Sustainable chemistry: imidazolium salts in biomass conversion and CO2 fixation
Yugen
Zhang
* and
Jin Young Gerentt
Chan
Institute of Bioengineering and Nanotechnology, 31 Biopolis Way, The Nanos, Singapore, 138669, Singapore. E-mail: ygzhang@ibn.a-star.edu.sg
First published on 17th November 2009
Abstract
The research and development into imidazolium salts (IMSs) and their derivatives (N-heterocyclic carbene, NHCs) for sustainability research in biomass conversion and CO2 capture/utilization is reviewed. Research into using IMSs/NHCs in green synthesis, separation, and green materials has increased steadily over the past few years. Recent work utilizing imidazolium ionic liquids in dissolving and converting biomass are assessed. IMSs are particularly useful for directly processing raw biomass, such as cellulose, as feedstock to produce 5-hydroxymethylfurfural (HMF) under mild conditions. Catalytic activities of N-heterocyclic carbenes in sugar conversion are also studied, with a view towards practical industrialization. Imidazolium ionic liquids for CO2 capture and CO2 utilizations are also assessed. Imidazolium salts (IMSs) and their derivatives provide a promising way for carbon dioxide capture and utilization.
 Yugen Zhang | Dr Yugen Zhang is a Team Leader and Principal Research Scientist at the Institute of Bioengineering and Nanotechnology (IBN), Singapore. He graduated in geochemistry from the University of Science and Technology of China (USTC), where he also received his PhD in chemistry in 1992. After his PhD, he joined USTC as a faculty member and was promoted to Professor in 1999. He worked at the Organometallic Lab in RIKEN, Japan from 1996 to 1997 as a Visiting Scientist and from 2000 to 2001 as a RIKEN Fellow. Before he joined IBN in 2004, he worked at Harvard University as a Research Associate (2002–2004). His main research areas are green catalysis and nanostructured materials. |
 Jin Young Gerentt Chan | Mr Gerentt Chan graduated from Carnegie Mellon University in 2008 with a BSc in chemistry on an A*STAR scholarship. He joined the Institute of Bioengineering and Nanotechnology at A*STAR after obtaining his degree, and was engaged in the development of biofuel synthesis using imidazolium salts. He will be proceeding to Stanford University for his PhD studies in Fall 2009. |
Broader contextThe threat of global climate change is substantial, and due to the non-renewable nature of our oil resources, the transformation of the chemical industry towards “greener” and sustainable methods and feedstocks is imperative. Currently, a significant proportion of industry's organic chemical needs are fed by petroleum and its derivatives; such non-renewable resources have to be supplanted without radically redesigning current feedstock processes. An obvious alternative sustainable carbon source would be biomass, where development of broad spectrum transformations into different industrially viable products would make replacement possible. Imidazolium salts and their carbene derivatives are at the core of recent research into such biomass conversions. Another critical sustainability research area that imidazolium salts are making an impact in is carbon dioxide capture and utilization. |
1. Introduction
Imidazoli(ni)um salts (IMSs) are well known as room-temperature ionic liquids (RTILs) that can be used as electrolytes or green solvents because of their low vapour pressure and wide-ranging chemical stability.1,2 In general, IMSs are composed of an asymmetric imidazoli(ni)um cation and an inorganic anion; the presence of the large organic cation allows for solubilization of organic compounds while their overall ionic-columbic interactions do so for inorganic polar compounds. Asymmetry in the cation weakens regular lattice packing and induces lower melting points, leading to their foremost property of being molten at or near room temperature.3,4 IMSs can be designed to have specific properties for particular applications; this versatility is due to the customization possible from its nearly unlimited structural variations (Scheme 1). Imidazoli(ni)um salts have been investigated as solvents in a variety of fields, such as biomass solubilization,5–7 organic synthesis reaction media,8–10 and phase separation and extraction.11,12 In addition, function-specific IMSs have been designed and synthesized, such as, N-doped carbon materials,13 porous materials,14 nano-particles15 and other materials,16,17 aqueous phase catalysts18 and heterogeneous catalyst supports.19 A degree of biodegradability present in imidazoli(ni)ums allows them to serve as green and environmentally friendly reagents with extensive applications.20 Specific imidazoliums have also been used in biological systems for, but not limited to, pharmaceutical activity,21 biosensors,22 and bioimaging.23 Polymeric imidazoli(ni)ums were also developed with different structures and forms. Side-chain poly-imidazoli(ni)um materials (A, Scheme 2) are mostly designed for catalyst immobilization.24,25 Main-chain poly-imidazolium materials are well designed and synthesized in particulate or bulky forms and they have demonstrated excellent properties in heterogeneous catalysis (B, Scheme 2).26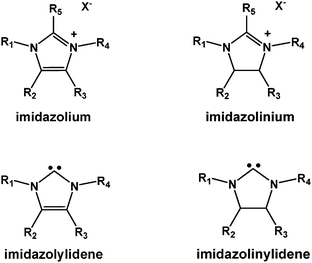 | ||
| Scheme 1 General structures of imidazoli(ni)ums (IMSs) and imidazol(in)ylidene N-heterocyclic carbenes (NHCs). | ||
 | ||
| Scheme 2 General structures for side-chain poly-IMSs (A) and main-chain poly-IMSs (B). | ||
One major application for imidazoli(ni)um salts is as precursors for stable imidazol(in)ylidene N-heterocyclic carbenes (NHC) or bisimidazolidines; these have tremendous applications in organic synthesis as catalysts or substrates (Scheme 1).27,28 In particular, N-heterocyclic carbenes (NHCs) have attracted considerable interest in recent years for their versatile chemistry. The striking chemical similarities between electron-rich organophosphanes (PR3) and NHCs (especially NHCs' excellent σ-donating properties) allow them to be employed both as nucleophile-type catalysts and as ligands of choice for a wide range of transition metal–ligand complexes.29 This has led to a lot of applications of NHC-based catalysts.27,28,30 There are already several excellent reviews on the general properties of imidazoliums and N-heterocyclic carbenes.1,2,8–12,27,28,31 Herein, this article will focus on the applications of imidazoliums and N-heterocyclic carbenes in sustainable research: biomass conversion and carbon dioxide capture and utilization.
2. Biomass conversion
Imidazolium ionic liquids in dissolving lignocellulose
Lignocellulose consists mainly of cellulose (35–50%), hemicellulose (20–35%) and lignin (5–25%).32 Thus, cellulose is the most abundant biopolymer on earth. Due to the large amount of intra and intermolecular hydrogen interactions, cellulose is insoluble in water and common solvents. Hemicellulose is an amorphous polymer with five different sugars. Lignin is an irregular polymer of several monoaromates and forms a three-dimensional network in which cellulose and hemicellulose fibers are embedded. Due to this complex structure, lignocellulose has a remarkable resistance against chemicals and hydrolysis.5 The central barrier for using lignocellulose is the separation of lignocellulose into its components for further enzymatic hydrolysis or chemical conversion. Several hydrothermal and chemical pretreatments for lignocellulose were developed.33 However, all of these methods feature various drawbacks, such as limited biomass resources, side-products which inhibit subsequent hydrolysis or fermentation, the need for extreme conditions and with toxic and hazardous pollutants.33 Therefore, ionic liquids (ILs) have been considered as a promising alternative as a green solvent. Imidazolium salts, as ionic liquids, have been investigated for utilization in biomass solubilization, especially for cellulose.5–7 The difficulties involved in cellulosic solubilization include overcoming the extensive intermolecular hydrogen bonding between adjacent cellulose molecules, and the non-ionic nature of the polymeric backbone. Imidazolium salts were found to elicit substantial dissolution of cellulose, with the solvation process involving hydrogen bonding between the carbohydrate hydroxyl groups and the anions.5,34 In 2002, Rogers’ group discovered that several imidazolium-based ILs can dissolve large amounts of cellulose.5 This prompted other groups to test a variety of ILs on their ability to dissolve cellulose. The cellulose regenerated from ILs was found to be essentially amorphous and porous, and was much more prone to degradation by cellulases or by chemical processes. Furthermore, imidazolium salts are also successful in dissolving other bio-mass macromolecules, such as lignin and lignocellulose (Table 1).35–38 This allows homogeneous chemical modifications of dissolved biomaterials.36 Different types of imidazolium ILs have been designed for different hydrolysis processes. IMSs containing chloride anions are good for chemical conversion of biomass,37 while enzyme-compatible IMSs, such as a glycol-substituted imidazolium cation and acetate anion, have also been developed and used in enzymatic transesterification of methyl methacrylate with glucose and cellulose.38 In general, imidazolium ILs for dissolving and pretreating biomass hold strong potential. However, their costs should be significantly reduced and in-depth exploration is still much needed.| Imidazolium saltsa | Carbonhydrate | Solubility |
|---|---|---|
| a BMIM = 1-butyl-3-methylimidazolium, EMIM = 1-ethyl-3-methylimidazolium, AMIM = 1-allyl-3-methylimidazolium, BDMIM = 1-butyl-2,3-dimethylimidazolium, AdMIM = 1-allyl-2,3-dimethylimidazolium, dca = dicyanamide, MoeMIM =1-methoxyethyl-3-methylimidazolium, MomMIM = 1-methoxymethyl-3-methylimidazolium, [(MeO)(R)PO2]− where R = H, Me or MeO. | ||
| [BMIM]Cl | Cellulose | 10 wt% (100 °C) |
| Wool keratin fibers | 11 wt% (130 °C) | |
| Eucalyptus pulp | ≥ 13.6% (85 °C) | |
| Solucell 1175 cellulose | 16 wt% (100 °C) | |
| [EMIM]Cl | Eucalyptus pulp | ≥ 15.8% (85 °C) |
| [AMIM]Cl | Cellulose | 8–14.5 wt% (80 °C) |
| Solucell 1175 cellulose | 10 wt% (100 °C) | |
| KZO3 (1085) cellulose | 12.5 wt% (100 °C) | |
| [BDMIM]Cl | Eucalyptus pulp | ≥ 12.8% (85 °C) |
| [AdMIM]Br | Cellulose | 4–12% (80 °C) |
| [BMIM][dca] | β-D-Glucose | 145 g L−1 (25 °C) |
| Sucrose | 195 g L−1 (25 °C), 282 g L−1 (60 °C) | |
| Lactose | 225 g L−1 (75 °C) | |
| β-Cyclodextrin | 750 g L−1 (75 °C) | |
| Amylose | 4 g L−1 (25 °C) | |
| [MoeMIM][dca] | β-D-Glucose | 91 g L−1 (25 °C) |
| Sucrose | 220 g L−1 (25 °C) | |
| [MomMIM][dca] | Sucrose | 249 g L−1 (25 °C), 352 g L−1 (60 °C) |
| [AMIM][HCOO] | Cellulose | 10–20 wt% (60–85 °C) |
| [EMIM][OAc] | Eucalyptus pulp | ≥ 13.5% (85 °C) |
| [BMIM][OAc] | Eucalyptus pulp | ≥ 13.2% (85 °C) |
| [EMIM][(MeO)(R)PO2] | Microcrystalline cellulose | 10 wt% (45–65 °C) |
| [MoeMIM][BF4] | β-D-Glucose | 5 mg mL−1 (55 °C) |
Imidazolium salts in cellulose dehydration
A core development of biomass conversion into biofuels or chemicals is the hydrolysis of cellulose to fermentable sugars. In general, hydrolysis of cellulose requires severe conditions, such as use of dilute sulfuric acid at high temperatures. However, these conditions are not suitable for further catalytic sugar dehydration processes. Therefore, acid assisted hydrolysis of biomass in ionic liquids has been widely studied. Zhao and Li reported a method to hydrolyse cellulose in an imidazolium ionic liquid with mineral acids as catalysts at 100 °C.39 This direct process achieved 20–44% of glucose yield and 50–77% of total reducing sugars. Schüth's group found that cellulose can be efficiently depolymerised by using solid acid catalysts, like Amberlyst, in imidazolium ionic liquids.40 Solid acids, especially acidic resins with large pores, are highly suitable catalysts for the depolymerisation of cellulose, even microcrystalline cellulose or wood. Different products, such as cellulo-oligomers or sugars could be selectively produced by varying the reaction time. This process is especially suitable for combining with metal catalyst/IMS systems to directly convert cellulose into 5-hydroxymethylfurfural (HMF), Scheme 3.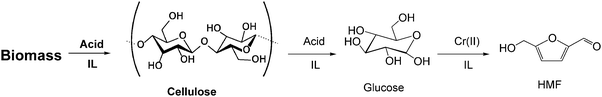 | ||
| Scheme 3 | ||
Raines' group developed a direct process to produce HMF from cellulose by using CrCl2 (25 mol%) and HCl (6 mol%) as catalyst in [EMIM+]Cl− or DMA-LiCl/[EMIM+]Cl−. 53% of HMF yield was achieved from cellulose in this process in 1–2 h at 140 °C.41 Lignocellulosic biomass has also been converted into HMF with 34–37% yield in this process with the same reaction conditions. Zhang's group developed a one step process for converting cellulose into HMF by using a pair of CuCl2/CrCl2 catalysts in imidazolium ionic liquid and a similar HMF yield (54%) was achieved in 8 h at 80–120 °C.42 In our group, we use zeolite (H–Y) as a solid acidic catalyst combined with a chromium catalyst in [BMIM+]Cl− for direct cellulose hydrolysis.43 HMF is selectively produced in 48% yield in 3–6 h at 120 °C. The common attribute between these processes is that cellulose is first dehydrated into sugars by acidic catalysts, such as mineral acid, solid acid or Lewis acid. The following step involves chromium-halide-catalyzed isomerisation of the glucose monomer into fructose and the dehydration of fructose to form HMF (see Scheme 3). Approximately 50% HMF yield from cellulose is the current benchmark and further improvements still need to be done.
Although most research is focused on biomass hydrolysis under acidic conditions, controlled pyrolysis of cellulose using non-acidic imidazolium salts was also studied. Sheldrake's group reported using molten dicationic imidazolium salts as a reuseable medium for controlled pyrolysis of cellulose without acid additives to give anhydrosugars, primarily levoglucosenone, at 180 °C (Scheme 4). It was found that the distance between two imidazole rings is important to the pyrolysis process.44
 | ||
| Scheme 4 | ||
Imidazolium ionic liquids in sugar conversion
Biomass-derived carbohydrate molecules can contain varied functionalities for utilization as fuels or chemicals, and one challenge is to develop methods to control the functionality in the final product.45,46Recent efforts have been devoted towards converting biomass to HMF, a versatile and key intermediate in biomass chemistry and the petroleum industry.47 HMF and its 2,5-disubstituted furan derivatives can replace key petroleum-based building blocks, such as 2,5-furandicarboxylic acid, which can replace terephthalic acid for plastic synthesis.48 Traditional processes to produce HMF involve the use of acid catalysts and are mainly limited to fructose as feed.46–49 A drawback with acid catalysts is that undesired by-products, such as levulinic acid and formic acid, are formed through in situ acid-catalyzed rehydration of the HMF product. Although various acid catalysts have been studied in the presence of different reaction systems, such as water, organic solvents, and biphasic systems, the industrial production of HMF is impeded by high costs of manufacturing.46–49
Recent developments in this field involve using imidazolium salt ILs as reaction media or catalysts to selectively produce HMF in high yield. Moreau's group first made use of imidazolium salts in the process of dehydration of fructose into HMF.50 In the systems of both [BMIM+][BF4−]/DMSO and [BMIM+][PF6−] with solid acid as catalyst (Amerlyst-15), moderate to good HMF yields were obtained. When the system was modified to using [HMIM+]Cl− as both solvent and acid catalyst, HMF yields as high as 92% were achieved within 15–45 min.51 Compared to other catalytic systems, a remarkable feature is that HMF is stable in this imidazolium system, and can then be separately or continuously extracted in a straightforward manner by using organic solvents. This stabilization of HMF from decomposition accords great potential to imidazolium salt ILs in highly selective and efficient biomass-derived HMF synthesis. Additionally, the authors also demonstrated good recyclability of the ionic liquid after water removal. The same system was also applied to the dehydration of sucrose, in which the fructose moiety was quantitatively converted into HMF, while the glucose moiety remained unreacted (see Fig. 1).
![Dehydration of sucrose in [HMIM+]Cl−.51](/image/article/2010/EE/b914206a/b914206a-f1.gif) | ||
| Fig. 1 Dehydration of sucrose in [HMIM+]Cl−.51 | ||
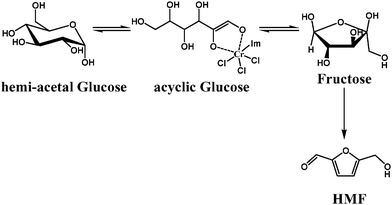
One breakthrough in this field is the introduction of metal chlorides in imidazolium ionic liquid systems as catalysts, which led to the conversion of glucose to HMF in high yield; this has never been achieved in other systems. Zhang's group reported a [metal chloride][alkyl imidazolium chloride] system in which chromium(II) chloride was found to be uniquely effective to glucose (68% HMF yield).52 It was found that catalytic amounts of certain metal chlorides appear to play a role in stabilizing HMF. The proposed mechanism involves the opening and isomerisation of the 6-membered glucose ring by CrCl3− to form fructose before further conversion to HMF (see Scheme 5). Other studies are focused on the development of biphasic reaction systems and mild reaction conditions to increase viability for industrialization.
 | ||
| Scheme 5 | ||
Cost-effective synthesis methods need to be developed to implement large-scale production of HMF. Biphasic continuous systems have more potential for industrial applications.45,47,53 The imidazolium ionic liquid-metal catalyst system has excellent stability and selectivity in HMF conversion from carbohydrates. However, it is limited to batch reaction protocols due to the incompatibility of the high reaction temperatures (80–120 °C) and the commonly reported extraction method using diethyl ether, a low boiling point solvent.50–52 Our group reported an imidazolium chloride-tungsten salt catalyst system that can effectively convert fructose to HMF at a much lower temperature (room temperature to 50 °C).54 A [BMIM+]Cl−-tetrahydrofuran (THF) biphasic system was also developed with the tungsten salt catalyst; this system offers a feasible large-scale continuous HMF production protocol under lower temperatures and ambient pressure. There are many advantages to this system; first, the lower reaction temperature produces fewer by-products, which are a major cause of system contamination; second, it allows lower boiling point solvents to be used as the mobile-extraction phase for biphasic systems to facilitate HMF product recovery by solvent distillation; third, the mild conditions lower the energy consumption and give rise to a longer system lifetime, which is important for developing a sustainable biomass conversion system. It was demonstrated that the continuous biphasic system steadily produced high yields of HMF from fructose (about 80% yield) over multiple runs (see Scheme 6). The IL and tungsten catalyst remained active for long reaction periods. This could be due to the continual extraction of both HMF and water by the THF mobile-extraction phase to push the dehydration reaction forward and suppress by-product generation. The mild reaction conditions could be another factor for the long lifetime of the system.
![Continuous process for fructose conversion to HMF at low temperature in the THF-[BMIM]Cl biphasic system with tungsten catalyst.](/image/article/2010/EE/b914206a/b914206a-s6.gif) | ||
| Scheme 6 Continuous process for fructose conversion to HMF at low temperature in the THF-[BMIM]Cl biphasic system with tungsten catalyst. | ||
In Scheme 5, it was proposed that the CrCl3− anion acted as the catalyst during glucose/fructose isomerisation and HMF production. However, in the CrCl2/IMS system, a NHC–Cr complex could be formed under the reaction conditions used and could then act as a catalyst.55 In our group, we have pre-synthesized N-heterocyclic carbene (NHC)-metal complexes as catalysts for the sugar dehydration reaction (Scheme 7).56 The NHC ligands offer a great deal of flexibility towards modifying the catalytic activity by varying the stereo and electronic properties of NHCs. The conversions of fructose and glucose were tested over 1-butyl-3-methyl imidazolium chloride ([BMIM+]Cl−) with different catalysts. The new NHC–Cr/ionic liquid system achieved excellent selectivity and efficiency. The HMF yields with Ipr-CrCl2 catalyst were as high as 96% and 81% for fructose and glucose, respectively. The new system also allowed for ease of recycling of the catalyst/ionic liquid phase, provided a sole HMF product with simple extraction, and was tolerant towards high substrate loading. These results suggest that NHC-CrClx complexes play a key role in glucose dehydration in [BMIM+]Cl−. Bulky NHC ligands prevent chromium from forming multiple NHC coordinations in [BMIM+]Cl− and give better activity profiles.
 | ||
| Scheme 7 | ||
Polymeric imidazolium salts (PIMS) and polymeric NHCs (PNHC) were also used in this study.57 Stable main-chain poly-imidazolium salt (PIMS) particles were synthesized from 1,4-bis(imidazol-1-ylmethyl)benzene and 2,4,6-tris(bromomethyl)mesitylene at controlled monomer ratios and suitable reaction conditions.26 These materials were further converted to stable poly-N-heterocyclic carbene (PNHC) particles and poly-N-heterocyclic carbene chromium particles (PNHC-CrCl2) (see Scheme 8). In the mixture of PIMS and [BMIM+]Cl−, fructose could be converted to HMF in 60–65% yield at 80–100 °C. In pure [BMIM+]Cl−, the HMF yield is below 40% under the same reaction conditions. In the microstructure of PIMS, significant amounts of terminal imidazole groups exist. The equilibrium between imidazolium and terminal imidazole could generate more acidic terminal imidazole-HBr that promote fast fructose dehydration (Scheme 8). Based on this assumption, acidified PIMS-HCl was synthesized by dipping PIMS in dilute acid solution (HCl) and followed by washing and drying (see Scheme 8). As expected, the acidified PIMS-HCl/[BMIM+]Cl− system gave better HMF yields (79–84% at 80–100 °C). The whole system can be reused at least 11 times without any deactivation. PIMS or PIMS-HCl showed little activity toward glucose conversion. However, poly-heterocyclic carbene supported chromium catalyst (PNHC-CrCl2) demonstrated reasonable activity in glucose feedstock. It gave 66% of HMF yield in 6 h at under 100 °C in a [BMIM+]Cl− system.
 | ||
| Scheme 8 The synthesis of main-chain poly-IMSs and poly-NHC-CrCl2 catalysts. | ||
An acidic IL, 1-ethyl-3-methylimidazolium hydrogen sulfate ([EMIM][HSO4]), was developed for the one-pot hydrolysis of mono/di/polysaccharides into furfurals. [EMIM][HSO4] is effective in converting xylose and fructose or related polysaccharins into furural (e.g. from xylose: 84% yield in 6 h) or HMF (e.g. from fructose: 88% yield in 30 min), but not glucose and polymers containing these units. The imidazolium ionic liquid could be recovered and reused without significant activity drop.58
3. Imidazolium ionic liquids for CO2 capture
IMSs for CO2 absorption
Carbon dioxide (CO2) is a non-toxic, non-combustible and non-flammable gas. It is the foremost component among greenhouse gases in terms of annual anthropogenic generation and total amount present in the atmosphere. The capture, storage and utilization of carbon dioxide are some of the most critical challenges towards creating a sustainable society. Selective physisorption of CO2 on porous inorganic and organic adsorbents like zeolites, silica gels, aluminas, metal–organic frameworks (MOFs) and activated carbons has been intensively studied.59,60 Ionic liquids, which exhibit higher CO2 solubility than conventional organic solvents, have attracted considerable attention as new non-volatile and renewable CO2 absorbents.61 In imidazolium ionic liquid systems, CO2 is absorbed by occupying the free space between the ions through physical absorption mechanisms.62 Molecular dynamics simulations suggest that the cations and anions of imidazolium salts are arranged to form a relatively rigid network. On addition of CO2 into IMSs, the ionic rearrangement produces a large amount of free volume available to accommodate CO2 molecules.63 The CO2 solubility of the IMSs is thus dependent on their cations, anions, and substituents, with the anions playing a major role. The effect of the anions on the CO2 solubility was studied by Brennecke et al. for several common IMSs consisting of a [BMIM]+ cation and different anions.64 It was found that the solubility dependency of CO2 in the IMSs are: NO3− < DCA− < BF4− = PF6− < TfO− < Tf2N− < Tf3C−, where TfO−, Tf2N− and Tf3C− contain one, two, and three CF3 groups, respectively (Scheme 9). | ||
| Scheme 9 Structures of anions. | ||
Although various IMSs with different anions, cations and substitutions have been studied for CO2 absorption, the low CO2 absorption capacity is still the major hurdle for practical applications.61 The solubility of CO2 in conventional IMSs is up to only 3.5 mol% (0.5 wt%) at ambient temperature and pressure. Other efforts have also been tested for improving CO2 absorption capacity, such as impregnating IMSs into porous materials for developing supported ionic liquid membranes and immobilizing IMSs onto polymer chains.65 However, no significant improvement for CO2 absorption capacity has been achieved.
Due to the limitations in absorption capacity for IMSs, several research groups have initiated a new concept with the incorporation of functional groups to allow enhanced (chemical) binding of CO2 to the absorbent. Davis’ group was one of the first to combine imidazoliums with amine groups.66 These ILs consist of an imidazolium-based cation that is tethered to an amine group through a (variable) alkyl chain (Scheme 10). The amine functionalized ILs show a molar up-take of CO2 of nearly 0.5 at 0.1 MPa over a period of 3 h. This is significantly increased compared to [HMIM][PF6]. One of the disadvantages of this material is the relatively high viscosity compared with other IL solutions.67 Sanchez et al.68 verified this concept by measuring the CO2 absorption of several functionalized [BMIM]+ cations carrying (i) a primary amine group, (ii) a tertiary amine group, or (iii) a hydroxyl group. The functionalized derivatives display a sharp increase in CO2 absorption up to 0.1 MPa, followed by a steady increase between 0.1 and 1 MPa, exhibiting a typical chemical absorption profile. The material containing a tertiary amine function displays a far lower, but still sharp, increase. This observation demonstrates that the reactivity of higher substituted amines is reduced compared with simple primary amines.69 Beyond 0.1 MPa, the absorption capacity of all of the amine-functionalized ILs still increases steadily after the involved amine functions reach saturated absorption. This is attributed to the physical absorption mechanism.69 These ILs can be regenerated by heating under vacuum at 80 °C.
![Carbamate formation between CO2 and [aminopropylmim][BF4].69f](/image/article/2010/EE/b914206a/b914206a-s10.gif)
Compared with amine/ammonia systems, IMSs have certain advantages as CO2 absorbents. In general, IMSs are thermally and chemically stable; they are non-volatile and non-flammable, and less energy is required to remove the absorbed CO2 from IMSs in the regeneration step. The major drawback for IMSs is their low CO2 absorption capacity and that is mostly related to their high viscosity. Novel solid IMS materials with porous structures could be a way to solve this problem.
4. Conversion of CO2 using imidazolium ionic liquids
The coupling of epoxide with CO2 to form carbonates or polycarbonates is one of the most promising technologies in the utilization of CO2 (Scheme 11).70 Many types of catalysts have been intensively studied for this kind of reaction; however there are some disadvantages, such as low catalytic activity, severe reaction conditions, difficult recycling of the catalysts and so on.70 Recently, IMSs have been investigated as catalysts or promoters for this kind of reaction. It was reported that metal catalysts combined with imidazolium salts show significant advantages over the conventional catalysts, such as high catalytic efficiency (turn over frequency, TOF), mild reaction conditions, non-toxic reagents and recycling of the IL catalysts, although the cycloaddition between propylene oxide and CO2 could not be effectively catalyzed by solely using IMSs.71 The catalytic system comprised of zinc chloride (ZnCl2) and 1-butyl-3-methylimidazolium bromide ([BMIM]Br) achieved 95% yield, >98% selectivity and 5410 h−1 TOF under mild reaction conditions without any cosolvents.72 The reaction mechanism was proposed in (Scheme 12). ZnCl2 and [BMIM]Br react to generate the zinc imidazolium complex (1), which coordinates the epoxide. At the same time, nucleophilic attack of the dissociated [BMIM]Br on the less sterically hindered carbon atom of the coordinated epoxide occurs (2) to form the active species (3). The insertion of CO2 into the Zn–O bond of (3) would give a zinc carbonate active species (4), which eventually forms the cyclic carbonate. ILs can be used in the synthesis of alkylidene carbonates from CO2 and propargyl alcohols by combining with metal catalysts (for example, the synthesis of methylene cyclic carbonate catalyzed by CuCl with [BMIM]PhSO3).73 Imidazolium ionic liquids have also been applied as solvents or promoters in the conversion of CO2 with amines to synthesize symmetric urea derivatives74 and to hydrogenate CO2 to form formic acid.75 | ||
| Scheme 11 | ||
![Proposed mechanism for the reaction of epoxides and CO2 catalyzed by ZnX2/[BMIM]Br.69f](/image/article/2010/EE/b914206a/b914206a-s12.gif) | ||
| Scheme 12 Proposed mechanism for the reaction of epoxides and CO2 catalyzed by ZnX2/[BMIM]Br.69f | ||
5. N-heterocyclic carbenes (NHCs) in CO2 utilization
Carbon dioxide is an attractive C1 building block in organic synthesis, as it is a highly functional, abundant, renewable carbon source and an environmentally friendly chemical reagent.70 Significant efforts have been devoted towards exploring technologies for CO2 transformation, whereby metal catalysts played a key role.76 NHCs have been well established as organo-catalysts in organic synthesis.27–30 NHCs behave as nucleophiles due to the presence of a lone electron pair. Recently, NHCs have demonstrated great potential in CO2 utilization research. Rogers et al. first reported the synthesis of carbene-CO2 adducts through a reaction between dimethyl carbonate (DMC) and 1-methylimidazole at 120 °C (Scheme 13).77 Following that, Louie et al. reported the process of reversible carboxylation of N-heterocyclic carbenes, demonstrating that NHCs can activate CO2 to form zwitterionic products (Scheme 14).78 The reversibility of NHC-CO2 indicates the potential application of NHCs as a CO2 carrier. Initially, the novel NHC-CO2 adducts were only used in transcarboxylation reactions in stoichiometric ratios. They have been used in the synthesis of carboxylates, cyclic carbonates, and halogen-free ionic liquids from CO2. NHC-CO2 adducts have also been used as NHC ligand transferring reagents for the synthesis of different organometallic catalysts.79 However, the most attractive application for NHC-CO2 could be using NHCs as catalysts to fix CO2 and/or transfer CO2 to other chemicals. The release of CO2 from the zwitterionic salts and the closing of a catalytic cycle with NHCs may supply a new and exciting metal-free protocol for CO2 utilization. In principle, the NHC-CO2 adducts could be utilized as a conventional CO2 carrier to accomplish CO2 fixation through this nucleophilic incorporation. | ||
| Scheme 13 | ||
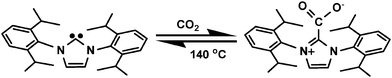 | ||
| Scheme 14 | ||
Lu and co-workers reported 1,3-bis(2,6-diisopropylphenyl)imidazoli(ni)um-2-carboxylate (IPr-CO2 (SIPr-CO2)) catalyzing the coupling of CO2 with epoxides under 2 MPa CO2 atmosphere and 120 °C.80 Under suppressed CO2 atmosphere, NHC-CO2 adducts are thermally stable and are assumed to be the catalyst in epoxide ring opening and the following CO2 attack. Ikariya and co-workers reported an interesting carboxylative cyclization reaction of propargylic alcohols catalyzed by NHCs or NHC-CO2.81 The reaction was conducted under 4.5–10 MPa CO2 at 60–100 °C. This reaction demonstrated that NHCs and NHC-CO2 adducts serve as potent organocatalysts, providing straightforward methods for carbonate synthesis. The cyclic carbonate formations from propargylic alcohol using NHC-CO2 adduct was explained with a mechanism involving the nucleophilic addition of the imidazolium-2-carboxylate to the C≡C bond and subsequent intramolecular cyclization of the alkoxide intermediate (Scheme 15).
 | ||
| Scheme 15 Suggested reaction pathway for carboxylations catalyzed by NHCs. | ||
During the studies by our group on CO2 fixation to produce highly valuable chemicals or fuels, we developed the first organocatalyst catalyzed CO2 reduction reaction: the catalytic reduction of CO2 to methanol by NHCs with hydrosilane as reductant and hydrogen source (Scheme 16).82 Remarkably, the reaction was conducted at room temperature and ambient pressure conditions and proceeded smoothly even with dry air as CO2 feedstock. The reaction was not appreciably poisoned by the presence of O2. Compared to transition metal catalysts in CO2 hydrosilylation reactions,83 NHCs present superior efficiencies and allow the use of far milder and more flexible reaction conditions. This approach offers a very promising chemical CO2 activation and fixation protocol. This procedure demonstrated a novel possibility for converting CO2 to fuel under mild conditions. However, industrial applications may be limited due to the expensive hydrosilane reagent.84
 | ||
| Scheme 16 Converting CO2 to methanol with hydrosilane and catalyzed by NHCs. | ||
6. Conclusion
The use of imidazoliums and their derivatives in sustainable research has not been limited to biomass conversion and CO2 capture/utilization. Work using IMSs/NHCs in green synthesis, separation, and green materials has increased steadily over the past few years. With their unique properties, IMSs have demonstrated great potential in non-acidic, less energy-intensive biomass conversion processes. IMSs are particularly useful for directly processing raw biomass, such as cellulose, as feedstock to produce HMF under mild conditions. Imidazolium ionic liquids have been studied as a green CO2 absorbent. However, this has been limited by their low absorption capacities. Imidazolium salts are also a very promising solvent and may serve as catalyst or promoter in many CO2 conversion reactions. CO2 utilization typically involves metal catalysts and severe reaction conditions. However, NHCs or NHC-CO2 adducts, as powerful organocatalysts, provide a promising new way to catalytically utilize CO2 in very mild conditions.Acknowledgements
This work was supported by the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore).References
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- A. E. Visser, R. P. Swatloski and R. D. Rogers, Green Chem., 2000, 2, 1 RSC.
- A.-L. Rollet, P. Porion, M. Vaultier, I. Billard, M. Deschamps, C. Bessada and L. Jouvensal, J. Phys. Chem. B, 2007, 111, 11888 CrossRef CAS.
- A. Mele, C. D. Tran and S. H. De Paoli Lacerda, Angew. Chem., Int. Ed., 2003, 42, 4364 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbery and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974 CrossRef CAS.
- D. A. Fort, R. C. Remsing, R. P. Swatloski, P. Moyna, G. Moyna and R. D. Rogers, Green Chem., 2007, 9, 63 RSC.
- O. A. El Seoud, A. Koschella, L. C. Fidale, S. Dorn and T. Heinze, Biomacromolecules, 2007, 8, 2629 CrossRef CAS.
- (a) P. Sledz, M. Mauduit and K. Grela, Chem. Soc. Rev., 2008, 37, 2433 RSC; (b) J. H. Davis, Jr, Chem. Lett., 2004, 33, 1072 CrossRef.
- (a) P. Wasserscheid, T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2003 Search PubMed; (b) J. Dupont, R. F. De Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS.
- (a) Z. Zhang, Y. Xie, W. Li, S. Hu, J. Song, T. Jiang and B. Han, Angew. Chem., Int. Ed., 2008, 47, 1127 CrossRef CAS; (b) X. Han and D. W. Armstrong, Acc. Chem. Res., 2007, 40, 1079 CrossRef CAS.
- J. E. Bara, T. K. Carlisle, C. J. Gabriel, D. Camper, A. Finotello, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2009, 48, 2739 CrossRef CAS.
- S.-G. Lee, Chem. Commun., 2006, 1049 RSC.
- J. S. Lee, X. Wang, H. Luo, G. A. Baker and S. Dai, J. Am. Chem. Soc., 2009, 131, 4596 CrossRef CAS.
- R. E. Morris, Chem. Commun., 2009, 2990 RSC.
- R. Tatumi and H. Fujihara, Chem. Commun., 2005, 83 RSC.
- A. J. Boydston, C. S. Pecinovsky, S. T. Chao and C. W. Bielawski, J. Am. Chem. Soc., 2007, 129, 14550 CrossRef CAS.
- P. H. J. Kouwer and T. M. Swager, J. Am. Chem. Soc., 2007, 129, 14042 CrossRef CAS.
- S. Luo, X. Mi, L. Zhang, S. Liu, H. Xu and J. Cheng, Angew. Chem., Int. Ed., 2006, 45, 3093 CrossRef CAS.
- (a) Z. Fei, T. J. Geldbach, D. Zhao and P. J. Dyson, Chem.–Eur. J., 2006, 12, 2122 CrossRef CAS; (b) Y. Zhang, L. Zhao, P. K. Patra and J. Y. Ying, Adv. Synth. Catal., 2008, 350, 662 CrossRef CAS.
- J. R. Harjani, J. Farrell, M. T. Garcia, R. D. Singer and P. J. Scammells, Green Chem., 2009, 11, 821 RSC.
- (a) L. Zhao, C. Zhang, L. Zhuo, Y. Zhang and J. Y. Ying, J. Am. Chem. Soc., 2008, 130, 12586 CrossRef CAS; (b) C. Zhang, Z. Ding, N. M. Suhaimi, Y. Kng, Y. Zhang and L. Zhuo, Free Radical Res., 2009, 43, 899 CrossRef CAS.
- W. Zheng and L. He, J. Am. Chem. Soc., 2009, 131, 3432 CrossRef CAS.
- V. W. Yam, J. K. Lee, C. Ko and N. Zhu, J. Am. Chem. Soc., 2009, 131, 912 CrossRef CAS.
- (a) J. S. Kingsbury and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 127, 4510 CrossRef CAS; (b) M. Mayr, B. Mayr and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2001, 40, 3839 CrossRef CAS.
- (a) P. D. Stevens, J. D. Fan, H. M. R. Gardimalla, M. Yen and Y. Gao, Org. Lett., 2005, 7, 2085 CrossRef CAS; (b) C.-G. Dong and Q.-S. Hu, J. Am. Chem. Soc., 2005, 127, 10006 CrossRef CAS.
- (a) Y. Zhang, L. Zhao, P. K. Patra, D. Hu and J. Y. Ying, Nano Today, 2009, 4, 13 CrossRef; (b) M. X. Tan, Y. Zhang and J. Y. Ying, Adv. Synth. Catal., 2009, 351, 1390 CrossRef CAS.
- For recent reviews on NHC organo-catalysis, see: (a) N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS; (b) N-Heterocyclic Carbenes in Synthesis; ed. S. P. Nolan, Wiley-VCH, Weinheim, 2006 Search PubMed; (c) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS.
- For recent reviews on NHC-ligated organometallic catalysis, see: (a) D. Bourissou, O. Guerret, F. P. Gabbar and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef; (b) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (c) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef CAS; (d) V. Cesar, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (e) F. E. Hahn, Angew. Chem., Int. Ed., 2006, 45, 1348 CrossRef CAS; (f) S. Diez-Gonzalez and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874 CrossRef CAS.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS.
- (a) J. Seayad, P. K. Patra, Y. Zhang and J. Y. Ying, Org. Lett., 2008, 10, 953 CrossRef CAS; (b) F. T. Wong, P. K. Patra, J. Seayad, Y. Zhang and J. Y. Ying, Org. Lett., 2008, 10, 2333 CrossRef CAS; (c) Y. Zhang, K. Ngeow and J. Y. Ying, Org. Lett., 2007, 9, 3495 CrossRef CAS.
- (a) B. Jastorff, R. Stoermann, J. Ranke, K. Moelter, F. Stock, B. Oberheitmann, W. Hoffmann, J. Hoffmann, M. Nuechter, B. Ondruschka and J. Filser, Green Chem., 2003, 5, 136 RSC; (b) J. Ranke, S. Stolte, R. Stoermann, J. Arning and B. Jastorff, Chem. Rev., 2007, 107, 2183 CrossRef CAS; (c) D. Kralisch, D. Reinhardt and G. Kreisel, Green Chem., 2007, 9, 1308 RSC.
- L. R. Lynd, P. J. Weimer, W. H. Van Zyl and I. S. Pretorius, Microbiol. Mol. Biol. Rev., 2002, 66, 506 CrossRef CAS.
- C. E. Wyman, B. E. Dale, R. T. Elander, M. Holtzapple, M. R. Ladisch and Y. Y. Lee, Bioresour. Technol., 2005, 96, 1959 CrossRef CAS.
- R. C. Remsing, R. P. Swatloski, R. D. Rogers and G. Moyna, Chem. Commun., 2006, 1271 RSC.
- (a) D. M. Phillips, L. F. Drummy, D. G. Conrady, D. M. Fox, R. R. Naik, M. O. Stone, C. Trulove, H. C. De Long and R. A. Mantz, J. Am. Chem. Soc., 2004, 126, 14350 CrossRef CAS; (b) H. Xie, S. Li and S. Zhang, Green Chem., 2005, 7, 606 RSC; (c) M. Zavrel, D. Bross, M. Funke, J. Buchs and A. C. Spiess, Bioresour. Technol., 2009, 100, 2580 CrossRef CAS.
- H. Xie, A. King, I. Kilpelainen, M. Granstrom and D. S. Argyropoulos, Biomacromolecules, 2007, 8, 3740 CrossRef CAS.
- (a) C. Li, Q. Wang and Z. K. Zhao, Green Chem., 2008, 10, 177 RSC; (b) L. Vanoye, M. Fanselow, J. D. Holbery, M. P. Atkins and K. R. Seddon, Green Chem., 2009, 11, 390 RSC.
- (a) H. Zhao, G. A. Baker, Z. Song, O. Olubajo, T. Crittle and D. Peters, Green Chem., 2008, 10, 696 RSC; (b) H. Zhao, C. L. Jones, G. A. Baker, S. Xia, O. Olubajo and D. Peters, J. Biotechnol., 2009, 139, 47 CrossRef CAS.
- C. Li and Z. K. Zhao, Adv. Synth. Catal., 2007, 349, 1847 CrossRef CAS.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047 CrossRef CAS.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS.
- Y. Su, H. M. Brown, X. Huang, X. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117 CrossRef CAS.
- M. X. Tan, L. Zhao and Y. Zhang, manuscript in preparation.
- G. N. Sheldrake and D. Schleck, Green Chem., 2007, 9, 1044 RSC.
- (a) Special issue on Sustainability and Energy, Science 2007, 315, 781 Search PubMed; (b) A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef; (c) M. Parikka, Biomass Bioenergy, 2004, 27, 613 CrossRef; (d) D. L. Klass, in Encyclopedia of Energy, ed. C. J. Cleveland, Elsevier, Dordrecht, 2004, vol. 1 Search PubMed; (e) L. R. Lynd, J. H. Cushman, R. J. Nichols and C. E. Wyman, Science, 1991, 251, 1318 CrossRef CAS; (f) Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982 CrossRef CAS.
- For recent reviews, see: (a) J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS; (b) G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184 CrossRef CAS; (c) B. Kamm, Angew. Chem., Int. Ed., 2007, 46, 5056 CrossRef CAS; (d) J. O. Metzger, Angew. Chem., Int. Ed., 2006, 45, 696 CrossRef CAS; (e) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS; (f) J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123, 59 CrossRef CAS; (g) M. Bicker, J. Hirth and H. Vogel, Green Chem., 2003, 5, 280 RSC; (h) A. Gandini and M. N. Belgacem, Prog. Polym. Sci., 1997, 22, 1203 CrossRef CAS; (i) C. Moreau, M. N. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11 CrossRef CAS.
- (a) Y. Roman-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933 CrossRef CAS; (b) J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342 RSC; (c) G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446 CrossRef CAS; (d) B. M. F. Kuster, Starch/Staerke, 1990, 42, 314 Search PubMed; (e) C. Moreau, R. Durand, S. Razigade, J. Duhamet, P. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211 CrossRef CAS; (f) X. Qian, M. R. Nimlos, M. Davis, D. K. Johnson and M. E. Himmel, Carbohydr. Res., 2005, 340, 2319 CrossRef CAS; (g) M. J. Antal, Jr., T. Leesomboon, W. S. Mok and G. N. Richards, Carbohydr. Res., 1991, 217, 71 CrossRef; (h) D. Mercadier, L. Rigal, A. Gaset and J. P. Gorrichon, J. Chem. Technol. Biotechnol., 1981, 31, 489 Search PubMed; (i) H. H. Szmant and D. D. Chundury, J. Chem. Technol. Biotechnol., 1981, 31, 135 Search PubMed.
- (a) F. W. Lichtenthaler, Acc. Chem. Res., 2002, 35, 728 CrossRef CAS; (b) T. Werpy, G. Petersen, Top Value Added Chemicals from Biomass: Vol. 1 – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Report No. NREL/TP-510–35523, National Renewable Energy Laboratory, Golden, CO, 2004 Search PubMed.
- H. E. van Dam, A. P. G. Kieboom and H. van Bekkum, Starch/Staerke, 1986, 38, 95 Search PubMed.
- C. Lansalot-Matras and C. Moreau, Catal. Commun., 2003, 4, 517 CrossRef CAS.
- C. Moreau, A. Finiels and L. Vanoye, J. Mol. Catal. A: Chem., 2006, 253, 165 CrossRef CAS.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597 CrossRef CAS.
- (a) R. M. West, Z. Y. Liu, M. Peter, C. A. Gartner and J. A. Dumesic, J. Mol. Catal. A: Chem., 2008, 296, 18 CrossRef CAS; (b) M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924 CrossRef CAS; (c) R. M. Musau and R. M. Munavu, Biomass, 1987, 13, 67 CrossRef CAS; (d) F. Salak Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2006, 45, 2163 CrossRef CAS; (e) D. W. Brown, A. J. Floyd, R. G. Kinsman and Y. Roshan-Ali, J. Chem. Technol. Biotechnol., 1982, 32, 920 Search PubMed; (f) K. Seri, Y. Inoue and H. Ishida, Bull. Chem. Soc. Jpn., 2001, 74, 1145 CrossRef CAS; (g) S. Hu, Z. Zhang, Y. Zhou, B. Han, H. Fan, W. Li, J. Song and Y. Xie, Green Chem., 2008, 10, 1280 RSC.
- G. J. Y. Chan and Y. Zhang, ChemSusChem, 2009, 2, 731 CrossRef.
- T. Weskamp, V. P. W. Bohm and W. A. Herrmann, J. Organomet. Chem., 2000, 600, 12 CrossRef CAS.
- G. Yong, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2008, 47, 9345 CrossRef CAS.
- Y. Zhang, unpublished result.
- S. Lima, P. Neves, M. M. Antunes, M. Pillinger, N. Lgnatyev and A. A. Valente, Appl. Catal., A, 2009, 363, 93 CrossRef CAS.
- (a) F. M. Orr, Jr., Energy Environ. Sci., 2009, 2, 449 RSC; (b) S. Sircar, Adsorption by Carbons, ed. E. Bottani, J. Tascon, Elsevier, New York, 2008 Search PubMed; (c) G. E. Keller, R. A. Anderson, C. M. Yon, Handbook of Separation Process Technology, ed. R. W. Rousseau, Wiley Interscience, New York, 1987 Search PubMed.
- (a) J. D. Seader, E. J. Henley, Separation Process Principles, John Wiley & Sons, Toronto, ON, 1998 Search PubMed; (b) R. F. Service, Science, 2004, 305, 963 CrossRef CAS; (c) X. Ma, X. Wang and C. Song, J. Am. Chem. Soc., 2009, 131, 5777 CrossRef CAS; (d) C. Chen, S. Yang, W. Ahn and R. Ryoo, Chem. Commun., 2009, 3627 RSC.
- J. Huang and T. Ruther, Aust. J. Chem., 2009, 62, 298 CrossRef CAS and the references therein.
- J. Jacquemin, P. Husson, V. Mayer and I. Cibulka, J. Chem. Eng. Data, 2007, 52, 2204 CrossRef CAS.
- X. Huang, C. J. Margulis, Y. Li and B. J. Berne, J. Am. Chem. Soc., 2005, 127, 17842 CrossRef CAS.
- S. N. V. K. Aki, B. R. Mellein, E. M. Saurer and J. F. Brennecke, J. Phys. Chem. B, 2004, 108, 20355 CrossRef CAS.
- (a) P. Scovazzo, J. Kieft, D. A. Finan, C. Koval, D. L. DuBois and R. D. Noble, J. Membr. Sci., 2004, 238, 57 CrossRef CAS; (b) J. Tang, H. Tang, W. Sun, H. Plancher, M. Radosz and Y. Shen, Chem. Commun., 2005, 3325 RSC; (c) J. Tang, H. Tang, W. Sun, M. Radosz and Y. Shen, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5477 CrossRef.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. J. Davis, J. Am. Chem. Soc., 2002, 124, 926 CrossRef CAS.
- X. Li, M. Hou, Z. Zhang, B. Han, G. Yang, X. Wang and L. Zou, Green Chem., 2008, 10, 879 RSC.
- L. M. Galán Sánchez, G. W. Meindersma and A. B. Haan, Chem. Eng. Res. Des., 2007, 85, 31 CrossRef.
- (a) A. Chakma, Energy Convers. Manage., 1997, 38, S51 CrossRef CAS; (b) M. D. Soutullo, C. I. Odom, B. F. Wicker, C. N. Henderson, A. C. Stenson and J. H. J. Davis, Chem. Mater., 2007, 19, 3581 CrossRef CAS; (c) Y.-Y. Jiang, G.-N. Wang, Z. Zhou, Y.-T. Wu, J. Geng and Z.-B. Z. Zhang, Chem. Commun., 2008, 505 RSC; (d) T. Yamada, P. J. Lukac, T. Yu and R. G. Weiss, Chem. Mater., 2007, 19, 4761 CrossRef CAS; (e) G. Song, Y. Cai and Y. Peng, J. Comb. Chem., 2005, 7, 561 CrossRef CAS; (f) S. Zhang, Y. Chen, F. W. Li, X. Lu, W. Dai and R. Mori, Catal. Today, 2006, 115, 61 CrossRef CAS.
- (a) H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953 CrossRef CAS; (b) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS; (c) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS; (d) C. M. Rayner, Org. Process Res. Dev., 2007, 11, 121 Search PubMed; (e) G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487 CrossRef CAS.
- (a) J. J. Peng and Y. Q. Deng, New J. Chem., 2001, 25, 639 RSC; (b) M. Alvaro, C. Baleizao, D. Das, E. Carbonell and H. García, J. Catal., 2004, 228, 254 CrossRef CAS; (c) M. Alvaro, C. Baleizao, E. Carbonell, M. El Ghoul, H. García and B. Gigante, Tetrahedron, 2005, 61, 12131 CrossRef CAS.
- F. W. Li, L. F. Xiao, C. G. Xia and B. Hu, Tetrahedron Lett., 2004, 45, 8307 CrossRef CAS.
- Y. L. Gu, F. Shi and Y. Q. Deng, J. Org. Chem., 2004, 69, 391 CrossRef CAS.
- F. Shi, Y. Q. Deng, T. L. SiMa, J. J. Peng, Y. L. Gu and B. T. Qiao, Angew. Chem., Int. Ed., 2003, 42, 3257 CrossRef.
- Z. Zhang, Y. Xie, W. Li, S. Hu, J. Song, T. Jiang and B. Han, Angew. Chem., Int. Ed., 2008, 47, 1127 CrossRef CAS.
- (a) N. Eghbali and C.-J. Li, Green Chem., 2007, 9, 213 RSC; (b) P. G. Jessop, F. Joó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425 CrossRef CAS; (c) G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636 CrossRef CAS; (d) D. H. Gibson, Chem. Rev., 1996, 96, 2063 CrossRef CAS; (e) T. Aida and S. Inoue, Acc. Chem. Res., 1996, 29, 39 CrossRef CAS; (f) T. Ohishi, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 5792 CrossRef CAS.
- J. D. Holbrey, W. M. Reichert, I. Tkatchenko, E. Bouajila, O. Walter, I. Tommasi and R. D. Rogers, Chem. Commun., 2003, 28 RSC.
- H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 112 RSC.
- (a) J. D. Holbrey, W. M. Reichert, I. Tkatchenko, E. Bouajila, O. Walter, I. Tommasi and R. D. Rogers, Chem. Commun., 2003, 28 RSC; (b) A. Voutchkova, M. Feliz, E. Clot, E. Eisentein and R. H. Crabtree, J. Am. Chem. Soc., 2007, 129, 12834 CrossRef CAS; (c) M. Smiglak, J. D. Holbrey, S. T. Griffin, W. M. Reichert, R. P. Swatloski, A. R. Katrizky, H. Yang, D. Zhang, K. Kirichenko and R. D. Rogers, Green Chem., 2007, 9, 90 RSC; (d) I. Tommasi and F. Sorrentino, Tetrahedron Lett., 2005, 46, 2141 CrossRef CAS; I. Tommasi and F. Sorrentino, Tetrahedron Lett., 2006, 47, 6453 CrossRef CAS; I. Tommasi and F. Sorrentino, Tetrahedron Lett., 2009, 50, 104 CrossRef CAS; (e) A. Tudose, A. Demonceau and L. Delaude, J. Organomet. Chem., 2006, 691, 5356 CrossRef CAS.
- H. Zhou, W. Zhang, C. Liu, J. Qu and X. Lu, J. Org. Chem., 2008, 73, 8039 CrossRef CAS.
- Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 4194 CrossRef CAS.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322 CrossRef CAS.
- (a) H. Koinuma, F. Kawakami, H. Kato and H. Hirai, J. Chem. Soc., Chem. Commun., 1981, 213 RSC; (b) T. G. Eisenschmid and R. Eisenberg, Organometallics, 1989, 8, 1822 CrossRef CAS; (c) P. Deglmann, E. Ember, P. Hofman, S. Pitter and O. Walter, Chem.–Eur. J., 2007, 13, 2864 CrossRef CAS; (d) T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2006, 128, 12362 CrossRef CAS.
- S. K. Ritter, C & E News, 2009, 87, 39 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |