Engineering nanostructured electrodes and fabrication of film electrodes for efficient lithium ion intercalation
Dawei
Liu
and
Guozhong
Cao
*
Department of Materials Science and Engineering, University of Washington, Seattle, WA, USA 98195. E-mail: gzcao@u.washington.edu
First published on 16th June 2010
Abstract
Lithium ion batteries have been one of the major power supplies for small electronic devices since the last century. However, with the rapid advancement of electronics and the increasing demand for clean sustainable energy, newer lithium ion batteries with higher energy density, higher power density, and better cyclic stability are needed. In addition, newer generations of lithium ion batteries must meet the requirements of low and easy fabrication cost and be free of toxic materials. There have been many novel approaches to gain high energy storage capacities and charge/discharge rates without sacrificing the battery cyclic life. Nanostructured electrodes are seemingly the most promising candidate for future lithium ion batteries. Modification of the electrode surface chemistry and the control of appropriate crystallinity are also reported to improve the electrode intercalation capabilities. The study of appropriately designed nanostructures, interfaces and crystallinity has also promoted and is accompanied with the development of thin film electrodes without the addition of binders and conductive carbon that are typically used in the fabrication of traditional lithium ion battery electrodes, simplifying the electrode fabrication process and enhancing electrode storage density. In this perspective, we summarize and discuss the efforts of fabricating nanostructures, modifying surface chemistry and manipulating crystallinity to achieve enhanced lithium ion intercalation capacities, rate capabilities and cyclic stability, as well as the direct fabrication of binderless film electrodes with desirable nano- and microstructures.
 Dawei Liu | Mr. Dawei Liu is a PhD candidate under the supervision of Prof. Guozhong Cao in Department of Materials Science and Engineering at University of Washington, Seattle, WA. He has published five first-authored refereed papers and one book chapter. His specific research project is focused on nanostructured electrodes for efficient lithium ion intercalation. |
 Guozhong Cao | Dr Guozhong Cao is Boeing–Steiner Professor of Materials Science and Engineering at the University of Washington, Seattle, WA. He has published over 250 refereed papers, written and edited five books and monographs. His research has led to the creation of two spin-off companies on energy conversion and storage. His recent research is focused mainly on nanomaterials for solar cells, lithium ion batteries, supercapacitors, and hydrogen storage. |
Broader contextIn the new century, clean and renewable energy storage devices have become the foci of both the building industry and research development. Lithium ion batteries, as one of the most promising battery technologies, have attracted much attention due to their fast boom of market share. However, the commercialized lithium ion battery has not been good enough to satiate the public need and theoretically there is much room for improvement. Research has thus been focused on developing electrode materials with high discharge capacity, large charge/discharge rate and long life cycles. To achieve these goals, a lot of effort has been devoted to fabricating structures that best facilitate the intercalation behavior of lithium ions. In this perspective, these efforts are summarized and reported. |
1. Introduction
1.1 Lithium ion battery as energy storage device background
Energy has been the central focus of human development since global industrialization. Fossil fuels have been and are still the major energy source with much improved energy conversion efficiency and significantly reduced environmental pollution as a result of combined technology advancement and the public awareness of the health and environmental challenges associated with fossil fuels. Although renewable or sustainable energy including solar, wind, and hydro-energy remains a negligible fraction of our total energy consumption today,1,2 energy security and environmental concerns have spurred great technical and political interests in developing advanced technologies to improve the energy utilization efficiency including smart electrical grid3,4 and light emitting materials and devices,5,6 to reduce and recover the “waste” heat through developing smart building materials and structure,7 and converting the waste heat to electricity using thermoelectrics,8,9 and to harvest the clean and sustainable energy such as solar, wind and tidal energy.10–12Advanced energy storage technologies for vehicle electrification and efficient use of renewable energy from the sun and wind are a critical part of renewable energy.13,14 Several technologies are currently under intensive study.15–17 Generating biofuels from biomass is one example of converting solar energy to chemical fuels.18,19 Storing hydrogen in the liquid or solid forms near ambient conditions is another example.20,21 However, effective utilization of variable and intermittent sources of renewable energy in meeting growing electricity demand requires much improved electrical energy storage technologies that are economically viable to enable large scale market penetration for electric vehicles and renewable electricity storage. Capacitors and supercapacitors, also known as ultracapacitors, offer high specific power and long cyclic stability and life time, and thus are excellent choices for applications requiring a burst energy source; however, they suffer from low specific energy.22,23 Batteries possess great specific energy, but suffer from low specific power and cyclic degradation.24,25 Significant breakthroughs are needed for both supercapacitors and batteries to gain broad market penetration.
Batteries are more than a century old technology and have played critical roles in many technologies and today's mobile life. Although many types of batteries have been developed with much improved energy storage performance, the fundamental structure of a battery remains the same, consisting of an anode and a cathode with electrolyte sandwiched in between.26 The advancement in battery technology has been relying on the development and use of different types of materials for electrodes and electrolytes and thus with different electrochemical reactions.27,28Fig. 1 compares different types of batteries;29 lithium-ion batteries offer a balanced combination of high power and energy density, long cyclic life, and stability. The commercialization of lithium ion batteries has witnessed the soaring market share in the energy industry, especially in powering small electronic devices such as laptops and cellular phones.30,31
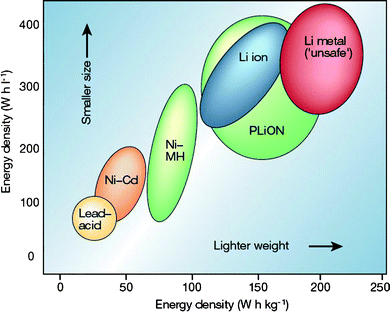 | ||
| Fig. 1 Comparison of the different battery technologies in terms of volumetric and gravimetric energy density. It is clear that for a given energy storage, lithium ion batteries are lighter and smaller. In addition, the lithium ion batteries are more environmental benign.29 | ||
A lithium ion battery, just like other types of batteries, consists of three major components: an anode, a cathode, and the electrolyte between them, and works by converting chemical potential to electric energy through Faradaic reactions, which include heterogeneous charge transfer process occurring at the surface of an electrode.32 Different from disposable or primary batteries, the lithium ion battery is a rechargeable or secondary battery, and the charging process involves the energy conversion from electric energy back to chemical potential.33 In a typical secondary lithium ion battery, Faradaic reactions are accompanied with both mass and charge transfer through the electrodes as well as dimension change; therefore, the surface area and the transport distance critically determine the performance of the battery in question.34 Chemical composition, crystal structure, and microstructure will have significant impacts on the reaction rate and transfer processes as well as its cyclic stability.29,35
1.2 The performance of lithium ion intercalation electrodes
| E = CV | (1) |
Obviously, a high energy storage density can be achieved by choosing or developing anodic and cathodic materials with high lithium-ion intercalation capacity with a large electrochemical potential difference. Although increasing the potential of cathodic materials and, thus increasing the cell voltage, with a given anode, is seemingly the most effective way to increase the energy storage density, the decomposition of electrolyte at the cathode surface becomes a problem.34 Before finding suitable electrolytes stable at high cathode potential, the research is focused mostly on the development of electrodes with high lithium ion intercalation capacity. Fig. 2 summarizes the electrochemical potential and the typical lithium ion storage capacities of both anodic and cathodic materials.29
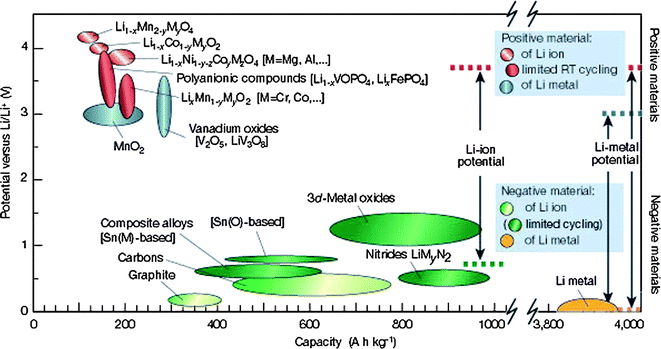 | ||
| Fig. 2 Voltage vs. capacity for positive and negative electrode materials presently used for under serious consideration for the next generation of rechargeable Li-based cells.29 | ||
While the lithium ion intercalation capacity of anodes, e.g. Sn can stabilize well above 500 mA h g−1 even after a big capacity loss following the initial cycle,36,37 the highest capacity of cathode materials is way below 500 mA h g−1. As a result, the performance of the cathode is a bottleneck for the improvement of the whole battery system.38,39 For example, the commercialized LiCoO2 has a mere capacity of ∼140 mA h g−1,40 and its potential alternatives LiMn2O4 and LiFePO4 have capacities of merely 150,41 and 170 mA h g−1, respectively.42 The cathode compounds demand more research efforts for new lithium ion batteries with much enhanced energy storage density.
1.3 Solutions by constructing specially designed structures
The lithium ion intercalation/de-intercalation process can also be considered as a phase transition process involving initial nucleation at the interface between the electrode and electrolyte and subsequent growth from the interface towards the bulk of electrode. Lithium ion intercalation occurs via redox or Faradaic reaction which entails the reduction of valence state of the transition metal ions in the electrode. The “formula” intercalation maximum can be calculated according to the highest valence reduction. Taking MnO2 for example, if the Mn4+ ions could be completely reduced to Mn2+, the specific capacity of MnO2 would be more than 600 mA h g−1. However, this high capacity has never been observed in experiments, due to other limitations that need to be considered. The very first consideration would be reversibility. The extraction of lithium ions may become very difficult or practically impossible if a maximal amount of lithium ions are inserted, due to large positive enthalpy associated with the phase transition. So practically achieved maximal insertion capacity is dependent on the reversible phase transition boundary and is always much lower than the theoretical limit calculated from maximal valence reduction.45 In addition, the insertion of lithium ions into the electrode host would induce a large volume change due to the change of crystal lattice and/or structure, resulting in a loss of mechanical integrity of the electrode.46,47In order to accommodate more lithium ions than the theoretical limit, the reversible phase transition boundary of the bulk materials needs to be extended. The Faradaic reaction and the phase transition associated with lithium ion intercalation and de-intercalation is dependent on the nature of the electrode materials including the arrangement of ions, the type and level of impurities and defects, and the surface energy. Electrodes away from equilibrium state may favor accommodating more lithium ions through reversible intercalation and de-intercalation process than the same materials in a thermodynamic equilibrium state. A number of ways could be used to create materials away from equilibrium. For example, nanostructured electrodes with huge surface energy are in a state far from thermodynamic equilibrium.48 As a result, the reversible phase transition limit of lithium ion intercalation and de-intercalation is often extended with much greater lithium ion storage capacity. In addition to the improvement of energy storage capacity, nanostructures also improve kinetics by providing a short diffusion path for lithium ions and enhance the rate capability.49,50 Nanostructures also permit freedom for volume change during long term cycles, alleviating the negative effect which could cause capacity degradation.51,52
Besides nanostructure, surface defects would contribute to lithium ion intercalation by shifting thermodynamics and improving kinetics. The presence of surface defects increases the surface energy and could possibly serve as nucleation center and, thus, facilitate phase transition. In addition, the presence of surface defects reduces the charge transfer resistance and improves the charge transfer kinetics while at the same time preventing active electrode dissolution in the electrolyte, which could improve cyclic stability.53 After the surface charge transfer process, lithium ion diffusion from the surface into interior depends on the crystal structure as well, so crystallinity is another factor to consider and manipulate to enhance lithium ion insertion capability.54 For some intercalation compounds, an amorphous structure appears to be more open to lithium ion diffusion due to the ability to withstand insertion stress with less well packed ions. Nanostructures, highly defective surfaces, and amorphous electrodes are all away from thermodynamically equilibrium states. Hence appropriately designed nano- and microstructures and the careful manipulation of surface and bulk chemistry will change the phase transition boundary, improve the transport kinetics, and permit more freedom for long-term cyclic stability.
In addition to searching and developing electroactive materials with higher lithium ion insertion capacity, rapid charge and discharge rate, and improved cyclic stability, research has been aimed at the development and fabrication of binderless electrode films (with directly incorporated conductive components) through solution-based chemical processing.55,56 In such an approach, the commonly used polymer binders to improve the mechanical integrity of the electrodes could be eliminated and, thus, enhance the energy storage capacity of a battery, as well as simplify the fabrication process and possibly reduce the fabrication cost. In addition, the possible electrolyte permeation path blocking problem induced by excess binder, which would severely reduce the active power electroactivity, could be avoided.57 Besides excluding binder from the electrode composition, some research groups also fabricated binderless and carbon-free thin film electrodes which promise a higher energy density because of the pure electroactivity of the electrode.58–60 These thin film electrodes, circumventing routine but tedious electrode casting fabrication, have a bright application future as microbattery electrodes, and could be used in vast number of research areas since they could power micro-electronic devices.61,62
2. Nanostructured lithium ion intercalation electrodes
2.1 Nanostructured LiFePO4 cathode and Li4Ti5O12 anode
Generally speaking, nanostructures refer to structures with one or more dimensions confined in the scale between molecular and microscopic scope, i.e. 0.1–100 nm. Nanostructure has been a hot topic and intensively studied for the past decade because of its scientific significance. Nanostructured materials offer the unusual mechanical, electrical and optical properties endowed by confining the dimensions of such materials and the overall behavior of nanostructured materials exhibit combinations of bulk and surface properties.63 Such materials have been applied in many engineering applications such as field-effect transistors,64 chemical and biological sensors65,66 and dye sensitized solar cells67,68 to improve device performance (e.g. efficiency, capacity etc.). Lithium ion batteries is one of these fields that has benefited from the introduction of nanostructures: the application of nanostructured electrodes has significantly improved the lithium ion intercalation capability, e.g. storage capacity, intercalation rate and cyclic stability.69,70 Considering the liquid/solid interface reaction characteristic of lithium ion intercalation followed by diffusion into electrode bulk, it is reasonable to expect that large surface area and short lithium ion diffusion path can ensure complete Faradaic reaction at the interface and facilitate the diffusion into the bulk. Thus nanostructured electrodes which meet these requirements are highly favorable as intercalation hosts instead of bulk electrodes consisting of micrometre sized particles.The conventional route for fabricating LiFePO4 powders was mainly through solid-state synthesis. The starting precursors consisted of stoichiometric amount of iron salt, a lithium compound and most commonly ammonium phosphate as phosphorus source. After heat treatment first at 300–400 °C to expel gases and followed by calcination at higher temperature up to 800 °C under inert or slightly reductive atmosphere, the LiFePO4 powders could be obtained.77 However, the obvious disadvantage of the conventional solid state method was the particle growth and agglomeration due to the high temperature employed and the resultant product always possessed very small specific surface area.78 To solve this problem, mechanochemical activation was introduced into the process and the resultant powders had a much larger specific surface area.79 There have also been efforts using microwave heating instead of the furnace heating which limited the calcination temperature to a lower value to avoid excessive particle growth.80 However, even with these modified methods, it was difficult to obtain LiFePO4 with particle sizes below hundreds of nanometres.
Solution based methods have been proved to be effective for producing nanostructured LiFePO4.81 Hydrothermal method and sol–gel method were the two main methods employed and the obtained nanostructured LiFePO4 exhibited noticeable intercalation capability improvement as compared with micrometre-sized LiFePO4.
Hydrothermal growth refers to crystallizing substances at elevated temperature (typically 100–200 °C) from aqueous solutions at high vapor pressures. The starting precursors for fabricating LiFePO4 were typically an iron salt such as FeSO4, phosphate acid H3PO4 and lithium base LiOH.82 Particles of sizes from tens of nanometres to hundreds of nanometres could be obtained by tuning the reaction time, temperature and pH value.83 Dumbbell-like LiFePO4 microstructures hierarchically constructed with two-dimensional nanoplates of ∼300 nm length and ∼50 nm thicknesses were fabricated via hydrothermal self-assembly and exhibited a stable discharge capacity of ca. 110 mA h g−1 over 70 cycles at a C/30 charge/discharge rate (Fig. 3).84 Recham et al. used a solvothermal–hydrothermal method to enable the growth of LiFePO4 with controlled size and morphology and the best sample with particle size of 300 and 500 nm could deliver a sustainable capacity of 150 mA h g−1 at a C/10 rate.85 LiFePO4 nanowires with diameters of a few hundred nanometres were fabricated by adding nitrilotriacetic acid and isopropanol to the precursors and could reach an initial discharge capacity of 150 mA h g−1 and retained still as high as 138 mA h g−1 after 60 cycles at a charge/discharge rate of 0.1 C.86
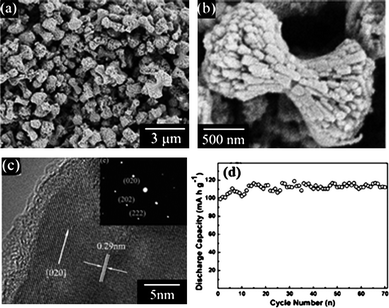 | ||
| Fig. 3 (a) Typical low-magnification SEM image of dumbbell-like LiFePO4, (b) an individual dumbbell-like LiFePO4 from the obverse side, (c) HRTEM image of the tip of an individual dumbbell shape and (d) discharge capacity vs. cycle number of dumbbell-like LiFePO4 at C/30 charge/discharge rate84 | ||
The sol–gel route is another commonly used solution based method to fabricate nanostructured LiFePO4. Porous nanostructured LiFePO4 powder with a particle size distribution of 100–300 nm was obtained by using an ethanol based sol–gel route with lauric acid as a surfactant. The nanoparticles could deliver a specific capacity of 157 mA h g−1 at a discharge rate of 1 C and still had 123 mA h g−1 delivered when the rate was increased to 10 C.87 Similar high rate capability for lithium ion intercalation was also reported on sol–gel prepared wired mesoporous LiFePO4.88
For the hydrothermal and sol–gel methods, there has also been quite a lot of reported work on nanostructured LiFePO4/C composites with carbon coating aiming at enhancement of electronic conductivity which had even better rate capability than single phase nanostructured LiFePO4.89–93 In addition to the hydrothermal and sol–gel method, co-precipitation,94,95 emulsion-drying96 and spray pyrolysis methods97 could also be employed to fabricate nanostructured LiFePO4 or LiFePO4/C composites.
For LiFePO4 cathode and Li4Ti5O12 anode as discussed above, both possess good capacities at low charge/discharge rate but capacities degraded severely as the rate was increased due to the poor electronic conductivity. The introduction of nanostructures improved the intercalation kinetics by providing larger electrode/electrolyte contact area and reduced path for electron transportation and lithium ion diffusion, resulting in noticeably improved capacities at high rates.
2.2 Nanostructured oxide electrodes
Template-based deposition involves deposition of the material of interest, or a precursor for that material, into the pores of a microporous template membrane. After deposition, the template will be eliminated by either chemical etching or thermal annealing. By using polycarbonate filtration membrane (PC) as template, Martin and co-workers have fabricated polycrystalline V2O5 nanorod arrays. In lithium ion intercalation property study, the resultant nanorods delivered three times the capacity of a thin-film V2O5 electrode at a high rate of 200 C.120 Wang et al. have successfully fabricated single-crystalline V2O5 nanorods, nanotubes and Ni–V2O5 nanocables by employing electrophoretic deposition.121–123 As-fabricated V2O5·H2O nanotube arrays demonstrated an initial high capacity of 300 mA h g−1, about twice that (140 mA h g−1) of the plain film of the same chemical composition. Ni–V2O5·nH2O core/shell nanocable arrays were prepared by a two-step electrodeposition method with Ni nanorod arrays fabricated by electrochemical deposition first and vanadium pentoxide shell deposited onto the surface of nickel nanorods through sol electrophoretic deposition. Compared with V2O5 nanorod arrays and sol–gel-derived V2O5 films, the specific power of the nanocable arrays was enhanced by 1–2 orders of magnitude as shown in Fig. 4. Similar template-based sol–gel deposition employing PC template was also used for the synthesis of MnO2 nanorods and the produced nanorods delivered an initial capacity of 183 mA h g−1 and stabilized on subsequent cycles to 134 mA h g−1.124
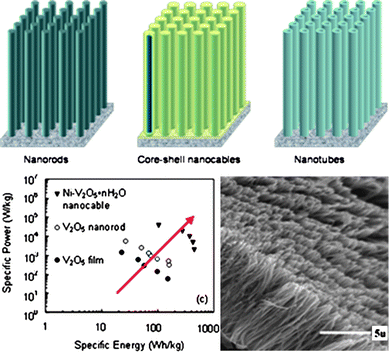 | ||
| Fig. 4 Schemes of nanorod, nanotube and nanocable array electrodes for energy storage (top). SEM images of oxide nanorod arrays (bottom right) and comparison of specific energy and specific power of vanadium pentoxide electrodes in the form of film, nanorod arrays and nanocable arrays (bottom left).123 | ||
The hydrothermal method is an effective method to fabricate one-dimensional nanostructures. Nanostructured α-, β- and γ-MnO2 have been synthesized through the solution and hydrothermal route.125 While α- and γ-MnO2 nanowire or nanorods exhibited good electroactivity to lithium ions by delivering capacities more than 200 mA h g−1, β-MnO2 nanostructures showed low capacity and poor cyclic stability. Other research indeed revealed that until reaching a small enough scale of particle size, the intercalation capacity of β-MnO2 was fairly low.126 TiO2 is one of the most “hydrothermally fabricated” compounds used for lithium ion intercalation application. TiO2 nanowires synthesized by hydrothermal method were reported by Armstrong et al. and the TEM images of the nanowires with the chronopotentiometric curves of lithium ion intercalation are shown in Fig. 5. The intercalation capacity was as high as 275 mA h g−1, corresponding to the lithiation of Li0.82TiO2.127,128 Nanorods and nanotubes of TiO2 were also fabricated by different groups and exhibited similar high capacities comparable to TiO2 nanowires.129–131
![(a) Low-resolution TEM image of the TiO2-B nanowire, (b) high-resolution lattice image viewed down the [100] projection (inset: electron diffraction pattern of a TiO2-B nanowire and (c) variation of potential (vs. 1 M Li+/Li electrode) with Li content (charge passed) for TiO2-B nanowires (solid line) and bulk TiO2-B (dashed line) cycled under identical conditions. Rate: 10 mA g−1 (10 mA of charge passed per gram of TiO2-B); voltage limits: +1 and +3 V; V = potential.127](/image/article/2010/EE/b922656g/b922656g-f5.gif) | ||
| Fig. 5 (a) Low-resolution TEM image of the TiO2-B nanowire, (b) high-resolution lattice image viewed down the [100] projection (inset: electron diffraction pattern of a TiO2-B nanowire and (c) variation of potential (vs. 1 M Li+/Li electrode) with Li content (charge passed) for TiO2-B nanowires (solid line) and bulk TiO2-B (dashed line) cycled under identical conditions. Rate: 10 mA g−1 (10 mA of charge passed per gram of TiO2-B); voltage limits: +1 and +3 V; V = potential.127 | ||
The fabrication of mesoporous nanostructures often involves the use of soft (surfactant) or hard templates. In the 1990s, surfactant-templated TiO2 was initially fabricated by using amphiphilic poly(alkylene oxide) triblock copolymer as the structure-directing agent in an ethanolic solution of TiCl4.135 Later application of this mesoporous structured compound as lithium ion intercalation electrodes revealed unusually fast capacitive and intercalation charging abilities.136 Besides the synthesis route just described, mesoporous TiO2 could also be synthesized by the evaporation-induced self-assembly procedure with novel poly(ethylene-co-butylene)-b-poly(ethylene oxide) polymer (KLE) used as template. The templated films after annealing at elevated temperatures transform to the anatase phase and can store a capacity of ca. 200–250 mA h g−1.137 The rate capability measurement was also carried out on mesoporous TiO2 fabricated by using amphiphilic molecule as the templating agent: the mesoporous samples delivered a capacity of 184 mA h g−1 at C/5 and 95 mA h g−1 at 30 C, possessing a much better rate capability than commercial samples.138 Lou et al. studied the storage properties of TiO2 mesoporous hollow particles and after the initial high capacity of 408 mA h g−1, the capacity stabilized at >170 mA h g−1 for 35 cycles.139
Mesoporous V2O5 was fabricated following similar surfactant-templated procedures by using VCl4 instead of TiCl4. At a very high charge/discharge rate of 50 C, the capacity delivered by mesoporous V2O5 could be as high as 125 mA h g−1, which promised its application as a low power capacitor or high power batteries owing to the good balance between specific power and specific energy.140
Besides using surfactant as soft template, mesoporous structure could also be obtained by using a hard template, e.g. mesoporous silica. Hollow LiFePO4 was fabricated by using the hard templates KIT-6 and had a large BET specific surface area of 103 m2 g−1 with pore size distribution centered at 5.6 nm.141 The mesoporous electrode demonstrated excellent rate capability and cyclic stability. At a rate of 15 C, the capacity was 153 mA h g−1, 95% of that at 0.2 C rate. In addition, no obvious capacity degradation was noticed after 80 cycles. Considering the well-known low rate capability of LiFePO4 due to its poor electronic conductivity, using mesoporous structured electrodes could be one of the possible solutions to solve this problem.
Besides the improved electrode cyclic stability and enhanced rate capability, for some compounds, the electroactivity was significantly enhanced after changing the bulk material into a mesoporous structured one. Successful fabrication of mesoporous β-MnO2 employing mesoporous silica KIT-6 was reported. While bulk β-MnO2 was for a long time assumed to be with extremely low intercalation capacity, i.e. below 60 mA h g−1,126 mesoporous β-MnO2 with a pore size centered at 3.65 nm exhibited a high capacity of 284 mA h g−1 and stabilized at 200 mA h g−1 after initial degradation at a current density of 15 mA g−1.142 The TEM and HRTEM images of the mesoporous β-MnO2 before and after cyclic reactions together with the electrochemical cyclic performance are shown in Fig. 6 and it could be clearly seen that the integrity of mesoporous structure was well maintained during the intercalation/de-intercalation cycles. This mesoporous electrode also possessed good rate capability by having 81% capacity remaining after the current density was increased to 300 mA h g−1. Mesoporous LiMn2O4 was made through a similar route by employing KIT-6 template but involved more solid-state reactions to evolve from Mn2O3 to LiMn2O4.143 Thus-obtained mesoporous LiMn2O4 nearly doubled the capacity of bulk LiMn2O4 and possessed a much better cyclic stability than nanoparticulate LiMn2O4. The surface area of these two mesoporous products were 127 and 90 m2g−1, respectively.
 | ||
| Fig. 6 TEM and high-resolution TEM (HRTEM) images of mesoporous β-MnO2: (a, b) as-prepared; (c, d) after first discharge; (e, f) end of discharge after 30 cycles; (g, h) end of charge after 30 cycles and (i) cyclic retention for mesoporous β-MnO2 cycled at (a) 15, (b) 30 and (c) 300 mAg−1; (d) bulk β-MnO2 cycled at 15 mA g−1.142 | ||
In conclusion, during lithium ion intercalation/de-intercalation, mesopores readily supplied ions from the electrolyte and acted as the buffering layers to alleviate the volume change experienced during lithium ion intercalation/de-intercalation. As a result, the cyclic stability of mesoporous structured electrodes was improved and the rate capability was enhanced. Using a template-based method to fabricate mesoporous structure could produce ordered pores with sizes controllable by template tuning. In general, the BET specific surface areas of template-derived mesoporous structures are >90 m2 g−1 and the pore sizes were centered from 3–6 nm. This structure is highly favorable for lithium ion intercalation as evidenced by noticeable improvement in capacities, cyclic stability and rate capabilities. However, the introduction of template into the synthesis processes induced much higher cost and the elimination of template after reactions was also a technical challenge. Recently, successful fabrication of high surface manganese dioxide following the template-free concept has been reported by Sinha et al. by means of a wet precipitation method144 and its application in extensive air purification turned out to be a great success. However, because of the unordered mesoporosity, manganese dioxide fabricated in this way was not suitable as a large lithium-ion capacity storage device since the electrolyte penetration would be very difficult in such thick oxide films with small pores. For unordered mesoporosity, the availability of macropores is crucial to facilitate complete electrolyte infiltration into the random distributed mesopores. Various attempts have been reported in fabricating a variety of macroporous metal oxides, e.g. NiO nanoflowers.145 The combination of macroporous and mesoporous structure was also reported in V2O5146 to improve lithium-ion intercalation kinetics. In our experiments, we have successfully deposited hierarchically mesoporous MnO2 nanowall arrays onto a platinum substrate to make thin film electrodes and obtained excellent capacities and cyclic stability which will be discussed in detailed in section 5.3
2.3 Problems of nanostructured electrodes
However, nanoscale is altogether positive and there are also some drawbacks associated with nanostructured electrodes which are extremely severe for certain electrodes. One problem was related with solid electrolyte interface (SEI) growth on the electrode surface. SEI was formed mainly by lithium containing organics and inorganics decomposed from the electrolyte solvents and salts before or in the initial cycles and could help stabilize the electrode/electrolyte interface by reducing the direct contact between the electrode and electrolyte and preventing further electrolyte decomposition.147,148 SEI was discerned on graphite anode as the electrode/electrolyte stabilizer and contributed to the capacity retention in long cycles despite that there was a large irreversible capacity caused by its formation.149,150 However, the SEI layer had very low ionic conductivity and electronic conductivity; the formation process was also accompanied by heat generation which would cause thermal shifting from the stable conditions.151 Excessive formation of an SEI layer on the electrode surface could cause thermal instability, e.g. temperature elevation and hinder intercalation kinetics, causing noticeable irreversible capacity (low coulombic efficiency).152 In addition, the excessive formation of an SEI layer could also consume considerable amount of lithium ions in the electrolyte, causing obvious capacity degradation.153,154 The most important factors determining the formation of SEI were the electrolyte used and electrode morphology.155,156 Owing to the large surface area and corresponding large electrode/electrolyte interface, SEI formation on nanostructured electrodes was often far more noticeable than on bulk electrodes and often accompanied by formation of thicker and more compact layers with much more heat produced.157 Taking LiCoO2 cathode for example, it was found that LiCoO2 with smaller particle size was identified with a thicker SEI layer which acted as a barrier for Li-ion diffusion and resulted in deteriorated rate capabilities at higher C rates.158 A similar problem was also identified on a LiMn2O4 cathode and the increase of SEI layer thickness directly caused capacity fading in long term cycles.159,160Another problem of nanostructured electrodes was related with the active metal ion dissolution in the electrolyte which would cause capacity degradation. LiMn2O4 was one typical victim: Mn ions in the electrode could easily dissolve in the electrolyte and the large electrode/electrolyte interface characteristic of nanostructures badly aggravated this problem.161,162 Despite that the discharge capacity and rate capability could possibly be improved, cyclic stability was obviously a big problem for nanostructured lithium manganese oxide and related manganese oxide electrodes.125,163
To solve the excessive SEI growth and active metal ion dissolution problems, special surface chemistry design is necessary for nanostructured electrodes as we are going to discuss next.
3. Surface chemistry engineering
3.1 Surface coating on electrodes
As just discussed, nanostructures have been proved highly effective in enhancing the electrode intercalation capability. The importance of the electrode and electrolyte interface was also recognized especially when the electrode dimension approached the nanoscale. However, for all the intercalation electrodes including nanostructured electrodes, there are two problems that need to be solved: (1) the first step of lithium ion intercalation into the electrode involved the charge transfer process of lithium ions from the electrolyte onto the surface of electrode and entailed redox reactions involving the participation of electrons. If the extra charge could not be readily transferred away after reaction, charge accumulation would occur to impede more reactions.164,165 To ensure complete redox reactions, good charge transfer conductivity must be guaranteed; (2) during the long-time repeated intercalation/de-intercalation cycles, the electrode surface could possibly dissolve into the electrolyte and this problem was more severe for nanostructured electrodes because of the large solid/liquid interface.166,167 The failure to maintain surface morphology integrity would directly lower intercalation capability, causing capacity degradation. Considering these two points, electrodes should possess good charge transfer conductivity and surface integrity over long-term cycles to ensure favorable intercalation capacity and cyclic stability.Introducing other elements into the electrode compound was adopted as one way to enhance the surface charge transfer conductivity. The element would either substitute the transition metal ions (doping) to modify the crystal structure168 or interact with the transition metal ions on the crystal surface without entering the lattice.169 The charge transfer resistances in both situations were significantly reduced and the rate capability was obviously improved. However, despite the enhancement of surface charge transfer conductivity, the dissolution of electrode in the electrolyte was still a challenge to the performance stability of the nanostructured electrode.
To protect the electrode from dissolution, the straightforward but essential concept was based on reducing the direct contact between electrode and electrolyte, which could be realized by the method of coating a layer of porous materials on the electrode surface. The commercialized LiCoO2 was used by many research groups as the model electrode to carry out the coating experiments and many oxides have been tested as the coating layer, e.g. Al2O3,170 AlPO4,171 CeO2,172 TiO2,173 Li4Ti5O12174etc.Fig. 7 shows a TEM image of coated LiCoO2 and compares the electrochemical cyclic performance comparison of the bare electrode and coated electrodes. It could also be seen that after coating, the cyclic stability of lithium ion intercalation measurements were obviously improved.175 More detailed experiments were also carried out by using different techniques to coat TiO2 onto the LiCoO2 electrode surface and it was found that the mechano-thermal coating from pre-formed nanoparticles were preferable to sol–gel coating from an alkoxide precursor, the cyclic stabilities in both situations were noticeably improved (5-fold for sol–gel coating and 12-fold for mechano-thermal coating).176 Besides acting as the protection layer to prevent electrode dissolution, the TiO2 coating was also found to help suppress the cycle-limiting hexagonal/monoclinic/hexagonal phase transitions accompanying the charge–discharge processes. In addition to oxides coating, polymer coating was also experimented with a combined model calculation.177 Electrochemical measurements showed that coating LiCoO2 with a poly-(2EHA-Co–F) film significantly decreased the activation energy for Li+ exchange which could improve reaction kinetics and facilitate the charge transfer process. Spinel LiMn2O4 or substituted LiMn2O4 is a promising cathode material but for a long time suffered rapid capacity fading during repeated charge/discharge cycles partly due to the dissolution of active Mn ions in the electrolyte especially when the electrodes were comprised of nanoparticles.178,179 The Mn dissolution phenomenon was even more severe at elevated temperatures,180,181e.g. 55 °C. It turned out that coating oxides onto the electrode surface to reduce the direct contact between spinel and the electrolyte was an effective method to improve the cyclic stability of LiMn2O4. Kannan and Manthiram coated LiMn2O4 with LixCoO2, LiNi0.5Co0.5O2, Al2O3 and MgO using solution-based coating followed by heat-treatment.182 All the surface coated (modified) samples showed much better capacity retention at both room temperature (25 °C) and elevated temperature (60 °C) than unmodified LiMn2O4. The LiNi0.5Co0.5O2-modified sample showed superior capacity retention with only 2.8% fade in 100 cycles at 60 °C with capacity around 110 mA h g−1; the Al2O3 modified sample showed a higher capacity of 130 mA h g−1 but with a faster fading rate (16% fade in 100 cycles at 60 °C); the Li0.75CoO2 modified sample showed the best combination of capacity (124 mA h g−1) and retention (8% fade in 100 cycles at 60 °C). Because of easy synthesis and chemical stability, Al2O3 was the most used coating compound onto electrodes. There have also been several coating methods of Al2O3 such as melting impregnation,183 reactive sputtering184 and solution soaking:185 noticeably improved cyclic stability of electrode being coated over uncoated one was observed. ZnO coating on LiMn2O4 was also reported and at elevated temperature of 55 °C, the coated LiMn2O4 showed capacity retention of 97%, significantly higher than the 55% capacity retention of the bare LiMn2O4.186 The ZnO coating collected HF from the electrolyte and better preserved the interfacial morphology and ensured the stable charge transfer process. A similar ZrO2 coating also improved the high-temperature cyclic stability by screening the acidic species from the active electrode;187 in addition, ZrO2 coating improved the rate capability up to the high rate of 10 C owing to the enhanced charge transfer conductivity because coated ZrO2 can act as a highly Li-conducting solid electrolyte interface and the strong bonding to LiMn2O4 which could tolerate the lattice stress resulting from the volume expansion during lithium ion intercalation.
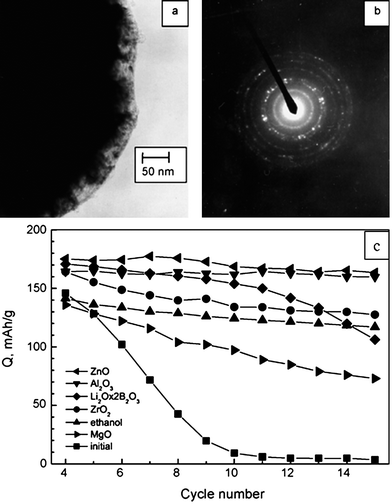 | ||
| Fig. 7 (a) TEM image of LiCoO2 surface modified with Al2O3, (b) electron diffraction of the coated LiCoO2 and (c) discharge capacity of bare and surface modified LiCoO2vs. cycle number.175 | ||
Compared with the coatings onto LiCoO2 and LiMn2O4 electrodes, the favorable coating compounds onto LiFePO4 electrodes were limited to conductive species. Because of the poor electronic conductivity of LiFePO4, the bare electrode could only be charged/discharged at a very low rate.188,189 Thus the adopted coating must be highly conductive in order not to aggravate the low rate performance problem of the electrode. The conductive coatings on LiFePO4 were mainly three kinds of conductive compounds: carbon coating,190,191 metal coating192,193 and conductive oxide coating.194Fig. 8 shows the TEM images and electrochemical cyclic performance at different charge/discharge rates of LiFePO4/C nanoplate coated by amorphous carbon.195 It could be seen that the coated electrodes exhibited excellent cyclic stability and the discharge capacity was still about 100 mA h g−1 at a high rate of 10 C, preserving more than 50% of the discharge capacity at 0.1 C.
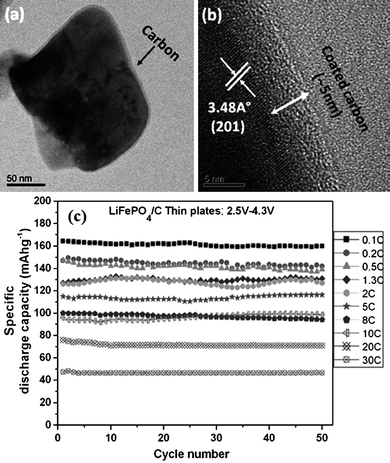 | ||
| Fig. 8 (a) TEM image showing a uniform coverage of amorphous carbon coating around the surface of a LiFePO4/C nanoplate, (b) HRTEM image showing a nearly 5 nm thick amorphous carbon layer around the surface of LiFePO4/C and (c) capacity vs. cycle number plots of LiFePO4/C thin nanoplates at various current rates of 0.1 to 30 C.195 | ||
For transition-metal oxide electrodes, carbon or metal was often used as coating layers, VOx coated with carbon prepared via reaction under autogenic pressure at elevated temperatures was reported.196 Both the reversibility and rate capability was much better than V2O5 nanoparticles without carbon coating. Metal layers were also coated onto nanostructured TiO2: the silver mirror reaction was used to coat Ag particles onto hydrothermally synthesized TiO2 nanotubes and cyclic stability and rate capability was found to be improved.197 A metal film of Cu or Sn was vacuum-deposited onto the surface of mesoporous anatase TiO2 electrodes and the electrode surface modification made by thin-film deposition improved the kinetics of Li intercalation/de-intercalation and remarkably enhanced the electrochemical performances in terms of capacity, stability and rate capability.198
Derived from the coating concept, recently a new method of surface grafting was developed to modify the surface chemistry, or say, to lower the interfacial chemical reactivity of side reactions (e.g. electrolyte decomposition) which was detrimental to long-term energy storage properties. Nitro-aryl groups were electrografted onto a Li1.1V3O8 surface by in situ diazonium chemistry during lithium ion intercalation when the electrode was potentiodynamically discharged.199 A homogeneous multilayer was formed whose thickness could be modulated. The multilayer did not impede charge transfer or limit the electrochemical reactivity. However, it did decrease the chemical reactivity of the material towards the electrolyte, resulting in significant improvements of the capacity retention.
To date, coating appears to be the most effective method to improve the surface charge transfer process. However, whatever kind of coating techniques was used, the coating process is often complicated and sometimes involved delicate equipment to achieve a homogeneous coating layer with good porosity. The basic requirement of the electrolyte permeability after coating was always challenging and tricky to meet. Considering all of these practical problems, we have been attempting to create a layer of surface defects on the intercalation electrodes to play a similar role as an exterior coating. The work included TiO2 nanotube arrays and V2O5 xerogel films annealed under reducing or inert gas flow and identified with surface defects (section 3.2 and 3.3)
3.2 Enhanced rate capability of TiO2 nanotube arrays with surface defects
The lithium ion intercalation and de-intercalation is a heterogeneous reaction which takes place at the interface between the solid electrode and liquid electrolyte; the surface chemistry and defects are, therefore, expected to play an important role in catalyzing or retarding the interface reaction and promote or prevent the nucleation. Appropriate surface chemistry and defects are expected to lead to enhanced lithium ion storage capacity and possibly improved kinetics.200 Surface chemistry and defects can be modified through various means such as surface coating, self-assembly of a monolayer, and injection of impurity species. Reacting the surface with reactive gas at elevated temperature would be a simple and easy way to accomplish such surface modification. TiO2 nanotube arrays annealed in CO gas serves as an example to illustrate the influence of surface defects on the lithium ion intercalation properties as described below.Titania nanotube arrays were synthesized by anodic oxidation method according to literature.201 Then they were annealed in reducing CO gas at 400 °C for 3 h.202 XPS study of the nanotube arrays annealed in CO gas confirmed carbon doping onto TiO2 surface in the form of a minor amount of Ti–C and the formation of Ti3+ point defects as shown in Fig. 9a. Compared with N2-annealed arrays which possessed an electrode resistance of 66 Ω and a charge-transfer resistance of 38 Ω, CO annealed arrays possessed an electrode resistance of 60 Ω and a reduced charge-transfer resistance around 26 Ω, indicating a higher charge-transfer rate of Li+ in the electrode. This improved charge-transfer conductivity of CO annealed TiO2 arrays could be attributed to the presence of surface Ti–C species and Ti3+ groups with oxygen vacancies which enhanced the surface conductivity of the electrode.
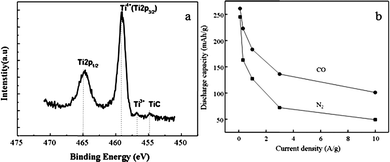 | ||
Fig. 9 (a) Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2p XPS spectra of TiO2 nanotube arrays annealed in CO gas at 500 °C with carbon doped Ti–C species and Ti3+ state available and (b) the initial discharge capacities of TiO2 nanotube arrays annealed in N2 and CO at 400 °C for 3 h as a function of applied discharge current densities. The measurements were carried out in a potential window between −0.6 and −2.1 V vs. Ag/AgCl as a reference electrode.202 2p XPS spectra of TiO2 nanotube arrays annealed in CO gas at 500 °C with carbon doped Ti–C species and Ti3+ state available and (b) the initial discharge capacities of TiO2 nanotube arrays annealed in N2 and CO at 400 °C for 3 h as a function of applied discharge current densities. The measurements were carried out in a potential window between −0.6 and −2.1 V vs. Ag/AgCl as a reference electrode.202 | ||
Fig. 9b summarizes and compares the relationship between the discharge current density and corresponding intercalation capacity of the TiO2 nanotube arrays annealed in N2 and CO, respectively. The lithium ion intercalation capacity of the N2 annealed nanotube arrays was found to be more sensitively dependent on the current density; the intercalation capacity reduced rapidly with the increased current density. At a current density of 100 mA g−1, the capacity of the N2 annealed TiO2 nanotube array was as high as 245 mA h g−1. However, when the current density was tripled to 320 mAg−1, the capacity decreased to 164 mA h g−1, losing one third of its discharge capacity. At a current density of 1 Ag−1, the capacity was further reduced to a value of 127 mA h g−1. In comparison, the CO annealed TiO2 nanotube arrays demonstrated less sensitive intercalation capacity. For example, an intercalation capacity of 261 mA h g−1 decreased to 223 mA h g−1, less than 20% reduction, when the current density increased from 100 to 320 mA g−1. It was clear that the CO annealed TiO2 nanotube arrays possessed much higher intercalation capacities, approximately double of that of N2 annealed TiO2 nanotube arrays at high current densities, i.e. possessing a capacity of 101 mA h g−1 at 10 A g−1.
The presence of defects may contribute to the improved intercalation capability of the CO annealed TiO2 nanotube arrays, as has been reported in other intercalation oxide electrodes such as V2O5.203 In the titania system, both intercalation and de-intercalation processes involve a phase transition between tetragonal TiO2 and orthorhombic LixTiO2 through the following reaction:
| xLi+ + xe− + TiO2 = LixTiO2 |
Phase transition occurs through nucleation at the interface and subsequent growth from the interface towards the interior. Besides improving the charge transfer conductivity, the presence of defects on the surface of TiO2 nanotubes could also serve as nucleation sites so as to promote the phase transition and enable more lithium ion intercalation and improve the rate capability.
3.3 Improved cyclic stability of V2O5 xerogel film with surface defects
Appropriately introduced surface defects could not only enhance the rate capability of electrodes but also improve the cyclic stability. The study of V2O5 xerogel films treated in nitrogen gas revealed the contribution of surface defects to improved cyclic stability.Sol–gel derived V2O5 films on FTO glass were annealed in N2 and air-flow atmosphere at 300 °C for 3 h under otherwise identical conditions.204 While air annealing did not change film color much; after N2 annealing, the originally yellow film turned dark green suggesting the presence of V4+ and V3+ species on the film surface. The charge transfer resistance of the N2-annealed film was two thirds that of the air-annealed film, reduced from 125 to 86 Ω. Fig. 10 compares the long term cyclic stability and corresponding XRD patterns of the air- and nitrogen-annealed films for lithium ion intercalation measured with a current density of 600 mA g−1 for 50 continuous discharge/charge cycles. The poor cyclic stability of air-annealed V2O5 film could be caused by the relatively larger grain size.205 For N2-annealed film, before reaching the highest discharge capacity value of 158 mA h g−1 at the 24th cycle, it started with a low discharge capacity of 68 mA h g−1. After 50 cycles, the capacity was still as high as 148 mA h g−1. While the mechanism causing such a change in storage capacity, with initially low followed by a sharp increase with increased lithium ion intercalation and de-intercalation cycles in the N2-annealed sol–gel derived V2O5 film electrodes, is not clear, the same or similar results or observation have been reported in literature and it could possibly be due to the surface defects.206,207 The crystallinity of these two kinds of films were also found to change differently during cycling: while the hydrous V2O5 peak degraded noticeably in the air-annealed film, such degradation was much less severe in the N2-annealed film, suggesting the disruption of layered structure. Along with defects on the film surface, the integrity of layered structure could be a further reason for the good cyclic stability of the N2-annealed film.
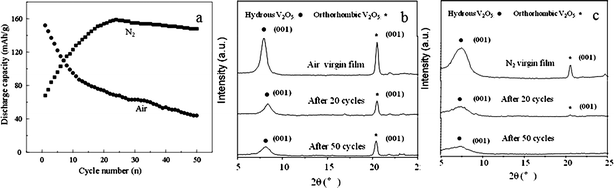 | ||
| Fig. 10 (a) The Li-ion intercalation discharge capacity of V2O5 films annealed in air and N2 at 300 °C for 3 h as a function of cyclic number. The measurements were carried out in a potential window between 0.6 and −1.4 V vs. Ag/AgCl as the reference electrode at a current density of 600 mA g−1; X-ray diffraction patterns of V2O5 films annealed in (b) air and (c) nitrogen before and after 20 and 50 cycles of lithium ion intercalation and de-intercalation measurements.204 | ||
The presence of surface defects could contribute significantly to the stability improvement which can be analyzed in three aspects: (1) the interfacial charge transfer abilities of the N2-annealed V2O5 film was improved because of the presence of more conductive surface defect species. As has been found in the optical absorption and impedance analysis, both the optical and electrical conductivity of the film annealed in N2 was improved compared with film annealed in air due to the presence of V4+, V3+ ions and associated oxygen vacancies on the film surface. It was found that the intercalation capability of lithium ions into the V2O5 xerogel film annealed at high temperature was mainly determined by the interfacial reactions at the electrolyte/electrode interface rather than the lithium-ion transport in the bulk oxide electrode.208 Since the enhanced charge transfer conductivity facilitated electron transportation during lithium ion intercalation/de-intercalation at the electrolyte/electrode interface which would obviously facilitate the lithium ion intercalation process,209 the cyclic stability and rate capability improvement could be explained. Similar improvement after N2 annealing was also observed in TiO2 nanotube arrays;210 (2) in addition to the conductivity enhancement contribution, the defect layer, like coating layers, also prevented the possible dissolution of V2O5 film in the electrolyte and ensured the integrity of film surface morphology upon cycling. One of the major causes of lithium ion intercalation capacity degradation in long-term cycles was the dissolution of the active material (electrode) in electrolyte. Building up a protecting layer on the surface will effectively reduce the possibility of electrode dissolution, thus improving the cyclic stability; (3) more than just a simple protecting layer, these surface defects of lower vanadium valency and oxygen vacancies could also serve as nucleation centers in the phase transformation process that occurred during lithium ions intercalation/de-intercalation.211 For this reason, the phase transformation process in the N2-annealed film was much easier and more reversible. The crystal structure was also more stable and the stability of lithium ion intercalation capacities was better than for air-annealed V2O5.
4. The crystallinity effect on lithium ion intercalation
4.1 Amorphous structure with high energy storage capacity
After initial interfacial charge transfer reactions, lithium ions will diffuse into the bulk through the open spaces inside the crystal structure of electrodes. With similar diffusion length, the diffusion kinetics is dependent on crystal structure order—the crystallinity—that could be estimated by examining X-ray diffraction patterns. Well-crystallized structures have long-range atom order while amorphous structures lack even short range order. Intuitively, long range ordered structure is good for lithium ion diffusion because of the absence of possible collisions with host atoms which impedes lithium ion transport; however, the rigid crystalline structure is also fragile to lattice expansion and means that the insertion of lithium ion, which definitely would induce strain, would cause irreversible deformation to the crystal structure and very detrimental to the reversibility of lithium ion intercalation/de-intercalation.212 In contrast, an amorphous structure with characteristic loose packing of atoms might possibly permit more diffusion freedom due to the ability to withstand more structure deformation.213The higher capacity of amorphous/low crystallinity substances over crystalline ones was reported and discussed as early as the study of intercalation cathode sulfides, e.g. MoS2. Whittingham et al. pointed out that this might be associated with either the more open lattice in amorphous compounds or the disordered framework which prevented the decomposition in some materials.214 In terms of nano-ionics, amorphous compounds have more interfaces within the crystal due to the small grain size and could store more lithium ions than well-crystallized compounds on the grain boundaries and interfaces. The low mobility of lithium ions inside the amorphous structure could be a limitation for high rate performance but the high energy storage capacity was indeed promising for powering small electronics. Especially given that the energy storage capacity of cathodes has not seen much impressive improvement in recent years, constructing appropriate amorphous structure might be a good direction to achieve high intercalation capacity and even high energy storage density.
Lithium transition metal oxides are often synthesized through high-temperature solid-state reactions, so the products were well crystallized structures like the layered structure of LiCoO2, spinel structure of LiMn2O4 and olivine structure of LiFePO4. However, for transition metal oxides which only consist of metal ions and oxygen, the variation of crystallinity was more controllable and the amorphous structure could be fabricated from low-temperature synthesis route.
Xu et al. synthesized amorphous manganese dioxide via a sol–gel route and the as fabricated material delivered a high capacity of 436 mA h g−1 and stored energy at a level of 1056 mW h g−1.215 However, the rate capability of this amorphous material was low due to the poor diffusion abilities of lithium ion inside the structure. To improve the rate capability, Xu et al. also made a cryogel from the sol–gel derived MnO2 aerogel via freeze drying. The nanoporous cryogel had a high specific surface area of 350 m2 g−1 and exhibited specific capacities of 289, 217 and 174 mA h g−1 at C/100, C/5 and 2 C rates, respectively, which demonstrated excellent rate capability.216 West et al. fabricated amorphous MnO2 nanowires arrays through a template-based electrodeposition and the cathode assembled from the crude material without adding any binder or conductive species exhibited a specific capacity of approximately 300 mA h g−1.217 Despite these good results obtained from amorphous MnO2, amorphous MnO2 often showed a quick fading rate during repeated cycles. Although the exact failure mechanism is not clear yet, the instability of the amorphous structure and the conglomeration of the small particles during cycling could be the possible reasons. There are also corresponding efforts to solve the problem by doping other elements such as Na,218 Cu219 or Bi220 to achieve improved cycling performance by stabilizing the local structure.
Amorphous intercalation compounds often exhibit sloping charge/discharge curves instead of possessing a well-defined plateau which was characteristic of well crystallized intercalation compound.221 As a result, the average intercalation voltage was lower. The energy density of a lithium ion battery is a product of capacity C and voltage V. The amorphous intercalation electrodes sacrifice the level of V for higher capacity C. In the case of amorphous MnO2 whose intercalation discharge curve is shown in Fig. 11,215 the sloping discharge curve possessed an average voltage of ca. 2.5 V, lower than 3 V reported from well-crystallized MnO2,124 however, the high capacity of more than 400 mA h g−1 was more than double the capacity of the well crystallized counterpart and the energy density was thus increased. In general, when calculating the energy density of amorphous intercalation electrodes, both voltage drop and capacity increase need to be considered.
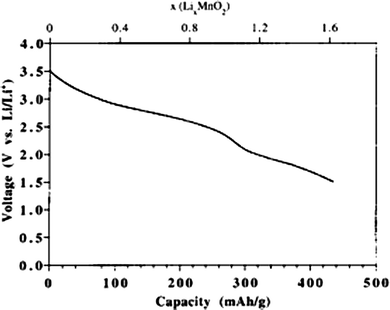 | ||
| Fig. 11 Variation of electrode potential with lithium ion content upon insertion into the oxide at a current density of 20 μA cm−2.215 | ||
4.2 Crystallinity effect on intercalation of capability of TiO2 nanotube arrays
After the discussion on the relationships between the lithium ion intercalation and the surface defects and amorphortized phases, one would reasonably wonder about the impact of perfection of crystal structure on the lithium ion intercalation properties. The lithium ion intercalation capacity and cyclic stability of TiO2 nanotube arrays with different crystallinity have been studied. Pristine TiO2 nanotube arrays fabricated by anodic oxidation were amorphous and the samples annealed at 400 °C for 3 h in nitrogen exhibited dominant anatase phase.210 Lithium ion intercalation measurements were carried out on these two samples together with two others treated at 300 °C and 500 °C and the cyclic performance was compared as shown in Fig. 12. It was found that the amorphous array possessed a high initial capacity of (202 mA h g−1). However, the cyclic stability was very poor and after 50 cycles the capacity was merely 40 mA h g−1. Upon crystallization, the cyclic stability was obviously improved. 300 °C annealed arrays started with a high capacity of 240 mA h g−1 and ended up at 148 mA h g−1. Well crystallized 400 °C arrays had a lower initial capacity of 163 mA h g−1 but the capacity after 50 cycles was still as high as 145 mA h g−1. Higher temperature annealed arrays exhibited similar stability but the absolute capacity further decreased. Amorphous TiO2 nanotube arrays might have higher capacities because of the more available sites to lithium ion intercalation (due to the defected and the disordered structure), However, because of the lithium diffusion limitation and the poor electronic conductivity, the cyclic stability of amorphous arrays was severely reduced as compared to well crystallized counterparts.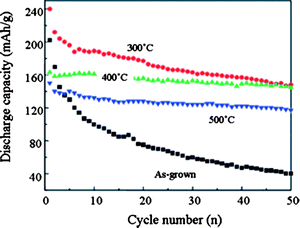 | ||
| Fig. 12 Li+-ion intercalation discharge capacity of amorphous as-grown TiO2 nanotube arrays and anatase TiO2 nanotube arrays annealed at 300, 400, and 500 °C in nitrogen for 3 h as a function of cyclic number. The measurements were carried out in a potential window between −0.6 and −2.1 V vs. Ag/AgCl as a reference electrode at a current density of 320 mAg−1.210 | ||
There were also some reports of composites having more amorphous structure than single oxides with good crystallinity due to the crystallization competition between oxides. TiO2 were added to V2O5 following the sol–gel route.222 While pure V2O5 xerogel exhibited typical hydrated V2O5 peaks, after introducing TiO2, no noticeable peaks were found and this suggested the amorphous state of the composites. Even after annealing at 500 °C for 1 h, the crystallinity was still poor. The intercalation capability of the amorphous composites were measured and compared with pure V2O5 film; it was found that all the composites films with different composition ratios exhibited a higher capacity and the composition of V/Ti = 80/20 exhibited a very high initial capacity of more than 400 mA h g−1. An et al. added amorphous NiO2 nanoparticles to a mixture of TiO2-B and anatase TiO2 nanotubes and both the cyclic stability and capacities at high current densities were improved.223
Silicon anode was another good system to demonstrate the advantage of amorphous electrodes. While crystalline silicon often suffers severe volume change during lithium ion intercalation/de-intercalation and possessed low cyclic stability,224 amorphous silicon showed a far less severe problem and could deliver stable high capacity over hundreds of cycles.225 Amorphous silicon thin film prepared by DC magnetron sputtering of silicon on stainless steel substrates showed a good performance with a stable capacity of about 3000 mA h g−1 and a relatively low irreversible capacity.226 Similar stability of high capacity was also reported on amorphous Si film made by pulsed laser deposition.227,228
It is somewhat arbitrary to conclude that amorphous structures deliver a higher capacity than well crystallized structure, or the opposite, because the situation varies depending on the intercalation compounds, e.g. the different story of TiO2 and V2O5 as discussed above. However, while the lithium ion storage capability of well crystallized structures is already well known and subject to little chance of ground-breaking improvement, appropriate manipulation of amorphous or low-crystallinity structures could be a good route to circumvent some of the limitations of the rigid structure and achieve significant advances.
5. Direct fabrication of nanostructured film electrodes
5.1 Development of binder- and carbon-free film electrodes
The electrodes for lithium-ion batteries are typically fabricated by mixing electroactive materials with conductive additive (typically 10–15% acetylene black in weight or volume) and binders (typically poly(vinylidene fluoride) 5%–10% in weight or volume) with n-methyl-2-pyrrolidone NMP as solvent to make a slurry, and then tape-casting into a film electrode. The as-obtained film electrode is then subjected to drying at 120 °C for 12 h or so.229–231 Such fabrication methods have been widely used in both industry and research laboratories and proved to be very successful. However, the addition of conductive additive and binder introduces extra processing steps and also compromises the amount of electroactive materials in electrodes.232,233 The use of binder incurs more problems related with side reactions during the working cycles of electrodes.234,235 Conventional admixing method may not be the best for obtaining the nanostructured electrodes. Direct fabrication of nanostructured film electrodes from solution chemical methods may offer some advantages. One justification for the introduction of conductive additive is for the favor of rate capability.236,237 However, when the film electrodes are appropriately designed and fabricated, e.g. nanostructure and surface defective film, the surface charge transfer and diffusion process could be improved and there would be no need to add conductive additive and binders.Preparation of binderless and carbon-free thin film electrodes were reported by several groups utilizing radio frequency (rf) magnetron sputtering,238,239 pulsed laser deposition (PLD),240,241 electrostatic spray deposition (ESD),242,243 sol–gel spin coating244,245 to directly deposit electroactive materials onto conductive substrates. These thin film electrodes without any additive exhibited comparable electrochemical performance to binder and carbon-added electrodes. In our experiments, we fabricated nanostructured thin film electrodes by sol–gel derived coating and electrodeposition. To improve the rate capability of thin film electrodes, we also modified the surface chemistry of electrodes and obtained good storage capacities under large charge/discharge current density.
5.2 Sol–gel derived films by drop coating or electrophoretic deposition
V2O5 sol–gel derived films were made by drop casting vanadium pentoxide sol onto conductive ITO glass.205 V2O5 coated ITO glass was used as the working electrode for the lithium ion intercalation study without any carbon or binder additive. The as-fabricated film without any post treatment was confirmed to be V2O5·1.6H2O and after thermal treatment of increasing temperatures, the xerogel film started to lose crystal water (Fig. 13a) and underwent crystallinity change. The crystallinity including crystal structure, interlayer distance and grain size was dependent on the annealing temperature as confirmed by XRD study. While the interlayer distance remained ∼11 Å when the annealing temperature was below 250 °C and crystal water content n ≥ 0.3, further increase of annealing temperature, i.e. 300 and 330 °C induced sharp shrinkage of interlayer distance to about 8 Å (Fig. 13b). In addition, well crystallized orthorhombic V2O5 phase started to form and the grain size became much larger (Fig. 13c). The lithium ion intercalation capacities in long cycles of films treated under different temperatures were measured and the well crystallized V2O5 showed quick capacity fading over all the cycles while the less crystallized 250 °C annealed film exhibited a high initial capacity of ∼275 mA h g−1 and stabilized at 185 mA h g−1 after 50 cycles (Fig. 13d).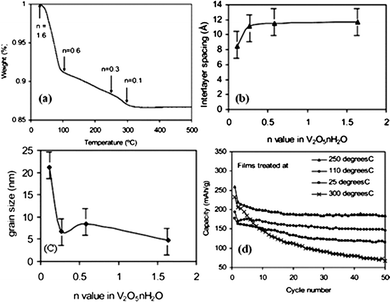 | ||
| Fig. 13 (a) TGA curve for V2O5·nH2O xerogels, (b) dependence of interlayer spacing on the n value in V2O5·nH2O, (c) dependence of grain size on the n value in V2O5·nH2O and (d) cycling performance at a current density of 100 μA cm−2 for V2O5·nH2O films obtained at 25, 110, 250 and 300 °C. The voltage ranges from 0.4 to −1.6 V vs. Ag/Ag+.205 | ||
V2O5 sol–gel derived film could also be synthesized through electrophoretic deposition. Electrophoretic deposition refers to the migration of colloidal particles suspended in a liquid medium under the influence of an electric field and deposition onto an electrode. It is a useful technique to deposit polycrystalline films with controlled crystalline texture and good porosity. In addition, it is simple and low cost with film thickness controllable by adjusting deposition conditions which are favorable for energy storage device electrodes. Zhitomirsky’s group has adopted electrophoretic deposition method to fabricate manganese oxide or manganese oxide–carbon nanotube composites films used as electrochemical supercapacitors with capacitances of 150 Fg−1 for pure manganese oxide and 650 F g−1 for composites being obtained.246,247 As a lithium ion battery electrode, V2O5 film could also be prepared by electrophoretic deposition from V2O5 sol using an anodic voltage 5 V followed by film calcination in air at 500 °C for 1 h.248 The resultant film exhibited an initial capacity of 250 mA h g−1 and increased to 310 mA h g−1 after 50 cycles in a potential window between 0.4 and −1.6 V.
5.3 Mesoporous MnO2 electrodes
Other solution-based methods have also been developed or used for the direct fabrication of porous nanostructure electrode films for lithium ion intercalation. For example, hierarchically structured mesoporous manganese dioxide nanowall arrays on cathodic substrates deposited by means of water electrolysis induced precipitation is an example for template-less fabrication of nanostructured electrodes.249,250 The formation of the mesoporous nanowall arrays was the result of water-electrolysis induced precipitation, as depicted in Fig. 14a: during the precipitation, the metal substrate surface consisted of two kinds of sites; one was where the nanoparticles of manganese hydroxide were precipitated and the other was where hydrogen gas bubbling occurred (no precipitation). This discontinuous precipitation on the substrate generated the macroporous nanowall arrays. In addition, in every precipitation cluster, in the space between neighboring nanoparticles were mesopores as found by TEM, and the nitrogen isotherm study revealed a large surface area ∼ 96 m2 g−1 and a pore size of ∼4.6 nm. In Fig. 14b and 14c, SEM and TEM images clearly indicated that the deposited hierarchical nanowall arrays were formed by closely stacked spherical nanoparticles with sizes around 50 nm. During the deposition, water electrolysis provided not only OH− which bonded with Mn2+ to precipitate manganese hydroxide nanoparticles but also generated H2 gas bubbles acting so as to prevent continuous precipitation on the whole substrate, thus producing the macropores of the nanowall arrays. Similar macroporous and mesoporous hierarchical structure was also found in cathodically deposited Co(OH)2.251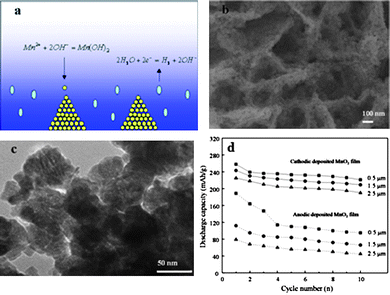 | ||
| Fig. 14 (a) Scheme showing the proposed growth mechanisms of hierarchically structured manganese hydroxide nanowall arrays on cathodes due to the increased pH value resulting from water electrolysis (blue area stands for high pH): precipitation of manganese hydroxide nanoparticles from the electrolyte accompanied with the release of hydrogen gas bubbles from the cathode surface, (b) SEM image of the hierarchically structured nanowall arrays reflecting the structure proposed in the growth mechanism scheme, (c) TEM image of stacked nanoparticles in a nanowall with voids (pores) and (d) comparison of discharge capacities of anodic deposited manganese dioxide and cathodic deposited manganese dioxide in the first 10 cycles. The measurements were carried out between 0.4 V and −1.4 V vs. Ag/AgCl at a current density of 30 mA g−1.249 | ||
The electrochemical properties of the as-fabricated nanowall arrays were directly measured on the platinum substrate without any electrode refinement and compared with an anodic oxidized MnO2 film. In the lithium-ion intercalation test, both the cathodic and anodic MnO2 were measured for different deposition thicknesses, i.e. 0.5, 1.5 and 2.5 μm and the long time cyclic performance of different thicknesses was compared in Fig. 14d. It was obvious that cathodic deposited manganese dioxide possessed a higher discharge capacity and better stability over anodic deposited manganese dioxide for each thickness. For the mesoporous nanowall arrays, when the film thickness was 0.5 μm, the initial capacity was as high as 256 mA h g−1 and after the thickness was increased to 2.5 μm, the initial capacity was still as high as 230 mA h g−1.
The above comparison clearly revealed that the cathodically deposited manganese dioxide films possessed a favorable hierarchically mesoporous structure with higher discharge capacities and better cycle stability than anodically deposited manganese dioxide. The cyclic stability improvement could be attributed to the mesoporous structure of cathodic deposited manganese dioxide nanowall arrays. As to the high discharge capacities at large deposition thickness, the macrostructure should be the key point. Besides the large surface area and shorter diffusion path provided for lithium-ion reaction, like tube arrays, this honeycomb macroporous structure facilitated the penetration of electrolyte to the bottom of the array even when thickness was large, thus minimizing the adverse effect of large deposition thickness, i.e. difficulty of electrolyte penetration. A similar phenomenon was also found when making a comparison of the thickness effect on vanadium oxide with and without macroporous structures.252,253 There are also other reports of porous thin film electrodes made by direct electrostatic spray deposition without binder and carbon additive introduction that exhibited excellent electrochemical performance, e.g. porous Fe2O3 film,254 porous Li4Ti5O12255 and porous NiO.256
Acidic anodization has been yet another method to fabricate electrode films for lithium ion intercalation. TiO2 nanotube arrays could be fabricated by anodization of Ti foil in a two-electrode electrochemical cell with platinum foil as a cathode at a constant potential at room temperature.257,258 Polished Ti foil was anodized in different types of electrolytes for 1 h to form nanotube arrays on Ti substrate. The nanotube diameters and length could be controlled by changing the anodization voltage and time. The as-fabricated TiO2 nanotube arrays exhibited excellent intercalation properties after appropriate heat treatment in nitrogen or carbon monoxide gas as discussed in section 3.2 and 4.2.
6. Concluding remarks
No era in human history has witnessed as fast a boom of the energy industry as the past decade. The invention of rechargeable lithium ion battery has actively changed the functionality of the modern life style by providing the uninterrupted power for electronic devices on the constant move such as cellular phones and laptops. Various kinds of intercalation electrodes have been studied and reported to have achieved high specific energy and high specific power as well as long lifetime. However, what was growing faster than the development of lithium ion battery was the human demand and social expectation supported by the rapid advancement of electronics. In addition, the increasing public awareness on environmental issues has been putting more pressures on clean sustainable energy including lithium ion batteries. In contrast to the ever-growing demands, the candidate pool of intercalation electrodes is limited. The introduction of nanostructures has achieved a huge success in the lithium ion battery field, as it did in many other fields. The essential contribution of nanostructure to the improvement of the intercalation capability lies not only in the increased specific surface area for interfacial Faradaic reactions and the reduced and favorable mass and charge diffusion path for lithium ions and electrons, but also in the modification of the surface thermodynamics and kinetics which facilitates the phase transition. The nanostructured electrodes have demonstrated their capabilities to accommodate an amount of lithium ions higher than that of their counterpart electrodes with micrometre-sized structure (referred to as bulk materials). Recently the thermodynamically non-equilibrium effects of nanostructured electrodes are being increasingly recognized. The presence of surface defects has also demonstrated to modify the surface thermodynamics and facilitate the phase transition boundary. The amorphous state could possibly store more lithium ions because of its more open structure; similar results have been reported in electrodes with poor crystallinity. Materials possessing nanostructures and surface and bulk defects and in poor crystallinity or amorphous state all lie away the from the equilibrium state. Such electrodes away from equilibrium state have demonstrated favorable lithium ion intercalation properties. The contribution of non-equilibrium state lies in three aspects: (1) enhancing the storage capacity by shifting the phase transition boundary; (2) improving the rate capability by introducing a fast mass and charge transport path; and (3) allowing longer cyclic stability by permitting more freedom for volume change accompanied by lithium ion intercalation and de-intercalation. The drawbacks of the electrodes away from equilibrium state is their chemical and structure stability; surface coating and passivation have been explored and studied to counter this challenge. The development of binderless and carbon-free film electrodes with appropriately designed nanostructures would be another exciting research direction, as the conventional electrode fabrication methods are not suitable for the retention of nanostructures. Film electrodes have the advantages of easy fabrication and high energy storage density but suffer from low rate capability because of absence of conductive species, however, nanostructured film electrodes could achieve comparable or higher rate capability when the appropriately designed nano and microstructures and surface chemistry are applied.Acknowledgements
D. W. L would like to acknowledge the graduate fellowship from the University of Washington Center for Nanotechnology (CNT). This work is also supported by NSF (DMI-0455994 and DMR-0605159), AFOSR (MURI, FA955006-1-0326), NCNT (Korea), WTC, PNNL and EnerG2.References
- M. Z. Jacobson, Energy Environ. Sci., 2009, 2, 148 RSC.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15731.
- P. Xie and R. B. Zhang, J. Mater. Chem., 2005, 15, 2529 RSC.
- G. Petrone, G. Spagnuolo, R. Teodorescu, M. Veerachary and M. Vitelli, IEEE Trans. Ind. Electron., 2008, 55, 2569 CrossRef.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556 CrossRef CAS.
- K. M. Coakley and M. D. McGehee, Chem. Mater., 2004, 16, 4533 CrossRef CAS.
- J. E. Fernandez, Science, 2007, 315, 1807 CrossRef CAS.
- M. S. Dresselhaus, G. Chen, M. Y. Tang, R. G. Yang, H. Lee, D. Z. Wang, Z. F. Ren and J. P. Fleurial, Adv. Mater., 2007, 19, 1043 CrossRef CAS.
- L. E. Bell, Science, 2008, 321, 1457 CrossRef CAS.
- I. B. Fridleifsson, Renewable Sustainable Energy Rev., 2001, 5, 299 CrossRef CAS.
- M. Asif and T. Muneer, Renewable Sustainable Energy Rev., 2007, 11, 1388 CrossRef.
- Q. F. Zhang, C. S. Dandeneau, X. Y. Zhou and G. Z. Cao, Adv. Mater., 2009, 21, 4087 CrossRef CAS.
- M. Granovskii, I. Dincer and M. A. Rosen, J. Power Sources, 2007, 167, 461 CrossRef CAS.
- D. P. Birnie III, J. Power Sources, 2009, 186, 539 CrossRef.
- V. Ananthachar and J. J. Duffy, Sol. Energy, 2005, 78, 687 CrossRef CAS.
- J. Linnemann and R. Steinberger-Wilckens, Int. J. Hydrogen Energy, 2007, 32, 1492 CrossRef CAS.
- Y. Qin, X. D. Wang and Z. L. Wang, Nature, 2008, 451, 809 CrossRef CAS.
- R. Yevich and J. A. Logan, Global Biogeochem. Cycles, 2003, 17, 1095 CrossRef.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542 RSC.
- R. H. Crabtree, Energy Environ. Sci., 2008, 1, 134 RSC.
- A. Feaver, S. Sepehri, P. Shamberger, A. Stowe, T. Autrey and G. Z. Cao, J. Phys. Chem. B, 2007, 111, 7469 CrossRef CAS.
- B. Batalla Garcia, A. M. Feaver, Q. F. Zhang, R. D. Champion, G. Z. Cao, T. T. Fister, K. P. Nagle and G. T. Seidler, J. Appl. Phys., 2008, 104, 014305 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271 CrossRef CAS.
- R. J. Brodd, K. R. Bullock, R. A. Leising, R. L. Middaugh, J. R. Miller and E. Takeuchi, J. Electrochem. Soc., 2004, 151, K1 CrossRef CAS.
- W. H. Meyer, Adv. Mater., 1998, 10, 439 CrossRef CAS.
- G. Y. Adachi, N. Imanaka and H. Aono, Adv. Mater., 1996, 8, 127 CAS.
- Y. Wang and G. Z. Cao, Chem. Mater., 2006, 18, 2787 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS.
- S. F. J. Flipsen, J. Power Sources, 2006, 162, 927 CrossRef CAS.
- J. Mader, IEEE AES Syst. Mag., 1996, 11, 17 Search PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS.
- J. M. Tarascon and D. Guyomard, Electrochim. Acta, 1993, 38, 1221 CrossRef CAS.
- P. Arora, R. E. White and M. Doyle, J. Electrochem. Soc., 1998, 145, 3647 CAS.
- H. Li, Z. X. Wang, L. Q. Chen and X. J. Huang, Adv. Mater. DOI:10.1002/adma.200901710.
- X. W. Lou, Y. Wang, C. L. Yuan, J. Y. Lee and L. A. Archer, Adv. Mater., 2006, 18, 2325 CrossRef CAS.
- F. M. Zhan, B. Y. Geng and Y. J. Guo, Chem.–Eur. J., 2009, 15, 6169 CrossRef CAS.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novak, Adv. Mater., 1998, 10, 725 CrossRef CAS.
- A. S. Aricò, P. G. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef CAS.
- M. J. Zou, M. Yoshio, S. Gopukumar and J. I. Yamaki, Chem. Mater., 2003, 15, 4699 CrossRef CAS.
- P. Strobel, S. Rohs and F. Le Cras, J. Mater. Chem., 1996, 6, 1591 RSC.
- J. B. Goodenough, J. Power Sources, 2007, 174, 996 CrossRef CAS.
- S. Ahn, Y. Kim, K. J. Kim, T. H. Kim, H. Lee and M. H. Kim, J. Power Sources, 1999, 81–82, 896 CrossRef CAS.
- L. J. Fu, T. Zhang, Q. Cao, H. P. Zhang and Y. P. Wu, Electrochem. Commun., 2007, 9, 2140 CrossRef CAS.
- J. Reed and G. Ceder, Chem. Rev., 2004, 104, 4513 CrossRef CAS.
- M. Nishizawa, H. Koshika and I. Uchida, J. Phys. Chem. B, 1999, 103, 192 CrossRef CAS.
- F. S. Ke, L. Huang, H. H. Jiang, H. B. Wei, F. Z. Yang and S. G. Sun, Electrochem. Commun., 2007, 9, 228 CrossRef CAS.
- J. W. Long, B. Dunn, D. R. Rolison and H. S. White, Chem. Rev., 2004, 104, 4463 CrossRef CAS.
- Y. X. Gu, D. R. Chen and M. L. Jiao, J. Phys. Chem. B, 2005, 109, 17901 CrossRef CAS.
- K. H. Reiman, K. M. Brace, T. J. Gordon-Smith, I. Nandhakumar, G. S. Attard and J. R. Owen, Electrochem. Commun., 2006, 8, 517 CrossRef CAS.
- J. Hu, H. Li, X. J. Huang and L. Q. Chen, Solid State Ionics, 2006, 177, 2791 CrossRef CAS.
- H. J. Liu, S. H. Bo, W. J. Cui, F. Li, C. X. Wang and Y. Y. Xia, Electrochim. Acta, 2008, 53, 6497 CrossRef CAS.
- L. J. Fu, H. Liu, C. Li, Y. P. Wu, E. Rahm, R. Holze and H. Q. Wu, Solid State Ionics, 2006, 8, 113 CAS.
- G. A. Wiegers, Prog. Solid State Chem., 1996, 24, 1 CrossRef CAS.
- K. Ui, S. Y. Funo, H. Nagase, Y. Idemoto and N. Koura, Electrochemistry, 2006, 74, 474 CAS.
- L. Ji and X. Zhang, Electrochem. Commun., 2009, 11, 1146 CrossRef CAS.
- C. C. Li, J. T. Lee and X. W. Peng, J. Electrochem. Soc., 2006, 153, A809 CrossRef CAS.
- N. Imanishi, K. Kanamura and Z. Takehara, J. Electrochem. Soc., 1992, 139, 2082 CAS.
- A. Eftekhari, J. Power Sources, 2003, 124, 182 CrossRef CAS.
- Q. Y. Li, S. J. Hu, H. Q. Wang, F. P. Wang, X. X. Zhong and X. Y. Wang, Electrochim. Acta, 2009, 54, 5884 CrossRef CAS.
- B. J. Neudecker, R. A. Zuhr and J. B. Bates, J. Power Sources, 1999, 81, 27 CrossRef.
- J. Song, X. Yang, S. S. Zeng, M. Z. Cai, L. T. Zhang, Q. F. Dong, M. S. Zheng, S. T. Wu and Q. H. Wu, J. Micromech. Microeng., 2009, 19, 045004 CrossRef.
- G. Z. Cao, Nanostructures and Nanomaterials, Synthesis, Properties and Applications, Imperial College Press, London, 2004 Search PubMed.
- A. Bachtold, P. Hadley, T. Nakanishi and C. Dekker, Science, 2001, 294, 1317 CrossRef CAS.
- Y. Cui, Q. Wei, H. Park and C. M. Lieber, Science, 2001, 293, 1289 CrossRef CAS.
- P. Xiao, D. W. Liu, B. B. Garcia, S. Sepehri, Y. H. Zhang and G. Z. Cao, Sens. Actuators, B, 2008, 134, 367 CrossRef.
- T. P. Chou, Q. F. Zhang, G. E. Fryxell and G. Z. Cao, Adv. Mater., 2007, 19, 2588 CrossRef CAS.
- Q. F. Zhang, T. P. Chou, B. Russo, S. A. Jenekhe and G. Z. Cao, Angew. Chem., Int. Ed., 2008, 47, 2402 CrossRef CAS.
- K. Takahashi, S. J. Limmer, Y. Wang and G. Z. Cao, J. Phys. Chem. B, 2004, 108, 9795 CrossRef CAS.
- P. G. Bruce, Solid State Ionics, 2008, 179, 752 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CAS.
- K. Zaghib, A. Mauger, F. Gendron and C. M. Julien, Chem. Mater., 2008, 20, 462 CrossRef.
- S. Y. Chung and Y. M. Chiang, Electrochem. Solid-State Lett., 2003, 6, A278 CrossRef CAS.
- P. S. Herle, B. Ellis, N. Coombs and L. F. Nazar, Nat. Mater., 2004, 3, 147 CrossRef CAS.
- Y. Xia, M. Yoshio and H. Noguchi, Electrochim. Acta, 2006, 52, 240 CrossRef CAS.
- N. J. Yun, H. W. Ha, K. H. Jeong, H. Y. Park and K. Kim, J. Power Sources, 2001, 97–98, 508.
- A. Yamada, S. C. Chung and K. Hinokuma, J. Electrochem. Soc., 2001, 148, A224 CrossRef CAS.
- M. Takahashi, Sh. Tobishima, K. Takei and Y. Sakurai, J. Power Sources, 2001, 97–98, 508–511.
- N. Kosova and E. Devyatkina, Solid State Ionics, 2004, 172, 181 CrossRef CAS.
- M. Higuchi, K. Katayama, Y. Azuma, M. Yukawa and M. Suhara, J. Power Sources, 2003, 119–121, 258 CrossRef CAS.
- D. Jugovic and D. Uskokovic, J. Power Sources, 2009, 190, 538 CrossRef CAS.
- J. J. Chen and M. S. Whittingham, Electrochem. Commun., 2006, 8, 855 CrossRef CAS.
- B. Ellis, W. H. Kan, W. R. M. Makahnouk and L. F. Nazar, J. Mater. Chem., 2007, 17, 3248 RSC.
- H. Yang, X. L. Wu, M. H. Cao and Y. G. Guo, J. Phys. Chem. C, 2009, 113, 3345 CrossRef CAS.
- N. Recham, L. Dupont, M. Courty, K. Djellab, D. Larcher, M. Armand and J. M. Tarascon, Chem. Mater., 2009, 21, 1096 CrossRef CAS.
- G. X. Wang, X. P. Shen and J. Yao, J. Power Sources, 2009, 189, 543 CrossRef CAS.
- D. W. Choi and P. N. Kumta, J. Power Sources, 2007, 163, 1064 CrossRef CAS.
- R. Dominko, M. Bele, J. M. Goupil, M. Gaberscek, D. Hanzel, I. Arcon and J. Jamnik, Chem. Mater., 2007, 19, 2960 CrossRef CAS.
- Y. Q. Wang, J. L. Wang, J. Yang and Y. N. Nuli, Adv. Funct. Mater., 2006, 16, 2135 CrossRef CAS.
- G. Meligrana, C. Gerbaldi, A. Tuel, S. Bodoardo and N. Penazzi, J. Power Sources, 2006, 160, 516 CrossRef CAS.
- A. V. Murugan, T. Muraliganth and A. Manthiram, J. Phys. Chem. C, 2008, 112, 14665 CrossRef.
- R. Dominko, M. Bele, M. Gaberscek, M. Remskar, D. Hanzel, J. M. Goupil, S. Pejovnik and J. Jamnik, J. Power Sources, 2006, 153, 274 CrossRef CAS.
- F. Yu, J. J. Zhang, Y. F. Yang and G. Z. Song, J. Mater. Chem., 2009, 19, 9121 RSC.
- G. Arnold, J. Garche, R. Hemmer, S. Strobele, C. Vogler and M. Wohlfahrt-Mehrens, J. Power Sources, 2003, 119–121, 247 CrossRef CAS.
- M. R. Yang, W. H. Ke and S. H. Wu, J. Power Sources, 2005, 146, 539 CrossRef CAS.
- T. H. Cho and H. T. Chung, J. Power Sources, 2004, 133, 272 CrossRef CAS.
- S. T. Myung, Sh. Komaba, N. Hirosaki, H. Yashiro and N. Kumagai, Electrochim. Acta, 2004, 49, 4213 CrossRef CAS.
- S. Y. Yin, L. Song, X. Y. Wang, M. F. Zhang, K. L. Zhang and Y. X. Zhang, Electrochim. Acta, 2009, 54, 5629 CrossRef CAS.
- D. Peramunage and K. M. Abraham, J. Electrochem. Soc., 1998, 145, 2609.
- J. L. Allen, T. R. Jow and J. Wolfenstine, J. Power Sources, 2006, 159, 1340 CrossRef CAS.
- A. Jaiswal, C. R. Horne, O. Chang, W. Zhang, W. Kong, E. Wang, T. Cherm and M. M. Doeff, J. Electrochem. Soc., 2009, 156, A1041 CrossRef CAS.
- H. W. Lu, W. Zeng, Y. S. Li and Z. W. Fu, J. Power Sources, 2007, 164, 874 CrossRef CAS.
- J. Y. Kim and J. P. Cho, Electrochem. Solid-State Lett., 2007, 10, A81 CrossRef CAS.
- J. Gao, J. R. Ying, C. Y. Jiang and C. R. Wan, J. Power Sources, 2007, 166, 255 CrossRef CAS.
- J. J. Huang and Z. Y. Jiang, Electrochim. Acta, 2008, 53, 7756 CrossRef CAS.
- L. Cheng, J. Yan, G. N. Zhu, J. Y. Luo, C. X. Wang and Y. Y. Xia, J. Mater. Chem., 2010, 20, 595 RSC.
- P. Heitjans and S. Indris, J. Phys.: Condens. Matter, 2003, 15, R1257 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS.
- J. Schoonman, Solid State Ionics, 2000, 135, 5 CrossRef CAS.
- J. Maier, Nat. Mater., 2005, 4, 805 CrossRef.
- J. Maier, Phys. Chem. Chem. Phys., 2009, 11, 3011 RSC.
- N. Meethong, H. Y. S. Huang, W. C. Carter and Y. M. Chiang, Electrochem. Solid-State Lett., 2007, 10, A134 CrossRef CAS.
- M. Wagemaker, W. J. H. Borghols, E. R. H. van Eck, A. P. M. Kentgens, G. J. Kearley and F. M. Mulder, Chem.–Eur. J., 2007, 13, 2023 CrossRef CAS.
- M. Wagemaker, W. J. H. Borghols and F. M. Mulder, J. Am. Chem. Soc., 2007, 129, 4323 CrossRef.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS.
- J. Yan, Z. Chen, J. Jiang, L. O. Tan and X. C. Zeng, Adv. Mater., 2009, 21, 314 CrossRef CAS.
- G. Z. Cao and D. W. Liu, Adv. Colloid Interface Sci., 2008, 136, 45 CrossRef CAS.
- I. Turyan and D. Mandler, J. Am. Chem. Soc., 1998, 120, 10733 CrossRef CAS.
- D. Q. Shi, J. P. Tu, Y. F. Yuan, H. M. Wu, Y. Li and X. B. Zhao, Electrochem. Commun., 2006, 8, 1610 CrossRef CAS.
- C. J. Patrissi and C. R. Martin, J. Electrochem. Soc., 1999, 146, 3176 CrossRef CAS.
- K. Takahashi, Y. Wang and G. Z. Cao, J. Phys. Chem. B, 2005, 109, 48 CrossRef CAS.
- Y. Wang, K. Takahashi, H. M. Shang, K. H. Lee and G. Z. Cao, J. Phys. Chem. B, 2005, 109, 3085 CrossRef CAS.
- Y. Wang, K. Takahashi, K. Lee and G. Z. Cao, Adv. Funct. Mater., 2006, 16, 1133 CrossRef CAS.
- M. Sugantha, P. A. Ramakrishnan, A. M. Hermann, C. P. Warmsingh and D. S. Ginley, Int. J. Hydrogen Energy, 2003, 28, 597 CrossRef CAS.
- F. Y. Cheng, J. Z. Zhao, W. Song, C. S. Li, H. Ma, J. Chen and P. Shen, Inorg. Chem., 2006, 45, 2038 CrossRef CAS.
- T. X. T. Sayle, R. R. Maphanga, P. E. Ngoepe and D. C. Sayle, J. Am. Chem. Soc., 2009, 131, 6161 CrossRef CAS.
- A. R. Armstrong, G. Armstrong, J. Canales and P. G. Bruce, Angew. Chem., Int. Ed., 2004, 43, 2286 CrossRef CAS.
- A. R. Armstrong, G. Armstrong, J. Canales, R. Garcia and P. G. Bruce, Adv. Mater., 2005, 17, 862 CrossRef.
- X. P. Gao, H. Y. Zhu, G. L. Pan, S. H. Ye, Y. Lan, F. Wu and D. Y. Song, J. Phys. Chem. B, 2004, 108, 2868 CrossRef CAS.
- J. R. Li, Z. L. Tang and Z. T. Zhang, Electrochem. Solid-State Lett., 1999, 3, 316 CrossRef.
- H. Zhang, G. R. Li, L. P. An, T. Y. Yan, X. P. Gao and H. Y. Zhu, J. Phys. Chem. C, 2007, 111, 6143 CrossRef CAS.
- J. Y. Luo, J. J. Zhang and Y. Y. Xia, Chem. Mater., 2006, 18, 5618 CrossRef CAS.
- F. Cheng, Z. Tao, J. Liang and J. Chen, Chem. Mater., 2008, 20, 667 CrossRef CAS.
- A. S. Yu and R. Frech, J. Electrochem. Soc., 2002, 149, A99 CrossRef CAS.
- P. D. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152 CrossRef CAS.
- L. Kavan, J. Rathousky, M. Gratzel, V. Shklover and A. Zukal, J. Phys. Chem. B, 2000, 104, 12012 CrossRef CAS.
- D. Fattakhova-Rohlfing, M. Wark, T. Brezesinski, B. M. Smarsly and J. Rathousky, Adv. Funct. Mater., 2007, 17, 123 CrossRef CAS.
- P. Kubiak, J. Geserick, N. Husing and M. Wohlfahrt-Mehrens, J. Power Sources, 2008, 175, 510 CrossRef CAS.
- X. W. Lou and L. A. Archer, Adv. Mater., 2008, 20, 1853 CrossRef CAS.
- P. Liu, S. H. Lee, C. E. Tracy, Y. F. Yan and J. A. Turner, Adv. Mater., 2002, 14, 27 CrossRef CAS.
- S. Lim, C. S. Yoon and J. Cho, Chem. Mater., 2008, 20, 4560 CrossRef CAS.
- F. Jiao and P. G. Bruce, Adv. Mater., 2007, 19, 657 CrossRef CAS.
- F. Jiao, J. L. Bao, A. H. Hill and P. G. Bruce, Angew. Chem., Int. Ed., 2008, 47, 9711 CrossRef CAS.
- A. K. Sinha, K. Suzuki, M. Takahara, H. Azuma, T. Nonaka and K. Fukumoto, Angew. Chem., Int. Ed., 2007, 46, 2891 CrossRef CAS.
- X. M. Ni, Y. F. Zhang, D. Y. Tian, H. G. Zheng and X. W. Wang, J. Cryst. Growth, 2007, 306, 418 CrossRef CAS.
- P. Liu, S. H. Lee, C. E. Tracy, J. A. Turner, J. R. Pitts and S. K. Deb, Solid State Ionics, 2003, 165, 223 CrossRef CAS.
- B. V. Ratnakumar, M. C. Smart and S. Surampudi, J. Power Sources, 2001, 97–98, 137 CrossRef CAS.
- S. S. Zhang, K. Xu and T. R. Jow, Electrochem. Solid-State Lett., 2002, 5, A92 CrossRef CAS.
- F. Beguin, F. Chevallier, C. Vix-Guterl, S. Saadallah, V. Bertagna, J. N. Rouzaud and E. Frackowiak, Carbon, 2005, 43, 2160 CrossRef CAS.
- J. Yan, B. J. Xia, Y. C. Su, X. Z. Zhou, J. Zhang and X. G. Zhang, Electrochim. Acta, 2008, 53, 7069 CrossRef CAS.
- A. Lewandowski and A. Swiderska-Mocek, J. Power Sources, 2009, 194, 601 CrossRef CAS.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379 CrossRef CAS.
- M. S. Wu, P. C. J. Chiang and J. C. Lin, J. Electrochem. Soc., 2005, 152, A1041 CrossRef CAS.
- T. Yoshida, M. Takahashi, S. Morikawa, C. Ihara, H. Katsukawa, T. Shiratsuchi and J. I. Yamaki, J. Electrochem. Soc., 2006, 153, A576 CrossRef CAS.
- K. Edström, A. M. Andersson, A. Bishop, L. Fransson, J. Lindgren and A. Hussenius, J. Power Sources, 2001, 97–98, 87 CrossRef CAS.
- M. Itagaki, S. Yotsuda, N. Kobari, K. Watanabe, S. Kinoshita and M. Ue, Electrochim. Acta, 2006, 51, 1629 CrossRef CAS.
- Y. S. Park and S. M. Lee, Electrochim. Acta, 2009, 54, 3339 CrossRef CAS.
- M. Jo, Y. S. Hong, J. Choo and J. Cho, J. Electrochem. Soc., 2009, 156, A430 CrossRef CAS.
- J. L. Lei, L. J. Li, R. Kostecki, R. Muller and F. Mclarnon, J. Electrochem. Soc., 2005, 152, A774 CrossRef CAS.
- L. Yang, M. Takahashi and B. F. Wang, Electrochim. Acta, 2006, 51, 3228 CrossRef CAS.
- D. H. Jang, Y. J. Shin and S. M. Oh, J. Electrochem. Soc., 1996, 143, 2204 CAS.
- T. F. Yi, Y. R. Zhu, X. D. Zhu, J. Shu, C. B. Yue and A. N. Zhou, Ionics, 2009, 15, 779 CrossRef CAS.
- S. H. Ye, J. Y. Lv, X. P. Gao, F. Wu and D. Y. Song, Electrochim Acta, 2004, 49, 1623 CAS.
- D. Aurbach, M. D. Levi, E. Levi, H. Teller, B. Markovsky, G. Salitra, U. Heider and L. Heider, J. Electrochem. Soc., 1998, 145, 3024 CAS.
- C. L. Olson and I. Ballard, J. Phys. Chem. B, 2006, 110, 1828.
- Y. Yang, C. Xie, R. Ruffo, H. L. Peng, D. K. Kim and Y. Cui, Nano Lett., 2009, 9, 4109 CrossRef CAS.
- N. A. Chernova, M. Roppolo, A. C. Dillon and M. S. Whittingham, J. Mater. Chem., 2009, 19, 2526 RSC.
- D. C. Li, Y. Sasaki, K. Kobayakawa and Y. Sato, Electrochim. Acta, 2006, 51, 3809 CrossRef CAS.
- C. L. Tan, J. J. Zhou, W. S. Li, X. H. Hou, D. S. Lv, M. Q. Xu and Q. M. Huang, J. Power Sources, 2008, 184, 408 CrossRef CAS.
- G. T. K. Fey, J. G. Chen and T. P. Kumar, J. Power Sources, 2005, 146, 250 CrossRef CAS.
- B. Kim, C. Kim, T. G. Kim, D. Ahn and B. Park, J. Electrochem. Soc., 2006, 153, A1773 CrossRef CAS.
- H. W. Ha, N. J. Yun, M. H. Kim, M. H. Woo and K. Kim, Electrochim. Acta, 2006, 51, 3297 CrossRef CAS.
- Z. R. Zhang, Z. L. Gong and Y. Yang, J. Phys. Chem. B, 2004, 108, 17546 CrossRef CAS.
- N. Ohta, K. Takada, L. Q. Zhang, R. Z. Ma, M. Osada and T. Sasaki, Adv. Mater., 2006, 18, 2226 CrossRef CAS.
- N. V. Kosova and E. T. Devyatkina, J. Power Sources, 2007, 174, 959 CrossRef CAS.
- G. T. K. Fey, C. Z. Lu, T. P. Kumar and Y. C. Chang, Surf. Coat. Technol., 2005, 199, 22 CrossRef.
- M. Kaneko, M. Nakayama, Y. Wakizaka, K. Kanamura and M. Wakihara, Electrochim. Acta, 2008, 53, 8196 CrossRef CAS.
- Y. Shao-horn, S. A. Hackney, A. J. Kahaina, K. D. Kepler, E. Skinner, J. T. Vaughey and M. M. Thackeray, J. Power Sources, 1999, 81–82, 496 CrossRef CAS.
- K. M. Shaju and P. G. Bruce, Chem. Mater., 2008, 20, 5557 CrossRef CAS.
- G. Amatucci, A. Du Pasquier, A. Blyr, T. Zheng and J. M. Tarascon, Electrochim. Acta, 1999, 45, 255 CrossRef CAS.
- Z. X. Yang, W. S. Yang, D. G. Evans, Y. Y. Zhao and X. Wei, J. Power Sources, 2009, 189, 1147 CrossRef CAS.
- A. M. Kannan and A. Manthiram, Electrochem. Solid-State Lett., 2002, 5, A167 CrossRef CAS.
- J. Tu, X. B. Zhao, G. S. Cao, D. G. Zhuang, T. J. Zhu and J. P. Tu, Electrochim. Acta, 2006, 51, 6456 CrossRef CAS.
- A. Eftekhari, Solid State Ionics, 2004, 167, 237 CrossRef CAS.
- T. Okumura, T. Fukutsuka, Y. Uchimoto, K. Amezawa and S. Kobayashi, J. Power Sources, 2009, 189, 471 CrossRef CAS.
- J. Han, S. Myung and Y. Sun, J. Electrochem. Soc., 2006, 153, A1290 CrossRef CAS.
- M. M. Thackeray, C. S. Johnson, J. S. Kim, K. C. Lauzze, J. T. Vaughey, N. Dietz, D. Abraham, S. A. Hackney, W. Zeltner and M. A. Anderson, Electrochem. Commun., 2003, 5, 752 CrossRef CAS.
- S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 2002, 1, 123 CrossRef CAS.
- S. F. Yang, Y. N. Song, K. Ngala, P. Y. Zavalij and M. S. Whittingham, J. Power Sources, 2003, 119–121, 239 CrossRef CAS.
- N. Ravet, J. B. Goodenough, S. Besner, M. Simoneau, P. Hovington and M. Armand, Electrochem. Soc. Abstr., 1999, 99–2, 172 Search PubMed.
- I. Belharouak, C. Johnson and K. Amine, Electrochem. Commun., 2005, 7, 983 CrossRef CAS.
- F. Croce, A. D. Epifanio, J. Hassoun, A. Deptula, T. Olczac and B. Scrosati, Electrochem. Solid-State Lett., 2002, 5, A47 CrossRef CAS.
- J. Morales, R. Trocoli, E. Rodriguez-Castellon, S. Franger and J. Santos-Pena, J. Electroanal. Chem., 2009, 631, 29 CrossRef CAS.
- Y. S. Hu, Y. G. Guo, R. Dominko, M. Gaberscek, J. Jamnik and J. Maier, Adv. Mater., 2007, 19, 1963 CrossRef CAS.
- K. Saravanan, M. V. Reddy, P. Balaya, H. Gong, B. V. R. Chowdari and J. J. Vittal, J. Mater. Chem., 2009, 19, 605 RSC.
- A. Odani, V. G. Pol, S. V. Pol, M. Koltypin, A. Gedanken and D. Aurbach, Adv. Mater., 2006, 18, 1431 CrossRef CAS.
- B. Li. He, B. Dong and H. L. Li, Electrochem. Commun., 2007, 9, 425 CrossRef CAS.
- M. Mancini, P. Kubiak, J. Geserick, R. Marassi, N. Husing and M. Wohlfahrt-Mehrens, J. Power Sources, 2009, 189, 585 CrossRef CAS.
- F. Tanguy, J. Gaubicher, A. C. Gaillot, D. Guyomard and J. Pinson, J. Mater. Chem., 2009, 19, 4771 RSC.
- D. R. Rolison and B. Dunn, J. Mater. Chem., 2001, 11, 963 RSC.
- P. Xiao, B. B. Garcia, Q. Guo, D. W. Liu and G. Z. Cao, Electrochem. Commun., 2007, 9, 2441 CrossRef CAS.
- D. W. Liu, Y. H. Zhang, P. Xiao, B. B. Garcia, Q. F. Zhang, X. Y. Zhou, Y. H. Jeong and G. Z. Cao, Electrochim. Acta, 2009, 54, 6816 CrossRef CAS.
- K. E. Swider-Lyons, C. T. Love and D. R. Rolison, Solid State Ionics, 2002, 152–153, 99 CrossRef CAS.
- D. W. Liu, Y. Y. Liu, B. B. Garcia, Q. F. Zhang, A. Q. Pan, Y. H. Jeong and G. Z. Cao, J. Mater. Chem., 2009, 19, 8789 RSC.
- Y. Wang, H. M. Shang, T. Chou and G. Z. Cao, J. Phys. Chem. B, 2005, 109, 11361 CrossRef CAS.
- J. Cho, T. J. Kim and B. Park, J. Electrochem. Soc., 2002, 149, A288 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS.
- S. Pyun and J. S. Bae, J. Power Sources, 1997, 68, 669 CrossRef CAS.
- L. J. Fu, H. Liu, C. Li, Y. P. Wu, E. Rahm, R. Holze and H. Q. Wu, Solid State Sci., 2006, 8, 113 CrossRef CAS.
- D. W. Liu, P. Xiao, Y. H. Zhang, B. B. Garcia, Q. F. Zhang, Q. Guo, R. Champion and G. Z. Cao, J. Phys. Chem. C, 2008, 112, 11175 CrossRef CAS.
- H. S. Jung, H. Shin, J. R. Kim, J. Y. Kim, K. S. Hong and J. K. Lee, Langmuir, 2004, 20, 11732 CrossRef CAS.
- C. M. Julien, Mater. Sci. Eng., R, 2003, 40, 47 CrossRef.
- C. M. Park and H. J. Sohn, Adv. Mater., 2010, 22, 47 CrossRef CAS.
- M. S. Whittingham, R. S. Chianelli, A. J. Jacobson, in: D. W. Murphy, J. Broadhead, B. C. H. Steele (ed.), Materials for Advanced Batteries, Plenum Press, New York, 1980, p. 291 Search PubMed.
- J. J. Xu, A. J. Kinser, B. B. Owens and W. H. Smyrl, Electrochem. Solid-State Lett., 1999, 1, 1 CrossRef.
- J. J. Xu and J. S. Yang, Electrochem. Commun., 2003, 5, 230 CrossRef CAS.
- W. C. West, N. V. Myung, J. F. Whitacre and B. V. Ratnakumar, J. Power Sources, 2004, 126, 203 CrossRef CAS.
- J. J. Xu, G. Jain and J. Yang, Electrochem. Solid-State Lett., 2002, 5, A152 CrossRef CAS.
- J. J. Xu, J. Yang and G. Jain, Electrochem. Solid-State Lett., 2002, 5, A223 CrossRef CAS.
- J. S. Yang, T. B. Atwater and J. J. Xu, J. Power Sources, 2005, 139, 274 CrossRef CAS.
- H. Furukawa, M. Hibino and I. J. Honma, J. Electrochem. Soc., 2004, 151, A527 CrossRef CAS.
- K. Lee and G. Z. Cao, J. Phys. Chem. B, 2005, 109, 11880 CrossRef CAS.
- L. P. An, X. P. Gao, G. R. Li, T. Y. Yan, H. Y. Zhu and P. W. Shen, Electrochim. Acta, 2008, 53, 4573 CrossRef CAS.
- S. W. Song and S. W. Baek, Electrochem. Solid-State Lett., 2009, 12, A23 CrossRef CAS.
- L. B. Chen, J. Y. Xie, H. C. Yu and T. H. Wang, J. Appl. Electrochem., 2009, 39, 1157 CrossRef CAS.
- V. Baranchugov, E. Markevich, E. Pollak, G. Salitra and D. Aurbach, Electrochem. Commun., 2007, 9, 796 CrossRef CAS.
- M. S. Park, G. X. Wang, H. K. Liu and S. X. Dou, Electrochim. Acta, 2006, 51, 5246 CrossRef CAS.
- H. Xia, S. B. Tang and L. Lu, Mater. Res. Bull., 2007, 42, 1301 CrossRef CAS.
- T. J. Boyle, D. Ingersoll, T. M. Alam, C. J. Tafoya, M. A. Rodriguez, K. Vanheusden and D. H. Doughty, Chem. Mater., 1998, 10, 2270 CrossRef CAS.
- S. Gopukumar, K. Y. Chung and K. B. Kim, Electrochim. Acta, 2004, 49, 803 CrossRef CAS.
- M. W. Raja, S. Mahanty and R. N. Basu, J. Mater. Chem., 2009, 19, 6161 RSC.
- D. A. Totir, B. D. Cahan and D. A. Scherson, Electrochim. Acta, 1999, 45, 161 CrossRef CAS.
- M. G. Lazarraga, S. Mandal, J. Ibanez, J. M. Amarilla and J. M. Rojo, J. Power Sources, 2003, 115, 315 CrossRef CAS.
- E. P. Roth, D. H. Doughty and J. Franklin, J. Power Sources, 2004, 134, 222 CrossRef CAS.
- M. Yoo, C. W. Frank, S. Mori and S. Yamaguchi, Chem. Mater., 2004, 16, 1945 CrossRef CAS.
- S. Ahn, Electrochem. Solid-State Lett., 1999, 1, 111 CrossRef.
- X. M. Liu, Z. D. Huang, S. Oh, P. C. Ma, P. C. H. Chan, G. K. Vedam, K. Kang and J. K. Kim, J. Power Sources, 2010, 195, 4290 CrossRef CAS.
- Y. J. Park, K. S. Ryu, K. M. Kim, N. G. Park, M. G. Kang and S. H. Chang, Solid State Ionics, 2002, 154–155, 229 CrossRef CAS.
- H. K. Kim, T. Y. Seong and Y. S. Yoon, Electrochem. Solid-State Lett., 2002, 5, A252 CrossRef CAS.
- J. D. Perkins, C. S. Bahn, J. M. McGraw, P. A. Parilla and D. S. Ginley, J. Electrochem. Soc., 2001, 148, A1302 CrossRef CAS.
- N. Kuwata, J. Kawamura, K. Toribami, T. Hattori and N. Sata, Electrochem. Commun., 2004, 6, 417 CrossRef CAS.
- C. H. Chen, E. M. Kelder and J. Schoonman, Thin Solid Films, 1999, 342, 35 CrossRef CAS.
- M. Mohamedi, M. Makino, K. Dokko, T. Itoh and I. Uchida, Electrochim. Acta, 2002, 48, 79 CrossRef CAS.
- Y. J. Park, J. G. Kim, M. K. Kim, H. T. Chung and H. G. Kim, Solid State Ionics, 2000, 130, 203 CrossRef CAS.
- Y. H. Rho, K. Kanamura and T. Umegaki, J. Electrochem. Soc., 2003, 150, A107 CrossRef CAS.
- M. Cheong and I. Zhitomirsky, Surf. Eng., 2009, 25, 346 CrossRef CAS.
- Y. H. Wang and I. Zhitomirsky, Langmuir, 2009, 25, 9684 CrossRef CAS.
- Y. Wang and G. Z. Cao, Electrochim. Acta, 2006, 51, 4865 CrossRef CAS.
- D. W. Liu, Q. F. Zhang, P. Xiao, B. B. Garcia, Q. Guo, R. Champion and G. Z. Cao, Chem. Mater., 2008, 20, 1376 CrossRef CAS.
- D. W. Liu, B. B. Garcia, Q. F. Zhang, Q. Guo, Y. H. Zhang, S. Sepehri and G. Z. Cao, Adv. Funct. Mater, 2009, 19, 948.
- S. M. Zhu, Z. Y. Zhou, D. Zhang and H. H. Wang, Microporous Mesoporous Mater., 2006, 95, 257 CrossRef CAS.
- C. Navone, J. P. Pereira-Ramos, R. Baddour-Hadjean and R. Salot, J. Electrochem. Soc., 2006, 153, A2287 CrossRef CAS.
- K. Le Van, H. Groult, A. Mantoux, L. Perrigaud, F. Lantelme, R. Lindstroem, R. Badour-Hadjean, S. Zanna and D. Lincot, J. Power Sources, 2006, 160, 592 CrossRef CAS.
- L. Wang, H. W. Xu, P. C. Chen, D. W. Zhang, C. X. Ding and C. H. Chen, J. Power Sources, 2009, 193, 846 CrossRef CAS.
- Y. Yu, J. L. Shui and C. H. Chen, Solid State Commun., 2005, 135, 485 CrossRef CAS.
- X. H. Huang, J. P. Tu, X. H. Xia, X. L. Wang, J. Y. Xiang, L. Zhang and Y. Zhou, J. Power Sources, 2009, 188, 588 CrossRef CAS.
- P. Xiao, Y. H. Zhang, B. B. Garcia, S. Sepehri, D. W. Liu and G. Z. Cao, J. Nanosci. Nanotechnol., 2009, 9, 2426 CrossRef CAS.
- Y. H. Zhang, P. Xiao, X. Y. Zhou, D. W. Liu, B. B. Garcia and G. Z. Cao, J. Mater. Chem., 2009, 19, 948 RSC.
| This journal is © The Royal Society of Chemistry 2010 |