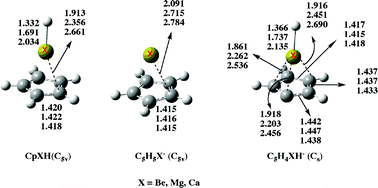
The structure and bonding of cyclopentadienylberyllium (CpBeH), magnesium (CpMgH), and calcium (CpCaH) hydrides as well as those of their deprotonated species have been investigated by means of B3LYP/6-311+G(3df,2p)//B3LYP/6-311+G(d,p) and B3LYP/6-311+G(3df,2p)//QCISD/6-311+G(d,p) density functional theory (DFT) calculations. The three compounds exhibit C5v equilibrium conformations in their ground states. For CpBeH the agreement between the calculated geometry and that determined by MW spectroscopy is excellent. CpMgH and CpCaH can be viewed almost as the result of the interaction between a C5H5− anion and a XH+ (X = Mg, Ca) cation. Conversely, for CpBeH the interaction between the C5H5 and the BeH subunits is significantly covalent. These compounds exhibit a significant aromaticity, usually named three-dimension aromaticity, in contrast with the unsubstituted cyclopentadiene compound. The CpBeH derivative behaves as a C acid in the gas phase and is less acidic than cyclopentadiene. More importantly, CpMgH and CpCaH, in spite of the X+δH−δ polarity exhibited by the X–H bond in the neutral systems, are predicted to be metal acids in the gas phase. Also surprisingly, both the Mg and the Ca derivatives are stronger acids than the Be analogue, and only slightly weaker acids than cyclopentadiene. This somewhat unexpected result is the consequence of two concomitant facts: the lower dissociation energy of the X–H (X = Mg, Ca) bonds with respect to the C–H bonds, and the significantly high electron affinity of the C5H5X˙ (X = Mg, Ca) radicals.

 Please wait while we load your content...
Please wait while we load your content...

