Cation and anion selectivity of zwitterionic salicylaldoxime metal salt extractants†
Ross S. Forgan, James E. Davidson, Francesca P. A. Fabbiani, Stuart G. Galbraith, David K. Henderson, Stephen A. Moggach, Simon Parsons, Peter A. Tasker* and Fraser J. White
School of Chemistry, University of Edinburgh, Edinburgh, UK EH9 3JJ. E-mail: peter.tasker@ed.ac.uk; Fax: (+44) 131 650 6453; Tel: (+44) 131 650 4706
First published on 7th January 2010
Abstract
3-Dialkylaminomethyl substituted salicylaldoximes are efficient metal salt extractants, and, in contrast to related “salen”-based reagents, are sufficiently stable to acid hydrolysis to allow commercial application in base metal recovery. Crystal structures show that metal salts are bound by a zwitterionic form of the reagents, with copper(II) nitrate, tetrafluoroborate and trifluoroacetate forming [Cu(L)2X2] assemblies in a tritopic arrangement with a trans-disposition of the anions outwith the coordination sphere. Copper(II) chloride, bromide and zinc(II) chloride form 1:1 assemblies, [Cu(L)X2], with the halides in the inner coordination sphere of the metal, leading to high chloride selectivity and very good mass transport efficiencies of CuCl2. Introduction of the anion-binding sites into the salicylaldoxime extractants changes their cation selectivities; the ligands co-extract small amounts of FeIII along with CuII from mixed metal aqueous feed solutions, an issue which will need to be addressed prior to industrial application.
Introduction
Solvent extractants which are capable of transporting both a metal cation and its attendant anion(s) into a water-immiscible solvent could underpin the development of new flowsheets in extractive hydrometallurgy.1,2 Major recent developments in leaching technology have defined a need for extractants to process high tenor pregnant leach solutions, which also have high concentrations of either sulfate or chloride.3 Conventional phenolic oxime extractants (Fig. 1) are very effective in recovering copper from heap leaching operations which generate low tenor feeds, acting as cation exchange reagents in pH-dependent load/strip equilibria (1) and ca. 25% of the world's copper is now recovered in this way.| 2Lorg + Cu2+⇌ [Cu(L-H)2]org + 2H+ | (1) |
 | ||
| Fig. 1 Generic structure for the Acorga and LIX extractants. Compound 1 represents Acorga P50™, an industrially used reagent.7 | ||
The efficiency of these extractants is reduced when processing high tenor feeds as transfer of copper to the organic phase leads to the build-up of high concentrations of proton in the aqueous phase. This is particularly problematic with chloride streams because high concentrations of chloride enhance proton activity.4,5 Interstage neutralisation of the aqueous solution by addition of a base or introducing base directly into mixer settlers is not desirable because this complicates circuit operation and has an adverse effect on the overall materials balances.6
Transferring metal values to an organic phase using metal salt extractants is not associated with a change in proton concentrations in the aqueous phase (2).
| MX2 + 2Lorg⇌ [ML2X2]org | (2) |
The salen-based reagents shown in Fig. 2, which carry pendant dialkylaminomethyl groups, have been shown to achieve this by binding the metal cation and attendant anion(s) in separated sites in a zwitterionic form of the extractant.8-12 The zwitterionic nature of the loaded ligand makes it possible to strip the cation and anions separately by contacting with acidic or basic aqueous solutions.6
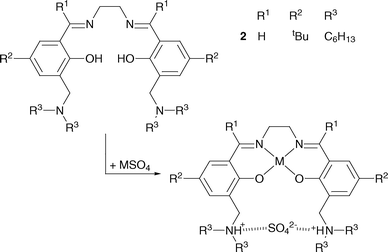 | ||
| Fig. 2 Loading of a metal(II) sulfate into the zwitterionic form of a “salen-type” metal salt extractant.6 | ||
Whilst these “salen-type” reagents meet many of the requirements for transporting metal salts, the imine bonds in these and similar reagents prove to be too sensitive to hydrolysis, particularly under the acidic conditions needed for metal stripping.13 Oxime derivatives are much more resistant to hydrolysis and have half lives of over 2.5 yrs in kerosenes in contact with acid solutions.7 Consequently we have investigated the use of the salicylaldoxime scaffold (see Fig. 3) in the development of metal salt extractants.
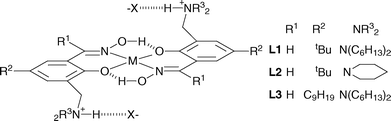 | ||
| Fig. 3 Formation of trans-complexes by the new 3-dialkylaminomethylsalicylaldoximes L1-L3 to give a tritopic binding motif for MX2 salts with potential for the metal cation to assist anion binding.14 | ||
5-tert-Butyl-2-hydroxy-3-dihexylaminomethylbenzaldehyde oxime (L1), 5-tert-butyl-2-hydroxy-3-piperidin-1-ylmethylbenzaldehyde oxime (L2) and 5-nonyl-2-hydroxy-3-dihexylaminomethylbenzaldehyde oxime (L3) have pendant dialkylaminomethyl arms, which are protonatable to form anion binding sites. As bis-salicylaldoximato complexes of CuII usually have a trans structure15 these sites will be well separated, forming a tritopic binding motif (Fig. 3) which may favour the transport of two monoanions. On these grounds it was postulated that they would be good candidates for transporting divalent metal chlorides, and in a preliminary report we have demonstrated that they can show remarkable transport efficiency in base metal recovery.16
Experimental
All solvents and reagents were used as received from Aldrich, Fisher and Acros. The syntheses of L1, L2, [Cu(L2)Cl2], [Cu(L2)2(NO3)2] and [Zn2(L2)2Cl4] have been described in a preliminary report.16L3 was prepared as exemplification for a patent relating to this work.17 The synthesis of (2) has been described previously.91H and 13C NMR were obtained using a Bruker AC250 spectrometer at ambient temperature. Chemical shifts (δ) are reported in parts per million (ppm) relative to internal standards. Fast atom bombardment mass spectrometry was carried out using a Kratos MS50TC spectrometer with a 3-nitrobenzyl alcohol (NOBA) matrix. Analytical data were obtained on a CE-440 Elemental Analyser by the University of Edinburgh Microanalytical Service and on a Carlo Erba CHNS analyser at the University of St Andrews. ICP-OES analysis was carried out using a Perkin Elmer Optima 5300DV spectrometer. The measurement of pH was carried out using a Fisher Scientific AR50 pH meter. X-Ray crystal structures were obtained by the University of Edinburgh Crystallography service. Chloride concentrations were measured with a chloride-selective Thermo Orion Ion Plus electrode.Synthesis
L2 was prepared as described previously.16 Crystals suitable for X-ray diffraction were grown by slow evaporation of a hexane/chloroform solution (for data see CSD code 774580). Crystal structure data: C17H26N2O2, Mr = 290.40, monoclinic, space group, C2/c, a = 20.1611(7), b = 8.5773(3), c = 20.6403(7) Å, β = 114.661(2) °, V = 3244.5(2) Å3, Z = 8, T = 150(2) K, 21572 reflections collected, 4287 independent reflections [R(int) = 0.064], R(F) = 0.0444, and wR2 = 0.1216.Hydrolytic stability
The hydrolytic stabilities of reagents were determined by stirring 0.01 M chloroform solutions (10 ml) with 0.8 M aqueous H2SO4/Na2SO4 solutions (10 ml) of varying pH for 16 h at room temperature. After phase disengagement, the pH of the aqueous phase was measured. The chloroform solutions were collected, dried with MgSO4 and then concentrated in vacuo. CDCl3 solutions of the residues were analysed by 1H NMR spectroscopy, and the integrals of the azomethine and aldehyde proton were compared to determine the extent of hydrolysis.Anion selectivity by solvent extraction
0.01 M [Cu(L1-H)2] in chloroform (10 ml) was added to a 0.8 M HX/NaX aqueous solution (10 ml) and stirred for 16 h at room temperature. After phase disengagement, the pH of the aqueous phase was measured, a 0.5 ml aliquot of the chloroform layer was taken for metal/sulfur analysis by ICP-OES and a 2.0 ml aliquot for chloride analysis. For chloride analysis, the aliquot was stirred with 10 ml of 0.1 M aqueous HNO3 overnight. After phase disengagement, a 5 ml aliquot of the aqueous phase was made up to 10 ml with 0.1 M NaOH and the chloride concentration was determined using a chloride selective electrode.Cation selectivity by solvent extraction
0.01 M chloroform solutions (5 ml) of L1 were contacted with aqueous solutions (5 ml) of the appropriate metal salt at concentrations of 0.01 M, prepared from 4 ml of 0.0125 M metal salt solution, to which was added 1 ml of 0.1 M sodium hydroxide/water or 1 ml of 2.5 M sulfuric acid/water solution to change pH. After vigorous stirring for 16 h at room temperature, the mixtures were separated and 0.5 ml aliquots of the organic phase removed for metal analysis by ICP-OES. The equilibrium pH of the aqueous phase was measured and plots of metal loading against equilibrium pH were used to determine the selectivity order. When the anion was to be in excess, the aqueous solution was prepared from 1 ml of 0.05 M metal salt solution, to which was added 4 ml of a mixture of 0.99 M HX and 0.99 M NaX, altering pH and keeping anion concentration constant at 0.8 M.Cation extraction from mixed metal feed solutions
0.01 M chloroform solutions (5 ml) of L1 were contacted with aqueous solutions (5 ml) of the appropriate metal salts at concentrations of 0.01 M. The aqueous solution was prepared from 1 ml of a solution containing each metal salt at a concentration of 0.05 M, to which was added 4 ml of a mixture of 0.99 M HX and 0.99 M NaX, altering pH and keeping anion concentration constant at 0.8 mol dm−3. In extraction from a mixed anion feed, the aqueous phase was prepared as described, but using 2 ml each of both a chloride and sulfate feed, to give 5 ml of an aqueous phase with metal concentrations of 0.01 M and sulfate and chloride concentrations of 0.4 M. After vigorous stirring for 16 h at room temperature, the mixtures were separated and 0.5 ml aliquots of the organic phase removed for metal analysis by ICP-OES. The equilibrium pH of the aqueous phase was measured and plots of metal loading against equilibrium pH were used to determine the selectivity order.Results and discussion
Ligand synthesis
The ligands were prepared in high yields on the gram scale by oximation of their salicylaldehyde precursors, whose syntheses have been described previously.8 The pendant dihexylaminomethyl arm of L1 imparts good solubility of both the ligand and its complexes in water-immiscible media, facilitating studies of the solvent extraction of base metal salts.Such studies can be undertaken with aqueous feed and strip solutions of low pH because the new oxime-containing L1-L3 are resistant to hydrolysis, significantly more so (see Fig. 4) than the prototype salen-based metal salt extractants, e.g., 2, which undergo greater than 95% hydrolysis to the parent aldehyde when chloroform solutions are contacted with aqueous sulfate solutions of pH < 2.5 for 16 h. By comparison, less than 10% hydrolysis is observed for the 5-t-butyl-substituted ligands L1 and L2 at the lowest pH and the most hydrophobic variant, L3, which carries a multibranched nonyl substituent, is almost as stable as the commercial copper extractant 5-nonylsalicylaldoxime (1).
 | ||
| Fig. 4 Percentage hydrolysis of the oxime (L1-L3 and 1) and di-imine (2) extractants as judged by the relative intensities of the oxime or azomethine 1H NMR signals (δ∼ 8) relative to the aldehyde (δ∼ 10) after contacting of 0.01 M chloroform solutions with aqueous sulfuric acid for 16 h. | ||
The pendant piperidinomethyl group of L2 ensures the crystallinity of the ligand and its metal salt complexes, allowing their study by X-ray diffraction. In the solid state, salicylaldoximes are known to form pseudomacrocyclic dimers or 1D chains via hydrogen bonding from the oxime OH to the phenol oxygen in a neighbouring molecule.15 The incorporation of a pendant amine arm provides an extra H-bond acceptor site in the neutral ligands, which leads to the formation of a different type of linear chain in the solid state structure of L2 based on oxime OH to piperidino nitrogen contacts (Fig. 5).
 | ||
| Fig. 5 Linear chains defined by oxime to pendant amine H-bonding (O2⋯N6′ = 2.723(2) Å) in the solid-state structure of L2. All H atoms not included in H-bonding have been removed for clarity. | ||
Selectivity of anion binding
The mechanism of extraction of copper and zinc salts was found to be very dependent on the nature of the anion. Halides (see below) are bound in the inner coordination sphere, whereas relatively weakly coordinating anions are present in the outer sphere and are hydrogen bonded to the protonated pendant amine groups. The isolation and X-ray structure determinations of the “copper-only” complex of L2, [Cu(L2-H)2], and a range of copper salt complexes, [Cu(L2)2X2], (X = NO3−, BF4− and CF3CO2−) allowed us to define structural changes which accompany uptake of anions and their binding motifs.Unusually for simple bis-salicylaldoximatocopper(II) complexes, the “copper-only” assembly [Cu(L2-H)2] is not centrosymmetric (see Fig. 6). The copper atom forms a weak contact with a phenolate oxygen atom in an adjacent complex, generating a centrosymmetric dimer which has an inversion centre at the centroid of the Cu2O2 bridging unit. The piperidine rings on each [Cu(L2-H)2] complex are displaced to the same side of the coordination plane, with N62A and N62B lying 0.605(2) and 1.340(2) Å above the least squares plane through the CuN2O2 moeity. The hydrogen bonding between the oxime OH and phenolate oxygen atom which generates the 14-membered pseudomacrocyclic structure is buttressed19 by interactions with the piperidino nitrogen atoms.
![The crystal structure of the [Cu(L2-H)2] dimer (Cu1⋯O1B′ = 2.516(1) Å) showing the typical 14-membered pseudomacrocycle15 defined by oxime to phenolate hydrogen bonds (O1A⋯O23B and O1B⋯O23A = 2.656(2) and 2.766(2) Å) and the buttressing18 of oxime to piperidino hydrogen bonds (O23B⋯N62A and O23A⋯N62B = 2.851(2) and 2.972(2) Å. All H atoms not involved in H-bonding have been removed for clarity.](/image/article/2010/DT/b916877j/b916877j-f6.gif) | ||
| Fig. 6 The crystal structure of the [Cu(L2-H)2] dimer (Cu1⋯O1B′ = 2.516(1) Å) showing the typical 14-membered pseudomacrocycle15 defined by oxime to phenolate hydrogen bonds (O1A⋯O23B and O1B⋯O23A = 2.656(2) and 2.766(2) Å) and the buttressing18 of oxime to piperidino hydrogen bonds (O23B⋯N62A and O23A⋯N62B = 2.851(2) and 2.972(2) Å. All H atoms not involved in H-bonding have been removed for clarity. | ||
The copper(II) nitrate, tetrafluoroborate and trifluoroacetate complexes (Fig. 7a, b and c, respectively) form tritopic assemblies of the type shown schematically in Fig. 3. The centrosymmetric nitrato and tetrafluoroborato assemblies are very similar, having planar CuN2O2 units and oxime OH to phenolate hydrogen bonds similar to those in the “copper-only” complex.
![Crystal structures showing the closest contacts of the anions (X) with the copper atom and the piperidinium NH groups in the tritopic assemblies a) [Cu(L2)2(NO3)2], X⋯Cu = 2.722(2), X⋯N6 = 2.842(2) Å; b) [Cu(L2)2(BF4)2], X⋯Cu = 2.638(2), X⋯N6 = 2.780(4) Å and c) [Cu(L2)2(CF3CO2)2], X⋯Cu = 2.673(5), X⋯N6 = 2.766(8) Å. All H atoms not involved in H-bonding have been removed for clarity.](/image/article/2010/DT/b916877j/b916877j-f7.gif) | ||
| Fig. 7 Crystal structures showing the closest contacts of the anions (X) with the copper atom and the piperidinium NH groups in the tritopic assemblies a) [Cu(L2)2(NO3)2], X⋯Cu = 2.722(2), X⋯N6 = 2.842(2) Å; b) [Cu(L2)2(BF4)2], X⋯Cu = 2.638(2), X⋯N6 = 2.780(4) Å and c) [Cu(L2)2(CF3CO2)2], X⋯Cu = 2.673(5), X⋯N6 = 2.766(8) Å. All H atoms not involved in H-bonding have been removed for clarity. | ||
In the trifluoroacetato complex the anion competes with the phenolate for the oximic hydrogen atom (Fig. 7c) and the N-O bonds of the oxime are displaced from the coordination plane; the oxime O atoms are located 0.369(4) Å above and below the least squares plane through the CuN2O2 coordination sphere. This results in a longer Cu-N bond (Cu-N = 1.975(5) Å) than the nitrato and tetrafluoroborato complexes (Cu-N = 1.946(1) and 1.943(1) Å, respectively).
The trifluoroacetate complex [Cu(L2)2(CF3CO2)2] was prepared by contacting a chloroform solution of the “copper- only” complex [Cu(L2-H)2] with an aqueous solution containing two molar equivalents of trifluoroacetic acid and an excess of its sodium salt. Monitoring the pH-dependence of anion uptake in this fashion, by titrating metal-only complexes with acid (3), is a convenient method to assess the relative strengths of anion binding.12
| [Cu(L-H)2]org + 2HX ⇌ [Cu(L)2X2]org | (3) |
The apparent basicity of the pendant amine groups in the metal-only complex is greater when the anion is more strongly bound, and thus the higher the pH0.5 (the pH at which 50% loading of the anion is observed) the stronger the anion-binding, and the selectivity of anion-binding is given by ΔpH0.5.
As the selectivity of chloride over sulfate uptake or vice versa is a very important issue in the commercial exploitation of new metal salt extractants, titrations with hydrochloric and sulfuric acid were carried out on chloroform solutions of [Cu(L1-H)2]. Fig. 8 shows that chloride is loaded at pH < 6 whereas for sulfate the pH has to be less than 4.5 for detectable uptake. The more favourable extraction of Cl− over HSO4−‡ or SO42− is consistent with the Hofmeister bias20-22 which predicts that charge-diffuse anions with less favourable free energies of hydration will be more readily transferred into low polarity solvents.
![Uptake of chloride or sulfate by a 0.01 M chloroform solution of [Cu(L1-H)2] after equilibration with 0.8 M aqueous solutions of NaCl or Na2SO4 of varied acidity. 100% loading corresponds to that expected for the formation of the tritopic assemblies [Cu(L1)2Cl2] and [Cu(L1)2SO4].](/image/article/2010/DT/b916877j/b916877j-f8.gif) | ||
| Fig. 8 Uptake of chloride or sulfate by a 0.01 M chloroform solution of [Cu(L1-H)2] after equilibration with 0.8 M aqueous solutions of NaCl or Na2SO4 of varied acidity. 100% loading corresponds to that expected for the formation of the tritopic assemblies [Cu(L1)2Cl2] and [Cu(L1)2SO4]. | ||
The pH-profile for chloride loading and other data (see below) suggest that chloride enters the inner coordination sphere displacing a salicylaldoximate ligand as in:
| [Cu(L1-H)2]org + 2HCl ⇌ [Cu(L1)Cl2]org + L1org, | (4) |
| L1org + HCl ⇌ [(L1+H)Cl]org | (5) |
Formation of the 1:1 complex [Cu(L1)Cl2] offers the possibility of doubling the mass transport efficiency over that delivered by the tritopic assembly [Cu(L1)2Cl2].
There is no evidence that sulfate uptake involves the formation of inner sphere complexes. The pH0.5 for sulfate loading by L1 alone is ∼2.8 (see supplementary information, Fig. S1†), which, compared to a value of ∼3.8 for [Cu(L1-H)2], indicates that the cooperativity of copper and sulfate binding is less pronounced9 than in the comparable salen ligand 2 shown in Fig. 2. This is consistent with the trans-configuration of the pendant aminomethyl groups in [Cu(L1-H)2] making it impossible for them, on protonation, to “chelate” the sulfate anion in the way that the cis-arrangement permits in the salen extractant 2 (Fig. 2). The small enhancement of sulfate binding by [Cu(L1-H)2] over L1 could arise from a weak interaction between the sulfate ion and copper(II) centre similar to the apical Cu⋯X interactions seen in the crystal structures of the tritopic assemblies shown in Fig. 7.
L1 was found16 to be a particularly efficient extractant for CuCl2 and ZnCl2, forming complexes with higher metal salt to ligand stoichiometries than predicted by the tritopic motif displayed in Fig. 1. Formation of 1:1 assemblies;
| Lorg + MCl2⇌ [MLCl2]org | (6) |
![Intracomplex H-bonding in the crystal structures of a) [Cu(L2)Cl2], showing the association of two complex units, Cu1A⋯O1B = 2.507(2), N62⋯O1 = 2.754(3), N62⋯Cl2 = 3.560(3), O23⋯Cl1 = 2.925(3) Å. b) [Zn2(L2)2Cl4], Zn1-O1A = 2.031(2), Zn1-O1B = 2.109(2), Zn2-O1A = 2.078(2), Zn2-O1B = 2.021(5), O23B⋯Cl1 = 2.987(3), N62B⋯Cl2 = 3.446(2), N62A⋯Cl3 = 3.379(3), O23A⋯Cl4 = 3.003(2) Å. c) [Cu(L2)Br2], which is isostructural to [Cu(L2)Cl2], Cu1A⋯O1B = 2.630(2), N62⋯O1 = 2.803(3), N62⋯Br2 = 3.642(3), O21⋯Br1 = 3.047(2) Å. All H atoms not involved in H-bonding have been removed for clarity.](/image/article/2010/DT/b916877j/b916877j-f9.gif) | ||
| Fig. 9 Intracomplex H-bonding in the crystal structures of a) [Cu(L2)Cl2], showing the association of two complex units, Cu1A⋯O1B = 2.507(2), N62⋯O1 = 2.754(3), N62⋯Cl2 = 3.560(3), O23⋯Cl1 = 2.925(3) Å. b) [Zn2(L2)2Cl4], Zn1-O1A = 2.031(2), Zn1-O1B = 2.109(2), Zn2-O1A = 2.078(2), Zn2-O1B = 2.021(5), O23B⋯Cl1 = 2.987(3), N62B⋯Cl2 = 3.446(2), N62A⋯Cl3 = 3.379(3), O23A⋯Cl4 = 3.003(2) Å. c) [Cu(L2)Br2], which is isostructural to [Cu(L2)Cl2], Cu1A⋯O1B = 2.630(2), N62⋯O1 = 2.803(3), N62⋯Br2 = 3.642(3), O21⋯Br1 = 3.047(2) Å. All H atoms not involved in H-bonding have been removed for clarity. | ||
Intracomplex hydrogen bonding appears to stabilise both complexes, and may be the driving force behind their formation instead of the expected tritopic motif (Fig. 3). In each case, the piperidinium proton forms H-bonds to the phenolate oxygen and may also form a long range contact with one of the chlorides, giving a bifurcated hydrogen bond. The oximic proton is involved in a close contact with the other chloride anion.
[Cu(L2)Cl2] molecules associate via an axial copper contact with a phenolate oxygen atom of an adjacent molecule in a similar fashion to [Cu(L2-H)2], generating a dimeric assembly with polar functionality located centrally and a hydrophobic exterior.
The zinc chloride complex has the dinuclear structure [Zn2(L2)2Cl4], with the phenolate groups forming similarly strong bonds to both metal atoms and the oximic N and two chloride ligands complete the distorted trigonal bipyramidal NO2Cl23− coordination sphere.
A bromide analogue of [Cu(L2)Cl2] was also isolated, and has a very similar structure in the solid state (see Fig. 9c) with the intracomplex H-bonding interactions in [Cu(L2)Br2] longer than those in [Cu(L2)Cl2], which is to be expected as Br− is larger than Cl−.23
The mass transport efficiency of L1 for copper, the mass of metal transported by 1 kg of ligand,7 was determined16 by solvent extraction of CuCl2. L1 has an observed mass transport efficiency of 143 g kg−1 per cycle when stripped with 220 g l−1 HCl, c.f. a theoretical maximum of 121 g kg−1 calculated for the commercial reagent 5-nonylsalicylaldoxime (1). This much improved copper transport could underpin the development of very efficient processes for metal recovery from chloride feeds. L1 also has the advantage that it functions in a zwitterionic form, with no release of protons to the aqueous phase on extraction and therefore no pH adjustment is needed to achieve high loadings.
As the nature of the anion present in the aqueous phase was found16 to have great influence on the cation-loading of the new reagents, the selectivity of extraction of a range of base metal cations of relevance to commercial processes was measured in the presence of both chloride or sulfate.
Selectivity of cation binding
The pH-dependence of cation loading by L1 from a range of base metal chloride feeds is displayed in Fig. 10. CuII and FeIII are extracted at lower pH than the other base metals, with pH0.5 values of 1.1 and 1.3 indicating that L1 is slightly selective for CuII under these conditions. An excess (0.8 M) of chloride in the aqueous phase in otherwise identical extraction experiments (see supplementary information, Fig. S2†) significantly increases CuII selectivity; FeIII loading has a pH0.5 of 0.9 but the value for CuII could not be calculated as loading is > 180% at the lowest accessible pH (∼0.25). This is a consequence of the formation of [Cu(L1)Cl2], which is clearly favoured at the high activities of chloride and proton found in high tenor feeds.3 The enhanced loading of FeIII with increased chloride concentration is consistent with formation of a neutral [Fe(L1)Cl3] species. However, we were unable to obtain X-ray quality crystals of an FeCl3 complex to determine the binding motif.![The pH dependency of uptake of metal cations by 0.01 M chloroform solutions of L1 after mixing with 0.01 M aqueous solutions of MIICl2 (except FeIIICl3) of varying acidity. 100% loading corresponds to that expected for the formation of [M(L1)2Cl2] assemblies, whilst 200% is consistent with the formation of [M(L1)Cl2].](/image/article/2010/DT/b916877j/b916877j-f10.gif) | ||
| Fig. 10 The pH dependency of uptake of metal cations by 0.01 M chloroform solutions of L1 after mixing with 0.01 M aqueous solutions of MIICl2 (except FeIIICl3) of varying acidity. 100% loading corresponds to that expected for the formation of [M(L1)2Cl2] assemblies, whilst 200% is consistent with the formation of [M(L1)Cl2]. | ||
Selectivity of cation loading was also studied by contacting a 0.01 M chloroform solution of L1 with mixed metal feeds of varying pH but constant (0.8 M) chloride concentration. At all pH values studied (see supplementary information, Fig. S3†) significant quantities of FeIII are extracted alongside CuII, which means that L1 may not be a suitable copper extractant as high CuII/FeIII selectivity is one of the main requirements for industrial use.2,7 The mechanism of FeIII co-extraction is unclear and complicated by the potential for extraction of chloridometallate anions such as FeCl4− in the anion binding site. Its elucidation may assist in solving the cation selectivity problem.
S-curves for base metal extraction from sulfate feeds (Fig. 11) show a selectivity order which is very different than that for chloride feeds. FeIII has a pH0.5 of 0.5 compared with 1.3 for CuII, indicating L1 is selective for FeIII. Iron loading occurs in two stages: a step in the loading profile (pH 0 to 2.0) at approximately 70% loading corresponds to formation of a 3:1 L:FeIII complex, and a second peak (pH 2.0 to 2.5) at 200% loading which would correspond to a 1:1 L:FeIII complex. At higher pH values, loading decreases due to precipitation of FeIII as iron oxyhydroxides. The higher pH0.5 of CuII loading from sulfate (1.3) compared to chloride (1.1) can be attributed to the greater stability of the [Cu(L1)Cl2] assembly.
![The pH dependence of uptake of metal cations by 0.01 M chloroform solutions of L1 after mixing with 0.01 M aqueous solutions of MIISO4 (except [(FeIII)2(SO4)3]) of varying acidity. 100% loading corresponds to that expected for the formation of [M(L1)2SO4] assemblies, whilst 200% would be consistent with the formation of [M(L1)SO4].](/image/article/2010/DT/b916877j/b916877j-f11.gif) | ||
| Fig. 11 The pH dependence of uptake of metal cations by 0.01 M chloroform solutions of L1 after mixing with 0.01 M aqueous solutions of MIISO4 (except [(FeIII)2(SO4)3]) of varying acidity. 100% loading corresponds to that expected for the formation of [M(L1)2SO4] assemblies, whilst 200% would be consistent with the formation of [M(L1)SO4]. | ||
An excess (0.8 M) of sulfate in the aqueous phase (see supplementary information, Fig. S4†) increases the selectivity of L1 for FeIII over CuII; pH0.5 values for loading are 0.3 and 1.5 respectively. Earlier studies24 have revealed an increase in pH0.5 for CuII loading by conventional, unsubstituted phenolic oxime extractants when sulfate concentration in the aqueous phase is increased.
The decrease in pH0.5 for FeIII loading indicates that the formation of the extracted iron(III) sulfate complex involves a cooperative interaction between the cation and anion. A recently characterised25 Fe6 cluster containing doubly deprotonated 2-hydroxyacetophenone oxime ligands has two sulfate anions each bridging three Fe atoms, and it may be that sulfate is bound in the inner coordination sphere of the Fe cations when extraction is carried out at higher pH, which will favour double deprotonation of the ligands and the formation of polynuclear complexes. In this respect, it is notable that increasing sulfate concentration extends the plateaux in the loading curve associated with the species which has the loading stoichiometry of 1FeIII:3L1 to a higher pH, again indicating that the complex formed involves a favourable interaction between the cation and the anion.
Due to the lower pH0.5 value for FeIII compared to CuII, it was expected that, when extracting from a mixed metal sulfate feed, L1 would favour FeIII uptake. Complete iron selectivity was confirmed (see supplementary information, Fig. S5†), with CuII extraction only occurring at pH values high enough for iron to be partially precipitated from the feed.
In an attempt to replicate conditions which might be found with feed solutions resulting from the leaching of sulfidic ores, we studied the selectivity of metal extraction in the presence of an excess of both sulfate and chloride. As expected from the results described above, only CuII and FeIII were loaded at pH < 3.0 (Fig. 12). Copper extraction is favoured, with loadings being approximately double those of iron but the selectivity would make the separation of these metals difficult on an industrial scale unless the iron has been removed by precipitation prior to solvent extraction.
 | ||
| Fig. 12 Loadings of chloroform solutions of L1 (5 ml, 0.01 M) from 5 ml of an aqueous mixed metal feed solution (0.01 M of each metal, containing 0.4 M sulfate and chloride as their sodium salts, total ionic strength of 0.8 M) at different equilibrium pH values. | ||
Conclusions
We have shown, as intended, that the incorporation of dialkylaminomethyl groups in the 3-position of salicylaldoximes transforms their properties from reagents which transport metal cations to reagents which transport metal salts in metal recovery circuits based on solvent extraction.A key design feature of the new reagents is that they operate in a zwitterionic form, binding metal salts to form a charge neutral assembly without a net transfer of protons to or from the aqueous phase. Consequently no pH adjustment of hydrometallurgical feeds is required to obtain high metal loadings.
With copper salts of weakly coordinating anions, tritopic assemblies, e.g., [Cu(L2)2(NO3)2], are formed with the anions being well separated as a consequence of the trans arrangement of the pendant dialkylammonium groups which results from the head-to-tail hydrogen bonding between the salicylaldoximate units. In marked contrast, halide salts form inner sphere complexes with a 1:1 ligand:metal salt stoichiometry, e.g., [Cu(L2)Cl2], and consequently the molar transport efficiency is double that observed for the conventional commercial phenolic oxime extractants which have no pendant dialkylaminomethyl groups.
Introducing an anion binding site to phenolic oxime ligands increases the number and complexity of the equilibria involved in extraction. One of the major challenges associated with the commercial exploitation of these new reagents will be to obtain high selectivity of both cation and anion transport. The co-operativity of cation and anion binding which has been observed in several cases in this paper may assist such a development programme.
Acknowledgements
The authors wish to thank Mr Ronald M. Swart and Mr John Campbell, Cytec Industries UK Ltd for useful discussions and the EPSRC, Cytec Industries UK Ltd and Infineum for funding.Notes and references
- S.G. Galbraith and P.A. Tasker, Supramol. Chem., 2005, 17, 191–207 CrossRef CAS.
- P.A. Tasker, P.G. Plieger and L.C. West, Compr. Coord. Chem. II, 2004, 9, 759–808 Search PubMed.
- K. Sole, Solvent Extr. Ion Exch., 2002, 20, 601–602 CAS.
- G. Senanayake, Miner. Eng., 2007, 20, 634–645 CrossRef CAS.
- D.M. Muir,Chloride Metallurgy 2002: Practice and Theory of Chloride/Metal Interaction, 32nd Annual Hydrometallurgy Meeting, Montreal, QC, Canada, 2002, 2, 759-791 Search PubMed.
- P.A. Tasker, C.C. Tong and A.N. Westra, Coord. Chem. Rev., 2007, 251, 1868–1877 CrossRef CAS.
- J. Szymanowksi, Hydroxyoximes and Copper Hydrometallurgy, CRC Press, London, 1993 Search PubMed.
- R.A. Coxall, L.F. Lindoy, H.A. Miller, A. Parkin, S. Parsons, P.A. Tasker and D.J. White, Dalton Trans., 2003, 55–64 RSC.
- S.G. Galbraith, P.G. Plieger and P.A. Tasker, Chem. Commun., 2002, 2662–2663 RSC.
- H.A. Miller, N. Laing, S. Parsons, A. Parkin, P.A. Tasker and D.J. White, J. Chem. Soc., Dalton Trans., 2000, 3773–3782 RSC.
- D.J. White, N. Laing, H.A. Miller, S. Parsons, S. Coles and P.A. Tasker, Chem. Commun., 1999, 2077–2078 RSC.
- S.G. Galbraith, Q. Wang, L. Li, A.J. Blake, C. Wilson, S.R. Collinson, L.F. Lindoy, P.G. Plieger, M. Schröder and P.A. Tasker, Chem.–Eur. J., 2007, 13, 6091–6107 CrossRef CAS.
- N. Akkus, J.C. Campbell, J. Davidson, D.K. Henderson, H.A. Miller, A. Parkin, S. Parsons, P.G. Plieger, R.M. Swart, P.A. Tasker and L.C. West, Dalton Trans., 2003, 1932–1940 RSC.
- J.W. Steed, Chem. Soc. Rev., 2009, 38, 506–519 RSC.
- A.G. Smith, P.A. Tasker and D.J. White, Coord. Chem. Rev., 2003, 241, 61–85 CrossRef CAS.
- R.S. Forgan, J.E. Davidson, S.G. Galbraith, D.K. Henderson, S. Parsons, P.A. Tasker and F.J. White, Chem. Commun., 2008, 4049–4051 RSC.
- R.S. Forgan, D.K. Henderson, and P.A. Tasker, UK Patent, P14523GB, 2006.
- A.L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- R.S. Forgan, P.A. Wood, J. Campbell, D.K. Henderson, F.E. McAllister, S. Parsons, E. Pidcock, R.M. Swart and P.A. Tasker, Chem. Commun., 2007, 4940–4942 RSC.
- F. Hofmeister, Arch. Exp. Path. Pharm., 1888, 24, 247–260 Search PubMed.
- K. Kavallieratos and B.A. Moyer, Chem. Commun., 2001, 1620–1621 RSC.
- A. Bianchi, K. Bowman-James and E. Garcia-Espana, Supramolecular Chemistry of Anions, 1997 Search PubMed.
- H.D.B. Jenkins, H.K. Roobottom, J. Passmore and L. Glasser, Inorg. Chem., 1999, 38, 3609–3620 CrossRef.
- V.I. Lakshmanan, G.J. Lawson and J.L. Tomliens, J. Inorg. Nucl. Chem., 1975, 37, 2181–2185 CrossRef CAS.
- I.A. Gass, C.J. Milios, A. Collins, F.J. White, L. Budd, S. Parsons, M. Murrie, S.P. Perlepes and E.K. Brechin, Dalton Trans., 2008, 2043–2053 RSC.
- J.M. Casas, F. Alvarez and L. Cifuentes, Chem. Eng. Sci., 2000, 55, 6223–6234 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed solvent extraction data and crystal structure files. CCDC reference numbers 660849, 744579–744582. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b916877j |
| ‡ At pH < 2, HSO4− predominates26 over SO42− and so sulfate loading values, based on the concentration of sulfur in the organic phase as measured by ICP-OES, can reach 200%. |
| This journal is © The Royal Society of Chemistry 2010 |