established from studies that fructose is more reactive than glucose under identical conditions. Hence, fructose is a more efficient and selective substrate than glucose in the synthesis of HMF. Glucose and fructose can be in their cyclic and acyclic forms in aqueous and non-aqueous media; hence, their dehydration to HMF has been proposed to be possible through either cyclic or acyclic pathways. Moreover, it has been experimentally demonstrated by Antal et al.12 that the mechanism of formation of HMF from hexose is via cyclic intermediates. It involves the sequential elimination of three molecules of water from the hexose substrate in its cyclic form (see Scheme 2). The elimination of the first water molecule is the rate-determining step. It leads to an enol intermediate and is hence tagged the ‘enolisation’ step. Fructose enolyses more readily than glucose, this accounts for why most studies on the synthesis of HMF involve the use of fructose as the substrate.
2.1 Catalytic site requirement for selective conversion of fructose to HMF
In this section, we attempt to identify and draw attention to the catalytic site requirements for the efficient and selective conversion of fructose to HMF. The dehydration of fructose is known to be catalysed by Brønsted acids as well as Lewis acids. The review by Lewkowski10 indicates that nearly a hundred inorganic and organic compounds positively qualify as catalysts for the synthesis of HMF. The catalysts are divided into five groups (see Table 1).Dehydration of fructose to HMF is traditionally carried out in aqueous media. The reaction is generally characterised by poor selectivity for HMF. However, a number of reports showed that selectivity for HMF is enhanced when the reaction is carried out in non-aqueous media, notably dimethylsulfoxide (DMSO) and ionic liquids.13
| Organic acids | Inorganic acids | Salts | Lewis acids | Others |
|---|---|---|---|---|
| oxalic acid | phosphonic acids | (NH4)2SO4/SO3 | ZnCl2 | ion-exchange resins |
| levulinic acid | sulfuric acid | pyrid/PO4−3 | AlCl3 | zeolites |
| maleic acid | hydrochloric acid | aluminium salts | BF3 | |
| p-TsOH | iodine or hydroiodic acid generated in situ | Th and Zr ions/zirconium phosphates | ||
| Ions: Cr, Al, Ti, Ca, In | ||||
| ZrOCl2 | ||||
| Vo(SO4)2, TiO2 | ||||
| Zr-, Cr-, Ti-porphyrin |
According to Hegarty and Dowling,15 enolisation of aldehydes in aqueous media is via concerted catalysis. These authors report that at high buffer concentrations the enolisation of aldehydes is markedly catalysed simultaneously by acidic and basic species. It was also asserted that concerted catalysis is largely independent of the buffering species, and that the process is overall base catalyzed.16 A similar phenomenon was reported to account for the efficiency and selectivity of the enzyme catalysed isomerisation of glucose to fructose. The catalytic activity of the enzyme was attributed to the synergistic activation of the substrate (glucose) by a Lewis acid and a weak Brønsted acid sites.17 The acid–base bi-functional model in aqueous media can be correlated with electrophilic and nucleophilic concept in non-aqueous media.
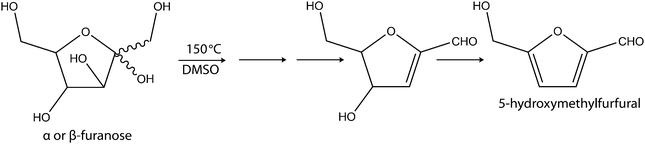
DMSO is a polar compound with a dipole on the S–O bond. The sulfur end of the bond is electrophilic while the oxygen end is nucleophilic. The observed increase of α-furanose at the expense of β-pyranose can be attributed to the isomerisation of β-pyranose to α-fructose (glucose to fructose) via synergistic activation by the nucleophilic and electrophilic parts or ends of the DMSO. As illustrated in the mechanism proposed by Ananda and associates14 (Scheme 4), the initiation step in dehydration of fructose to HMF in DMSO is via simultaneous activation of the OH group on the C2 of fructose by the electrophilic end of DMSO and H-bonding interaction between the nucleophilic end of DMSO and one of the hydrogen atoms on C1 of the fructose. Elimination of the first molecule of water is accomplished as a result of the ease of proton transfer and oxygen exchange with DMSO.
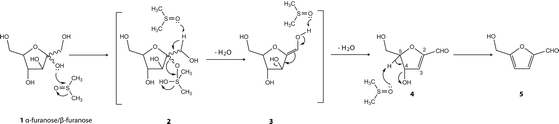 | ||
| Scheme 4 Mechanism for the dehydration of the furanose D-fructose forms 1 (α–furanose/β–furanose) to 5-hydroxymethylfurfural (5) in DMSO at 150 °C.14 | ||
The OH group at the anomeric centre of fructose attacks the electrophilic (sulfur) centre of DMSO. This brings about the formation of a covalent bond between the sulfur atom of DMSO and the oxygen atom at the anomeric centre of the substrate. This is also accompanied by a proton transfer from the OH of the anomeric centre of fructose to oxygen atom in DMSO. The H-bonding interaction between one of the hydrogen atoms on C1 and a nucleophilic centre of another DMSO molecule facilitates its elimination and the simultaneous cleavage of the C–O bond of the anomeric centre and the S–OH bond of DMSO in the DMSO–substrate complex/intermediate. An enol intermediate is formed through the exchange of an oxygen atom between DMSO and the OH oxygen atom at the anomeric centre of fructose and removal of the hydrogen atom from C1 and an OH group from the S–OH as water. The removal of the remaining two molecules of water is promoted by the nucleophilic centre (oxygen) of DMSO.
It can be said that the concerted catalysis at the initiation or enolisation step reduces the chances of condensation or polymerisation side reactions. Also, the absence of aqueous protons in the system prevents subsequent degradation of the HMF to levulinic acid. This demonstrates that bifunctional catalytic systems with readily accessible nucleophilic and electrophilic sites are required for the selective and efficient conversion of fructose to HMF.
Despite active investigations on the use of ionic liquids in the dehydration of fructose to HMF, the main focus of most of reports on the subject has been the development of efficient (low temperature) and selectivity (high yield of HMF) systems. There is a literature gap on the mechanism of dehydration of fructose to HMF in ionic liquid systems, as reported for DMSO systems. However, a critical analysis of the results presented in Table 2 reveals that the nature of the reaction medium used in the dehydration of fructose to HMF in ionic liquids is similar to that in DMSO. This suggests that the same mechanism may account for the catalytic actions in the two systems. It implies that the catalytic action of ionic liquids can also be explained in terms of concerted catalysis, as described earlier in DMSO. The catalytic action of ionic liquids can thus be rationalised in a similar manner as follows: The conversion of fructose to HMF is via a sequence of consecutive elementary steps, leading to the elimination of three molecules of water. The rate-determining step is the enolisation of fructose by the concerted activation of fructose at the C–OH bond of the anomeric carbon by the cation (electrophile) of the ionic liquid and at the C–H of C1 of the fructose via hydrogen bonding by an anion (nucleophile) of the ionic liquid. This will result in the elimination of the first molecule of H2O and formation of the enol intermediate. Again, subsequent elimination of the second and the third molecules of water is facilitated by hydrogen-bonding interaction of the anions of the ionic liquid with the hydrogen of the OH groups at C1 and the hydrogen at C5 of the intermediate respectively. Thus, it can be said that the catalytic action of the ionic liquid in the dehydration reaction will be enhanced by accessibility of the cation and the hydrogen-bonding strength of the anion.
| Ionic liquid | Reaction T/°C | Reaction time | HMF yield (%) | Ref. | |
|---|---|---|---|---|---|
| a HMIM+Cl−: 3-methylimidazolium chloride. b EMIM+Cl−: 1-ethyl-3-methylimidazolium chloride. c BMIM+BF4−: 1-butyl-3-methylimidazolium tetrafluoroborate. | |||||
| 1 | BMIM+BF4− | 80 | 3 h | 40–50 | 18 |
| 2 | BMIM+BF4− (with concurrent HMF extraction with toluene) | 80 | 3 h | 85 | 18 |
| 3 | HMIM+Cl− | 90 | 15–45 min | 92 | 19 |
| 4 | EMIM+Cl− | 80–120 | 63 | 20 | |
| 5 | EMIM+Cl− (with transition metal chloride) | 80–120 | 85 | 20 |
Therefore, the observed catalytic performance (low temperature of 90 °C and the high yield of 92%) of HMIM+Cl− can be attributed to concerted catalysis by the HMIM+ cation and Cl− anion and not due to HMIM+Cl− acting as a proton reservoir as suggested by Moreau and co-workers.19 The lower performance of EMIM+Cl− compared to HMIM+Cl− can be linked to the reduced accessibility of the cation in facilitating the enolisation step. The observed enhancement of the catalytic action of EMIM+Cl− on addition of metal halides can be attributed to the ability of the metal cations to substitute for the cation of the ionic liquid as a soft Lewis acid and/or readily accessible electrophile for the concerted catalysis step.
In a report by Qi and co-workers,22 1-butyl-3-methylimidazolium chloride, BMIM+Cl−, efficiently dehydrates fructose to HMF with a sulfonic ion-exchange resin as the catalyst. A fructose conversion of 98.6% and a HMF yield of 83.3% were reported after 10 min of reaction at 80 °C. The reaction time was reduced to 1 min when the temperature was increased to 120 °C which resulted in an HMF yield of 82.2% and nearly 100% fructose conversion. Comparison of the BMIM+Cl− and BMIM+BF4 systems, each with a sulfonic ion-exchange resin as the catalyst, indicates that the higher efficiency and selectivity observed in the BMIM+Cl− system can be attributed to a higher tendency towards concerted catalysis due to the greater hydrogen-bonding character, nucleophilicity or basicity of the chloride ion. Also, when the dehydration reaction in the BMIM+Cl−/sulfonic acid system is compared with that of EMIM+Cl− and EMIM+Cl−/CrCl2, it would have been expected that the less bulky EMIM+ should give a higher HMF yield compared to BMIM+. In contrast, the BMIM+Cl−/sulfonic acid system was reported to exhibit a higher efficiency than the EMIM+Cl− system. The result can be rationalised as arising from the presence of a sulfonic ion-exchange resin in BMIM+Cl− which generates protons in the system. The proton is a more readily accessible electrophile than EMIM+. In the above case, the anions are identical (Cl−) but the cations are different. The observed catalytic performance illustrates the importance of the accessibility of the electrophile as suggested earlier.
Moreover, a comparison of the EMIM+Cl−/CrCl2 system with the BMIM+Cl−/sulfonic ion-exchange resin shows that the two systems produce similar yields of HMF (83.0% and 83.3% respectively). Based on the fact that the addition of CrCl2 to EMIM+Cl− affords the dehydration of fructose to HMF at lower temperatures than in EMM+Cl− alone, it can be suggested that electrophilicity and/or nucleophilicity may have been enhanced or extra electrophilicity and/or nucleophilicity introduced into the system. More fundamental studies are required to clarify the factors responsible for this observation. The accessibility of the electrophile is essential to the efficiency of the dehydration process. It is also important to note that the conversion of fructose was observed to be affected by the presence of water above a threshold of 5% in the BMIM+Cl−/H+ resin system. This may be attributed to interference in the concerted catalysis as a result of the hydrogen-bonding interaction between the substrate (fructose) and water molecules. The report of Bao et al.21 also lends support to this hypothesis in respect of the influence of the hydrogen-bonding character of the anion. The cations of the ionic liquids used are identical but the anions are different. Ionic liquids with higher hydrogen-bonding strengths have been reported to be more effective than those with lower hydrogen-bonding strengths.
A recent report indicates that an ionic liquid–tungsten salt catalyst system can dehydrate fructose to HMF at much lower temperatures (less than 50 °C) and at ambient pressure.23 The hexahalides of tungsten, WCl6, was used as the catalyst. The unprecedented efficiency of this system calls for a critical analysis. Perhaps it could lead to more insight into the pathway of an economical process for the production of HMF. WCl6 is known to have a zero dipole moment, one of the rare examples of a charge-neutral hexachloride. It has octahedral geometry with equivalent W–Cl bond length (2.24–2.26 Å), which accounts for the zero value of its dipole moments. Although a chloride ion and a covalently bonded chlorine atom are nucleophilic, the chloride ion Cl− is basic, while a chlorine atom in a non-polar covalent bond is neutral. However, the basicity of a covalently bonded chlorine atom increases with increasing dipole moment of the bond. This could suggest that covalently bonded chlorine atoms interact with the hydrogens in the intermediate in such a manner that it more efficiently promotes the elimination of water than the chloride ion. This may explain the observed lowering of the dehydration temperature by about 30 °C in the dehydration of mono- and polysaccharides using a biphasic reactor system.24 The system comprised a reactive aqueous phase modified with DMSO and an organic extractive phase consisting of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (w/w) methyl isobutyl ketone (MIBK)/2-butanol mixture or dichloromethane (DCM). The reactions with the MIBK/2-butanol mixture were carried out at 443 K with mineral acids in a pH range of 1.0 to 2.0, while the reaction with DCM as the extracting solvent was conducted at 413 K and was reported not to require the use of an acid catalyst. The higher efficiency (in terms of decrease in dehydration temperature) reported with DCM as the extractive phase can be explained as being due to the presence of DCM in the reactive phase (even in catalytic amounts). Dehydration of fructose to HMF is known to occur in DMSO at reaction temperature in the absence of catalysts. DCM has two covalently bonded chlorine atoms and has a very low dipole moment; hence the covalently bonded chlorine atoms are nucleophiles that promote the elimination of water molecules from fructose and the reaction intermediates. Hence, the combined catalytic effects of DMSO and DCM account for the decrease in the dehydration temperature, even in the absence of a catalyst. It may therefore be suggested that non-polar halogen compounds, such as CCl4, CHCl3, CH3Cl, SiCl4, PCl5etc., would have a tendency to exhibit similar catalytic effects as WCl6 in the ionic liquid dehydration of fructose.
:3 (w/w) methyl isobutyl ketone (MIBK)/2-butanol mixture or dichloromethane (DCM). The reactions with the MIBK/2-butanol mixture were carried out at 443 K with mineral acids in a pH range of 1.0 to 2.0, while the reaction with DCM as the extracting solvent was conducted at 413 K and was reported not to require the use of an acid catalyst. The higher efficiency (in terms of decrease in dehydration temperature) reported with DCM as the extractive phase can be explained as being due to the presence of DCM in the reactive phase (even in catalytic amounts). Dehydration of fructose to HMF is known to occur in DMSO at reaction temperature in the absence of catalysts. DCM has two covalently bonded chlorine atoms and has a very low dipole moment; hence the covalently bonded chlorine atoms are nucleophiles that promote the elimination of water molecules from fructose and the reaction intermediates. Hence, the combined catalytic effects of DMSO and DCM account for the decrease in the dehydration temperature, even in the absence of a catalyst. It may therefore be suggested that non-polar halogen compounds, such as CCl4, CHCl3, CH3Cl, SiCl4, PCl5etc., would have a tendency to exhibit similar catalytic effects as WCl6 in the ionic liquid dehydration of fructose.
2.2 Production of HMF from glucose
The use of glucose as the substrate for the production of HMF faces the problem of the very low reactivity of glucose in most reaction systems in which fructose is readily converted to HMF. Despite this fact, the use of glucose is still desirable because of its lower price compared to fructose. For example, it was reported that glucose does not significantly react in HMIM+Cl−.19 Only 3% glucose was converted after 30 min at a temperature of 90 °C. Under the same conditions, a 42% HMF yield was obtained with fructose. A dramatic change in the reactivity of glucose in EMIM+Cl− at 100 °C was reported with the addition of a catalytic amount of CrCl2. An HMF yield of 68–70% was obtained with CrCl2. Other metal halides investigated gave much lower HMF yields. The unprecedentedly high yield obtained with CrCl2 was explained as being due to the formation of a complex, [EMIM]+[CrCl3]−, due to interaction of EMIM+Cl− and CrCl2. It was proposed that the CrCl3− anion promotes proton transfer and hydride shift in the substrate. Proton transfer facilitates the mutarotation of glucose in EMIM+Cl−, while hydride shift leads to isomerisation of glucose to fructose. The fructose is then subsequently dehydrated to HMF via concerted catalysis in EMIM+Cl−. Since the conversion of glucose to HMF was indicated to proceed via a fructose intermediate, we briefly examine glucose–fructose isomerisation before we continue on the conversion of glucose to HMFIn an alkali-catalysed system, the hydroxide ion tends to abstract a proton from the α-carbon of the carbonyl carbon to generate a carbanion. The carbanion intermediate is a reactive species; it is prone to self- or cross-condensation reactions. Moreover, the absence of a complementary catalytic site, e.g., a Brønsted acid site, which synergistically drives the reaction path to the ketose product also accounts for the low selectivity of the alkali-catalysed glucose–fructose isomerisation. A modification of the aqueous reaction medium has been shown to affect the isomerisation reaction rate. Kinetic studies of alkaline isomerisation of glucose and fructose in 0.1 M ethanol solution showed that the isomerisation of glucose to fructose, and vice versa, was accelerated markedly as the alcohol concentration increased. The reaction rates in 70% ethanol solution were found to increase 2.4 and 1.7 times the corresponding values in water solutions.28 The increased reaction rates could be attributed to the higher Brønsted acidity of ethanol compared to water which complements the hydroxide ion in promoting the isomerisation reaction. The reduced hydrogen-bonding strength of the reaction medium may also be a contributing factor. Reports on investigations on the use of solid bases as catalysts for glucose isomerisation have revealed that a number of solid bases displayed high selectivities for fructose but had low glucose conversion. The high fructose selectivities have links to the acid–base bifunctionality of the solid base catalyst.29–34
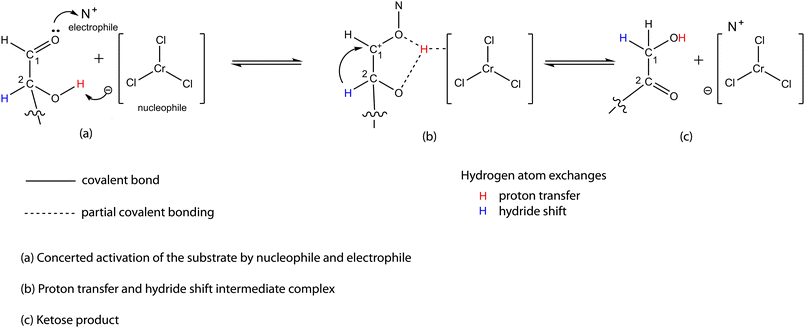 | ||
| Scheme 5 Isomerisation of glucose to fructose in non-aqueous medium via concerted catalysis. | ||
The chloride ion is less suitable as a facilitator of proton transfer because of its basicity. It tends to hold on to the proton too strongly to allow the proton transfer from C2 to C1. This accounts for the low reactivity of glucose in ionic liquid systems without CrCl2. A similar explanation is suggested for the inability of BMIM+Cl− to convert glucose to HMF as reported by Qi et al.22 However, glucose and related disaccharides were observed to be effectively converted to HMF in BMIM+Cl/CrCl3. The nucleophile here is [CrCl4]−. Using the principle of resonance, [CrCl4]− can also be assumed to have a very small dipole moment.
Furthermore, according to Dumesic et al.24 a biphasic system of a DMSO-modified aqueous phase and dichloromethane (DCM) affords an HMF yield of 53% in the dehydration of glucose in the absence of acid catalyst. As suggested earlier, DCM acts as a nucleophile to lower the temperature of dehydration of fructose to HMF by its participation in concerted catalysis. Again, the glucose is the substrate in the same system and the possibility of glucose isomerisation cannot be ruled out. The chloride atoms in DCM may facilitate a proton transfer in a similar manner to the chloride atoms in [CrCl3]− to promote the glucose isomerisation to fructose. Then the resulting fructose is further dehydrated to HMF through concerted catalysis.
2.3 Production of HMF from glucose and polysaccharides
The use of polysaccharides as the substrate for the production of HMF holds the advantage of availability and lower cost as the raw material in the future biorefinery. Efficient and selective conversion of polysaccharides especially cellulose are most desirable because of its abundance and because it is a non-food material. Starch and cellulose are made up of glucose units in the pyranose form. In starch, the pyranose rings are bonded via α-1,4-glycosidic linkages arranged in a helical structure such that the α-1,4-glycosidic linkages are easily accessible by chemicals and enzymes. However, in cellulose the pyranose rings are bonded via β-1,4-glycosidic linkages which gives a chain side-by-side arrangement. This arrangement favoured extensive intra- and inter-chain hydrogen bonding between the groups near the glycosidic bonds. Thus, the glycosidic bonds are not as readily accessible to reagents as those in starch. Therefore, the first hurdle at making cellulose chemically sensitive is the disruption of its network of hydrogen bonds. This entails solubilisation or depolymerisation of cellulose in pre-treatment reagents or systems that completely break or weaken the hydrogen bonds. Thus, the conversion of cellulose to HMF may involve three chemical processes, namely: hydrolysis, isomerisation and dehydration. A two-unit process operation could be envisaged which would consist of a hydrolysis unit and the glucose-to-HMF unit, since direct conversion of glucose to HMF has been demonstrated to be possible. In addition to these two chemical process units, a glucose and HMF isolation or purification unit may be required. But one-step conversion of cellulose to HMF will reduce the process units to two, cellulose-to-HMF and HMF isolation or purification units.One-step conversion of cellulose to HMF has been shown not only to be feasible but has also been demonstrated. This is because most ionic liquid reaction systems for conversion of glucose to HMF can sufficiently solubilise cellulose and hydrolyse cellulose in the presence of acid catalysts. Acidic ionic liquids can act as both solvent and catalyst. Examples of systems for direct conversion of cellulose to HMF are presented in Table 3.
| Solubilisation of cellulose (disruption of the hydrogen bond network) | Cleavage of the β-1,4-glycosidic bond | Isomerisation of glucose to fructose via concerted catalysis | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Reaction system | Solvent | Hydrolysis agent | Charge-neutral nucleophile | electrophile | Charged nucleophile | Reaction condition | HMF yield | Ref. | |
| Dehydration of fructose to HMF via concerted catalysis | |||||||||
| 1 | LiCl-HCl-CH2Cl CH2Cl | LiCl-HCl | HCl | CH2Cl-CH2Cl | Li+, H+ | Cl− | 65 °C, 18–30 h | 71% | 36 |
| 2 | [EMIM]Cl-CrCl2-CuCl2 | [EMIM]Cl | CrCl-CuCl2 | [EMIM]+ | Cl− | 80–120 °C | 55.4% | 37 | |
| 3 | DMA-LiCl-HCl-[EMIM]Cl-CrCl2 | DMA/LiCl(10%), [EMIM]Cl | HCl | CrCl2 | [Li(DMA)]+ [EMIM]+, Li+ | Cl− | 140 °C, 2 h | 54% | 38 |
| 4 | [AMIM]Cl-CrCl2 | [AMIM]Cl | CrCl2 | [AMIM]+ | Cl− | 50% | 39 | ||
| 5 | [BMIM]Cl-[EMIM][HSO4−]-CrCl2 | [BMIM]Cl-[EMIM][HSO4−] | [EMIM][HSO4−] | CrCl2 | [BMIM]+ [EMIM]+ | Cl− | 100 °C, 6 h | <13% | 40 |
From the results in Table 2, it is clear that the chloride ion is common to all the systems. It shows that the chloride ion is crucial to the dissolution of cellulose in the systems. The extent of solubilisation of cellulose can then be said to be a function of chloride ion concentration in the systems. The charge-neutral nucleophile components (in entries 2, 3, 4 & 5) are very similar but their electrophiles are different. So the yield of HMF and the efficiency of these systems can be attributed to the abilities of the electrophile to couple with their respective nucleophile in concerted catalysis of glucose to HMF. The lower yield of HMF in entry 5 could be attributed to inaccessibility of the charge centre in [BMIM] to the glucose due to steric effects. The system in entry 1 gives the highest HMF yield, but it is the least efficient considering its duration. The system does not give HMF directly but 5-(chloromethyl)furfural (CMF). CMF is hydrophobic, and although not itself a biofuel candidate, can be readily combined with ethanol to yield ethoxylmethylfurfural, a suitable fuel candidate compound. The chemical reactions involved here can be analysed as follows: solubilisation of the cellulose in LiCl and hydrolysis with HCl; the absence of water will yield chlorinated glucose. The chlorinated glucose is then isomerised and dehydrated to HMF in 1, 2-dichloroethane in the presence of more LiCl/HCl. The three reagents, LiCl, HCl and 1, 2-dichloroethane supply the catalytic species required for the direct conversion of cellulose to HMF.
From the above analyses, it clearly shows that the ionic liquid systems have the potential to make the goal of producing biofuels and biorefining competitive with fossil-derived fuels and petroleum refining respectively. However, the economic feasibility of the system is a function of the cost and efficiency of the ionic liquids for dissolution of cellulose. This efficiency is judged in term of the extent of dissolution and the solubilisation temperature of cellulose in the ionic liquid. It has been suggested that for efficient processing of cellulose biomass, no extra energy (for example heating) should be necessary in order to reduce the energy cost.41,42 The ability of ionic liquids to dissolve cellulose under mild conditions and the solubilisation temperature of cellulose appears to be linearly related to the viscosity of ionic liquids. Most of the common ionic liquids for dissolving cellulose are based on chloride salts which suffer from the disadvantage of high melting point and high viscosity. However, new classes of polar ionic liquids have been found to have lower melting points and viscosities than the chloride salt based ones. They are based on carboxylate salts and dimethylphosphate salts.43 The formate salts of l-ethyl-3-methylimidazolium, [EMIM] cation and 1-allyl-3-methylimidazolium [AMIM] cation were found to have lower viscosities than the corresponding chlorides salts. Cellulose was reported to dissolve in high concentration in 1-allyl-3-methylimidazolium formate ([AMIM][HCOO]−) under mild conditions. The formate-based ionic liquids have the drawback of poor thermal stability. On the other hand, [EMIM][(MeO)(Me)(PO2)] was reported to be thermally stable up to 260–290 °C and have viscosity values between 100–500 cP at 25 °C. The viscosity values are much lower than that of the chloride salts. The higher capacity of these ionic liquids to dissolve cellulose was attributed to their higher hydrogen bonding basicity over the chloride-based ones. Also the solubilisation temperatures of cellulose depend on the structure of the anions of the ionic liquid. It is expected that the use of these new ionic liquids will make possible higher yields of HMF in the direct cellulose-to-HMF process. But the acid test for the ionic liquids will be their suitability for the conversion of glucose to HMF.
In summary, the concerted catalysis principle was used to rationalise the conversion of common hexose substrates (fructose and glucose) to HMF in non-aqueous systems. The successful transformation required readily accessible nucleophiles and electrophiles. Two kinds of nucleophiles were identified, strongly charged nucleophiles that have high net dipole moments and charge-neutral nucleophiles that have very low values of net dipole moment. Conversion of glucose to HMF is via initial isomerisation to fructose before subsequent dehydration to HMF. The initial isomerisation step required the pairing of an electrophile and a charge-neutral nucleophile, however, fructose dehydration can be catalysed by both categories of nucleophiles. Addition of acid catalysts into ionic liquid systems that can efficiently convert glucose to HMF were shown to be suitable for the direct conversion of cellulose to HMF.
3. Transportation fuels or fuel intermediates from HMF and furfural
In this part of the paper, we examine efforts at transforming HMF and furfural to transportation fuels or fuel intermediates. We also discuss the conversion of HMF to a gasoline fuel and the need to increase the carbon chain length of HMF in order to obtain kerosene and diesel-range fuel intermediates. We appraise the updated strategies that have been employed in this respect and we suggest more potential reaction routes for obtaining kerosene and diesel-range fuel intermediates from HMF.3.1 Gasoline-range fuel from HMF and furfural
For a long time, lower alcohols (ethanol and butanol) were the prominent biofuels produced from carbohydrate biomass (Scheme 6). These biofuels are based on slow biochemical reactions and energy-demanding distillation processes which have generated numerous questions about the efficiency of their production.42,43
However, in recent times the range of biofuels obtainable from carbohydrate biomass has increased considerably. Part of these efforts is being championed by Dumesic and his co-workers.44 The group has demonstrated the possibility of producing hydrogen and hydrocarbons from carbohydrates and simple polyols. The process for producing hydrogen and hydrocarbons involves aqueous phase reforming (APR) and aqueous phase dehydration and hydrogenation (APD/H) respectively.44–47 The reactions opened up prospects of producing new varieties of chemicals and biofuels exclusively from carbohydrates. Among several efforts, Dumesic and co-workers demonstrated the strategies for the conversion of carbohydrate-based biomass to hydrocarbon (e.g. conversion of sorbitol to hexane) via aqueous-phase dehydration, hydrogenation and hydrogenolysis reactions. The conversion is achieved in a one-pot process with the use of a four-phase reactor. Oxygen removal through dehydration and hydrogenation transforms the hydrophilic substrate into a hydrophobic product with drastically reduced aqueous-phase solubility. The hydrophobic product readily separates out from the aqueous reaction system. These strategies afford about 90% of the energy of the carbohydrate and H2 feeds.46–48 These strategies lay the platform for the conversion of other carbohydrate substrates to hydrocarbons and new biofuel varieties. One of the applaudable contributions of Dumesic's group is the production of 2,5-dimethylfuran (DMF) from fructose via a two-step process.49 The fructose was dehydrated to HMF which was subsequently hydrogenolysed to DMF (Scheme 7).
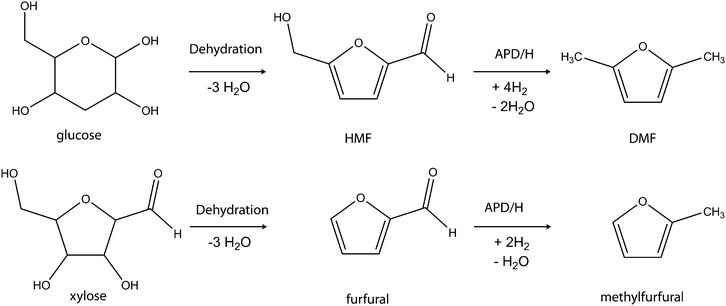 | ||
| Scheme 7 Chemical conversion of glucose to DMF and xylose to methylfurfural analogue. | ||
DMF has properties that are similar to current petroleum-based gasoline fuels and it is considered to be a suitable substitute or fuel additive for petrol engines due to its high octane number. It is also adjudged a better fuel than ethanol in terms of production efficiency, energy density, handling and storage.
3.2 Existing approaches to kerosene and diesel-range fuel intermediates from HMF and furfural
Kerosene and diesel are other important transportation fuels. These fuels have carbon chains and properties that are not met by hydrocarbons obtainable from one molecule of HMF or furfural. Thus, the production of diesel-range fuels based on the strategies described above will require self-condensation of two or more HMF or furfural molecules or their cross-condensation with other compatible compounds that are derived from renewable feedstocks. The success of this new task will require intelligent use of the intrinsic functionality already present in intermediates. The functionalities of these intermediates are –OH group, –CO group and the five-membered ring heterocyclic aromatic system. HMF has these three functionalities but the –OH group is absent in furfural. With these functionalities, HMF and furfural can undergo several self- and cross-condensation reactions. A superficial survey of the literature reveals that most of the reports on the formation of diesel-range fuel intermediates focus on cross-condensation approaches. The approaches involve the use complimentary feedstocks in order to extend the carbon chain of the resulting intermediates or biofuels. The cross-condensation strategies are discussed below.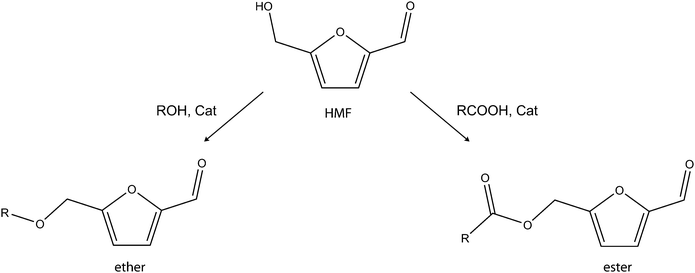 | ||
| Scheme 8 Formation of alkoxylmethylfurfural ethers and esters. | ||
In a similar carbanion reaction, malonic acid, an activated methylene compound that can be derived from biomass was suggested as a suitable coupling partner with HMF. Here, the malonic acid can link furfural or HMF by C–C bond formation via a Knoevenagel reaction, giving (C8 and C9) biofuel intermediates respectively (see Scheme 10).54
The resulting compounds in Schemes 9 and 10 are not fuels but fuel intermediates. They are rich in oxygen and will be hydrophilic. Subsequent hydrogenolysis is required to eliminate as much oxygen as possible to reduce the hydrophilicity to an acceptable level as in the case of HMF to DMF. Or complete elimination and saturation to yield the corresponding alkanes.
 | ||
| Scheme 9 Cross-aldol condensation of HMF with acetone. | ||
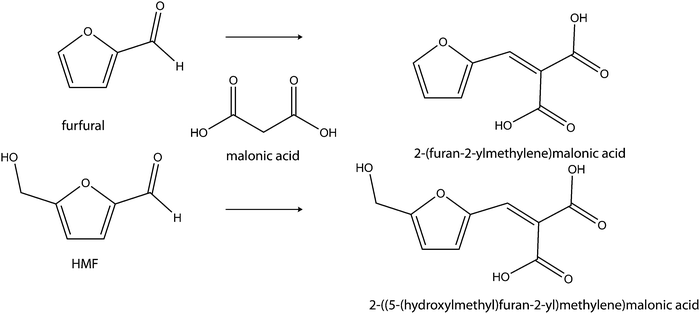 | ||
| Scheme 10 Cross-condensation of HMF and furfural with malonic acid through the Kneovenagel reaction. | ||
3.3 New strategies to kerosene and diesel-range fuel intermediates from HMF and furfural
The approaches described above for the production of kerosene and diesel-range fuel intermediates from HMF and furfural are viable routes in terms of reaction conversions and selectivities. However, the complimentary feedstocks (ethanol, acetone, ethanoic acid, malonic acid etc.) engaged in these processes may not be exclusively from renewable sources, or they may suffer from energy balance limitations inherent in their production methods. The need for complimentary feedstocks can be avoided if there are efficient reaction routes for self-condensation of HMF or furfural through the use of the same or alternative functionalities of these fuel intermediate molecules. That is, a reaction route that can link two furfurals or HMFs through their –OH groups, two HMFs through their –CO groups and two or more HMFs through the reaction between the –CO group of one with the –OH group of an other. Such reactions can lead to kerosene and diesel-range fuel intermediates that are not only obtained from single starting carbohydrate feedstocks but also from exclusively renewable sources. Within the limit of available evidence from the literature, there have yet to be any reports on the production of kerosene and diesel-range fuel intermediates that are based on self-condensation of HMF or furfural. The potential reaction routes for achieving this goal include: the benzoin reaction, the hetero Diels–Alder reaction, condensation of furfurylic alcohol and ketonisation reactions. It is important to add that the resulting compounds from the C–C coupling of HMF molecules may not be suitable for use as fuels directly. Subsequent hydrogenolysis will transform them into fuels of kerosene and diesel-range in the same manner as products in the cross-coupling approach. Below are the highlights of the proposed reaction routes for the self-condensation of HMF and furfural.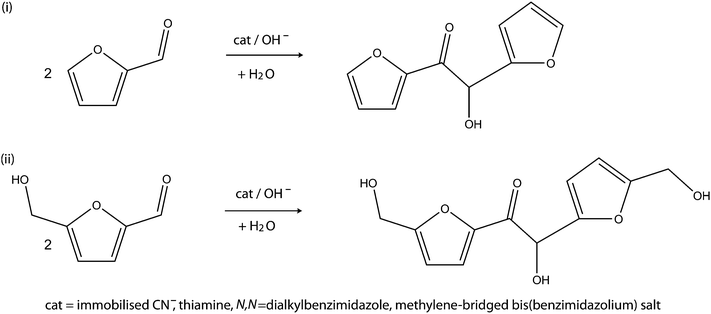
The reactivity of furan-based compounds as dienes is low because of their aromatic character, however, a number of [4 + 2] reactions involving furan have been reported in the literature. Although, Diels–Alder reactions can proceed without the need for catalysts, the reactions are slow and need to be accelerated by physical methods such as high pressure, high temperature or ultrasound ionisation. Also, Diels–Alder reactions are greatly enhanced by Lewis acid catalysis. Lewis-acid-catalysed Diels–Alder reactions proceed by coordinating to the activating group of the dienophile (for normal electron-demand Diels–Alder reactions), thereby lowering the LUMO dienophile–HUMO diene energy gap and accelerating the reaction. Specifically, Hafnium compounds have been identified as a suitable Lewis acid which provide the Diels–Alder reactions of furan in good yield.68,69 Other Lewis acids that are air stable and water tolerant have also been reported for the Diels–Alder reaction of furan with suitable dienophiles.70–72
The above highlight can be said to have established the platform for the application of Diels–Alder reactions for the formation of C12-biofuel intermediates exclusively from HMF. A C12 or C10 biofuel intermediate can be formed via the hetero Diels–Alder condensation of two HMF or furfural molecules respectively with a furan ring as the diene and the carbonyl group as the dienophile. Successive condensations of more than two units of HMF and furfural are also possible giving six- and five-carbon chain multiples of biofuel intermediates. Normal homogenous Lewis acid catalysts required to activate a carbonyl group containing a dienophile are faced with the problem of their use in excess. The process can be made greener by heterogenising the Lewis acid catalyst. This will also afford the advantage of the heterogeneous catalysts in the reaction in terms of operational simplicity, ability to be filtered and reusability.65
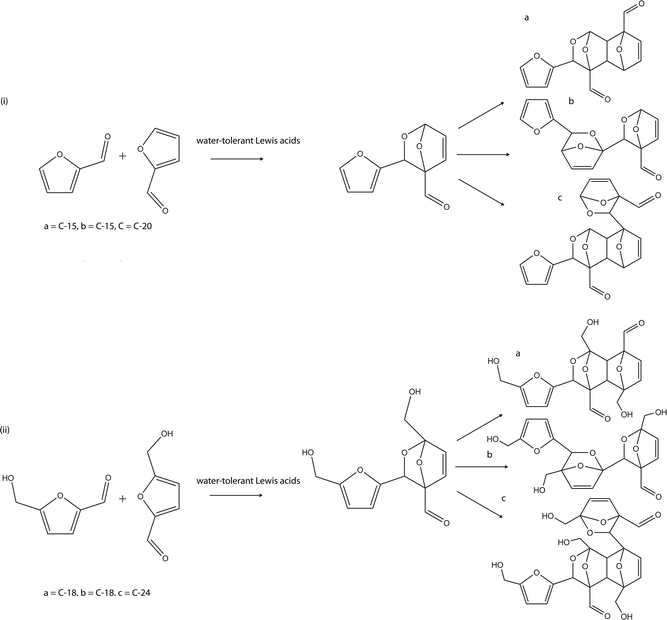 | ||
| Scheme 12 Proposed hetero Diels–Alder condensation of (i) furfural and (ii) HMF. | ||
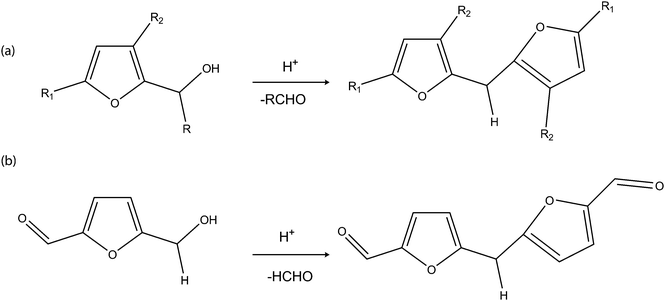 | ||
| Scheme 13 (a) Condensation of furfuryl alcohol. (b) Proposed acid condensation of HMF. | ||
The reactions in the Scheme 13 can be well adapted for the formation of C11 biofuel intermediates from HMF. Although, the atom economy in these reactions are lower when compared to that of the Diels–Alder condensation approach, the percentage carbon utilisation of biofuel intermediates is still reasonably high (91.7%) for the commercial production of diesel-range biofuel intermediates from HMF. In order to have an overall green process, the use of an immobilised, hydrophilic, strong Brønsted acid ionic liquid is suggested. The hydrophilic nature of the ionic liquid will allow close interaction of the acid site with the substrate, at the same time, the immobilisation will afford reusability of the acid and easy isolation of the product. This arrangement will not only make the process environmentally friendly but also afford intensification and optimisation of the catalyst and the solvent.
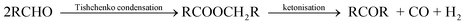 | (1) |
The ketone products will contain one carbon atom fewer than the number of carbon atoms in the substrate. The possibility of primary alcohol dehydrogenation with subsequent ketonisation had also been reported.81–84 The reaction has been found to proceed consecutively with the formation of aldehydes and esters as intermediates. Aldehydes and esters are the major products at lower temperatures while ketones are the predominant product at higher temperatures.84
 | (2) |
Furfural and HMF lack α-hydrogens to their carbonyl groups, HMF in addition has a primary hydroxyl group. They satisfy the functionality pre-requisite, hence, they are qualified as substrates for the transformation described above. Therefore, this unique reaction and catalysts can be extrapolated for the formation of longer carbon chain biofuel intermediates from furfural and HMF. The Tishchenko condensation of furfural and HMF may be sufficient enough to obtain the desired fuel product, provided the ester products have acceptable fuel properties (such as stability to oxidation, high cetane number, hydrophobicity etc.) like the alkoxylmethylfurfural esters and ethers. Otherwise, the C–C linkage via ketonisation of the esters then becomes necessary to give the ketone product (fuel intermediate).
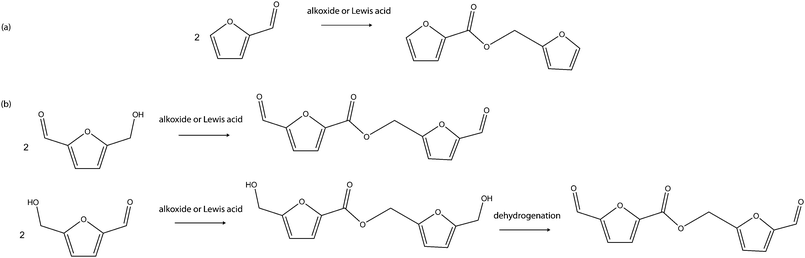 | ||
| Scheme 14 Proposed Tishcenko condensation of (a) furfural and (b) HMF. | ||
It has been showed that low acid–base activity of the catalyst is essential to the selectivity of the catalyst to ketone formation and also to minimise competitive side-reactions such as dehydration to alkene and ether formations. In line with this fact, iron oxide catalyst (100%Fe2O3, 2%Si, 1%Cr, 0.1%K) and ternary catalyst (Sn–Ce–Rh–O) have been found to have high ketonisation activities and selectivities for the reaction. Furfural and HMF contain at least one of the required functional groups for the ketonisation reaction, hence, their bimolecular condensation is expected to give C9 and C11 ketone products from furfural and HMF respectively. Furfural has just one of the required functional groups, an aldehyde group, hence, its bimolecular condensation is expected to give only the C9 ketone product. However, HMF has two functional groups, an aldehyde and a hydroxyl group, that can be engaged for the ketonisation process, multiple condensations are therefore possible in addition to the starting C11 ketone produced via a one stage bimolecular condensation like furfural. Thus, a C16, C21, C26, C31,… series of di, tri, tetra, penta,… ketone products is envisaged when HMF is the starting substrate.
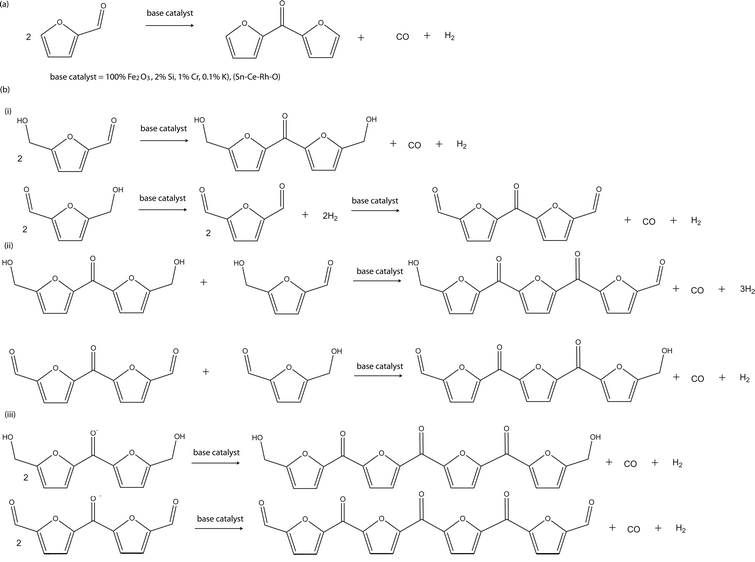 | ||
| Scheme 15 Proposed ketonisation reactions of (a) furfural and (b) HMF. | ||
Another important aspect of the application of the ketonisation reaction route for the production of longer carbon chain biofuel intermediates from HMF and furfural is the fact that ketone formation occurs in the gas phase at high temperatures. This is in contrast to the other self-condensation reaction routes suggested above as well as other cross-condensation reaction routes that have been explored for the same purpose where the reactions are carried out in the liquid phase. In those reaction routes, the reaction mixture HMF and furfural formation processes can be used directly without isolating them from their solution. In contrast, self-condensation by ketonisation will require the use of HMF and furfural in their pure forms. This places a demand of adopting HMF and furfural production routes from carbohydrate sources that enable the easy isolation at a reasonably high level of purity. An example of an HMF production route was reported by Hu et al.84 The process involves the conversion of fructose to HMF in an ethyl acetate/renewable ionic liquid (IL) biphasic system. The phase separation between the ethyl acetate and the renewable ionic liquid was shown to have no cross-contamination such that the renewable (IL) can be reused. In the same vein, the ethyl acetate containing the HMF product can be easily distilled from HMF and reused. This process affords a green process for the formation of HMF at a high level of purity. Therefore, the integration of this green process for the formation of HMF with the ketonisation reaction would enable another environmentally friendly process for the formation of longer carbon chain biofuel intermediates.
4. Conclusion
The production of transportation fuel from biomass is a world-wide strategy towards the reduction of CO2 emission from the continuous combustion of fossil fuels. This goal will naturally be actualised when the cost of producing biofuels is on a par with or cheaper than fuels from fossil sources. HMF and furfural are the key carbohydrate-derived intermediates for future biorefining. Hence, their efficient production from cheap and readily available feedstocks is paramount for achieving the goal of making biofuels economically competitive with fossil-derived fuels. Established processes for the production of HMF and furfural are based on fructose which is expensive and not readily available. Recent process developments in using readily available feedstocks (glucose and cellulose) for HMF and furfural production focused on non-aqueous homogenous catalytic processes. Efforts at producing diesel-range biofuels from carbohydrate biomass have focused on the cross-condensation of HMF or furfural with other lower carbon chain alcohols, carboxylic acids, aldehydes or ketones. The challenge envisaged here is that the feedstock for these processes may not be exclusively from renewable sources or it may have the disadvantages of poor energy balance inherent in the production of the co-reactants of the intermediates from renewable sources. Despite the availability of potential reaction routes for self-condensation of HMF and furfural from base organic chemistry, a superficial review of the literature reveals no effort in this direction. Therefore, four potential reaction routes (Schemes 11–15) which are yet to be exploited for the self-condensation of HMF and furfural were suggested and highlighted.Acknowledgements
Olusola O. James is grateful to TWAS and CSIR (India) for a TWAS-CSIR Postgraduate Fellowship Award. James is also thankful to Prof. K.O. Okonjo and T. A. Adeoye (Miss) for their encouragement and assistance.References
- S. Bilgen, S. Keleş, A. Kaygusuz, A. Sarı and K. Kaygusuz, Renewable Sustainable Energy Rev., 2008, 12(2), 372–396 CrossRef.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinasa and A. A. Romeroa, Energy Environ. Sci., 2008, 1, 542–564 RSC.
- Jens R. Rostrup-Nielsen, Science, 2005, 308, 1421–1422 CrossRef.
- Jurgen O. Metzger, Angew. Chem., Int. Ed., 2006, 45, 696–698 CrossRef CAS.
- Lanny D. Schmidt and Paul J. Dauenhauer, Nature, 2007, 447, 914–915 CrossRef CAS.
- B. Kamm, Angew. Chem., Int. Ed., 2007, 46, 5056–5058 CrossRef CAS.
- A. Faury, A. Gaset and J. P. Gorrichon, Inf. Chim, 1981, 214, 203 Search PubMed.
- B. M. F. Kuster, Starch/Staerke, 1990, 42, 314–321 Search PubMed.
- L. Cottier and G. Descotes, Trend. Heterocycl. Chem., 1991, 2, 233 Search PubMed.
- J. Lewkowski, Arkivoc, 2001,(i), 17–54.
- R. M. Musau and R. M. Munavu, Biomass, 1987, 13, 67 CrossRef CAS.
- M. J. Antal, W. S. L. Mok and G. N. Richards, Carbohydr. Res., 1990, 199, 91 CrossRef CAS.
- M. Bicker, J. Hirth and H. Vogel, Green Chem., 2003, 5, 280–284 RSC.
- A. S. Amarasekara, L. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021–3024 CrossRef CAS.
- A. F. Hegarty and J. Dowling, J. Chem. Soc., Chem., 1991, 996–997 Search PubMed.
- A. F. Hegarty, J. P. Dowling, S. J. Eustace and M. McGarraghy, J. Am. Chem. Soc., 1998, 120(10), 2290–229 CrossRef CAS.
- H. Hu, H. Liu and Y. Shi, Proteins: Struct., Funct., Genet., 1997, 27(4), 545–55 CrossRef CAS.
- C. Lansalot-Matras and C. Moreau, Catal. Commun., 2003, 4(10), 517–520 CrossRef CAS.
- C. Moreau, A. Finiels and L. Vanoye, J. Mol. Catal. A: Chem., 2006, 253(1–2), 165–169 CrossRef CAS.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS.
- Q. Bao, K. Qiao, D. Tomida and C. Yokoyama, Catal. Commun., 2008, 9(6), 1383–1388 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr., Green Chem., 2009, 11, 1327–1331 RSC.
- J. Y. G. Chan and Y. Zhang, ChemSusChem, 2009, 2(8), 731–734 CrossRef.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- S. Hu, Z. Zhang, Y. Zhou, B. Han, H. Fan, W. Li, J. Song and Y. Xie, Green Chem., 2008, 10, 1280–1283 RSC.
- C. Sievers, I. Musin, T. Marzialetti, O. M. B. Valenzuela, P. K. Agrawal and C. W. Jones, ChemSusChem, 2009, 2(7), 665–671 CrossRef CAS.
- S. Hu, Z. Zhang, Y. Zhou, J. Song, H. Fan and B. Han, Green Chem., 2009, 11, 873–877 RSC.
- R. S. Shallenberger and L. R. Mattick, Food Chem., 1983, 12(3), 159–165 CrossRef CAS.
- C. Moreau, R. Durand, A. Roux and D. Tichit, Appl. Catal., A, 2000, 193(1–2), 257–264 CrossRef CAS.
- J. Lecomte, A. Finiels and C. Moreau, Starch/Staerke, 2002, 54(2), 75–79 Search PubMed.
- M. Watanabe, Y. Aizawa, T. Iida, T. M. Aida, C. Levy, K. Sue and H. Inomata, Carbohydr. Res., 2005, 340(12), 1925–1930 CrossRef CAS.
- M. Watanabe, Y. Aizawa, T. Iida, R. Nishimura and H. Inomata, Appl. Catal., A, 2005, 295(2), 150–156 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr., Catal. Commun., 2008, 9(13), 2244–2249 CrossRef CAS.
- S. Lima, A. S. Dias, Z. Lin, P. Brandão, P. Ferreira, M. Pillinger, J. Rocha, V. Calvino-Casilda and A. A. Valente, Appl. Catal., A, 2008, 339(1), 21–27 CrossRef CAS.
- G. Yong, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2008, 47, 9345–9348 CrossRef CAS.
- M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924–7926 CrossRef CAS.
- Y. Su, H. M. Brown, X. Huang, X. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361(1–2), 117–122 CrossRef CAS.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131(5), 1979–1985 CrossRef CAS.
- J. Zhang, Y. Cao, L. Huiquan and M. Xinbin, 2009, http://aiche.confex.com/aiche/2009/webprogram/Paper169557.html.
- S. Lima, P. Neves, M. M. Antunes, M. Pillinger, N. Ignatyev and A. A. Valente, Appl. Catal., A, 2009, 363(1–2), 93–99 CrossRef CAS.
- G. Franceschin, A. Zamboni, F. Bezzo and A. Bertucco, Chem. Eng. Res. Des., 2008, 86(5), 488–498 CrossRef CAS.
- H. von Blottnitz and M. A. Curran, J. Clean. Prod., 2007, 15(7), 607–619 CrossRef.
- H. Ohno and Y. Fukaya, Chem. Lett., 2009, 38(1), 2–7 CrossRef CAS.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418(6901), 964–9 CrossRef CAS.
- J. W. Shabaker, G. W. Huber and J. A. Dumesic, J. Catal., 2004, 222(1), 180–191 CrossRef CAS.
- M. B. Valenzuela, C. W. Jones and P. K. Agrawal, Energy Fuels, 2006, 20(4), 1744–1752 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308(5727), 1446–1450 CrossRef CAS.
- J. O. Metzger, Angew. Chem., Int. Ed., 2006, 45, 696–698 CrossRef CAS.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- G. J. M. Gruter and F. Dautzenberg, Eur. Pat. 1834950, 2007.
- J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123(1–4), 59–70 CrossRef CAS.
- D. A. Simonetti and J. A. Dumesic, ChemSusChem, 2008, 1, 725–733 CrossRef CAS.
- R. M. West, Z. Y. Liu, M. Peter, C. A. Gärtner and J. A. Dumesic, J. Mol. Catal. A: Chem., 2008, 296, 18–27 CrossRef CAS.
- C. W. Jones, presented in part at ERE and GCEP Seminar, Stanford University Palo Alto, CA, December 2008 Search PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th edn, John Wiley & Sons,New Jersey, 2007, pp. 1396–1397 Search PubMed.
- G. Nagendrappa, Resonance, 2008, 13, 355–368 CrossRef CAS.
- Y. Yano, Y. Tamura and W. Tagaki, Bull. Chem. Soc. Jpn., 1980, 53(3), 740–744 CrossRef CAS.
- M. Breuer and B. Hauer, Curr. Opin. Biotechnol., 2003, 14(6), 570–576 CrossRef CAS.
- L.-W. Xu, Y. Gao, J.-J. Yin, L. Lyi and C.-G. Xia, Tetrahedron Lett., 2005, 46(32), 5317–5320 CrossRef CAS.
- J. M. D. Storey and C. Williamson, Tetrahedron Lett., 2005, 46(43), 7337–7339 CrossRef CAS.
- C. Moreau, A. Finiels and L. Vanoye, J. Mol. Catal. A: Chem., 2006, 253(1–2), 165–169 CrossRef CAS.
- K. Iwamoto, M. Hamaya, N. Hashimoto, H. Kimura, Y. Suzuki and M. Sato, Tetrahedron Lett., 2006, 47(40), 7175–7177 CrossRef CAS.
- K. Iwamoto, H. Kimura, M. Oike and M. Sato, Org. Biomol. Chem., 2008, 6, 912–915 RSC.
- The Diels–Alder Reaction: Selected Practical Methods, ed. F. Fringuelli and A. Taticchi, John Wiley & Sons, West Sussex, England, 2002 Search PubMed.
- E. B. Mubofu and J. B. F. N. Engberts, J. Phys. Org. Chem., 2004, 17, 180–186 CrossRef CAS.
- S. Otto and J. B. F. N. Engberts, Pure Appl. Chem., 2000, 72(7), 1365–1372 CrossRef CAS.
- E. B. Mubofu and J. B. F. N. Engberts, J. Phys. Org. Chem., 2007, 20, 764–770 CrossRef CAS.
- H. Yujiro, Nippon Kagakkai Koen Yokoshu, 2001, 79(2), 1079 Search PubMed.
- F. Fringuelli, R. Girotti, F. Pizzo, E. Zunino and L. Vaccaro, Adv. Synth. Catal., 2006, 348(3), 297–300 CrossRef CAS.
- D. Prajapati, D. D. Laskar and J. S. Sandhu, Tetrahedron Lett., 2000, 41(44), 8639–8643 CrossRef CAS.
- K. E. Litz, Molecules, 2007, 12, 1674–1678 Search PubMed.
- A. T. Balaban, A. Bota and A. Zlota, Synthesis, 1980, 136 CrossRef CAS.
- J. A. Marshall and X. Wang, J. Org. Chem., 1991, 56, 960 CrossRef CAS.
- T. A. Stroganova, A. V. Butin, L. N. Sorotskaya and V. G. Kul'Nevich, Arkivoc, 2000,(iv), 641–659.
- T. Seki, T. Nakajo and M. Onaka, Chem. Lett., 2006, 35(8), 824 CrossRef CAS.
- P. R. Stapp, J. Org. Chem., 1973, 38(7), 1433–1434 CrossRef CAS.
- T. Seki and H. Hattori, Catal. Surv. Asia, 2003, 7(2/3), 145–156 CrossRef CAS.
- M. M. Mojtahedi, E. Akbarzadeh, R. Sharifi and M. S. Abaee, Org. Lett., 2007, 9, 2791–2793 CrossRef CAS.
- A. Zuyls, P. W. Roesky, G. B. Deacon, K. Konstas and P. C. Junk, Eur. J. Org. Chem., 2008, 693–697 CrossRef CAS.
- H. Grabowska, R. Klimkiewicz, L. Syper and J. Wrzyszcz, Top. Catal., 2000, 11–12(1/4), 289–292 CrossRef.
- M. Gliński, W. Szymański and D. Łomot, Appl. Catal., A, 2005, 281(1–2), 107–113 CrossRef CAS.
- H. Teterycz, R. Klimkiewicz and B. W. Licznerski, Appl. Catal., A, 2001, 214(2), 243–249 CrossRef CAS.
- R. Klimkiewicz, H. Teterycz, E. Sikora, G. S. Szymański and J. Trawczyński, Kinet. Catal., 2007, 48(1), 67–73 CrossRef CAS.
- S. Hu, Z. Zhang, Y. Zhou, B. Han, H. Fan, W. Li, J. Song and Y. Xie, Green Chem., 2008, 10, 1280–1283 RSC.
| This journal is © The Royal Society of Chemistry 2010 |