DOI:
10.1039/B912219B
(Paper)
Dalton Trans., 2010,
39, 477-483
Reactivity and kinetic–mechanistic studies of regioselective reactions of rhodium porphyrins with unactivated olefins in water that form β-hydroxyalkyl complexes and conversion to ketones and epoxides†
Received 22nd June 2009, Accepted 19th September 2009
First published on 22nd October 2009
Abstract
This article reports on the selective oxidation of unactivated alkenes to ketones and epoxides through the intermediacy of β-hydroxyalkyl rhodium porphyrin complexes which are formed by reactions of terminal alkenes with tetra(p-sulfonatophenyl)porphyrin rhodium(III) complex. The β-hydroxyalkyl rhodium porphyrin complexes in water undergo β-C–H elimination to produce ketones in aqueous pH 9.0 solutions and O–H deprotonation in KOH/DMSO solutions resulting in the rapid and quantitative intramolecular nucleophilic displacement to form 1,2-epoxyalkanes.
Introduction
The activation of alkenes by coordination to metal centers is one of the most widely exploited synthetic methodologies in the functionalization of organic molecules,1-5 and has contributed significantly to advances in selective organic transformations.6-10Catalytic air oxidation of ethene to acetaldehyde by an aqueous Pd(II)/Cu(II) catalyst system is known as the Wacker process. Mechanistic studies for the Wacker reaction and closely related processes have shown that alkene binding with palladium(II) activates reactions of olefins with nucleophiles including water, alcohols, and amines to form β-substituted alkyl complexes.11 Subsequent C–H and O–H elimination reactions of the β-substituted alkyl complexes produce organic carbonyls, epoxides, heterocyclic molecules, and hydrofunctionalized alkenes.12-15 Several rhodium and iridium complexes have also been reported to mediate oxidation of olefins in hydrocarbon media,16-19 but relatively little attention has been given to complexes of transition metals other than palladium(II). The importance of olefin transformations and objectives of green chemistry justify the continuing search for new classes of catalyst materials that give oxidations in water and use dioxygen as the oxidant. This article reports on the observed stepwise reactions of rhodium porphyrins with olefins in water to form β-hydroxyalkyl complexes that react on to give stoichiometric oxidation of alkenes to ketones in aqueous media and epoxides in DMSO. Several objectives in olefin activation and oxidation have been advanced in this study through observation of product inhibited catalytic oxidation of olefins to organic carbonyls in water using dioxygen as the oxidant.
Results and discussion
Formation of β-hydroxyalkyl rhodium complexes in water
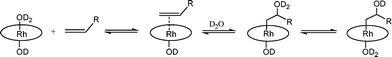
Tetra(p-sulfonatophenyl)porphyrin rhodium(III) diaquo complex [(TSPP)RhIII(D2O)2]3− (1) is a convenient entry point for aqueous (D2O) solution reactivity studies of rhodium porphyrins. The rhodium(III) diaquo complex occurs in D2O solution as a pH dependent equilibrium distribution with the mono- and bis-hydroxo complexes ([(TSPP)RhIII(OD)(D2O)]4− (2) and [(TSPP)RhIII(OD)2]5− (3)) where K1 = 1.4 ± 0.2 × 10−8 and K2 = 2.8 ± 0.3 × 10−12(eqn (1) and (2)).20| | | [(TSPP)RhIII(D2O)2]3−⇌ [(TSPP)RhIII(OD)(D2O)]4− + D+ | (1) |
| | | [(TSPP)RhIII(OD)(D2O)]4−⇌ [(TSPP)RhIII(OD)2]5− + D+ | (2) |
Reactions of (TSPP)RhIII complexes with a series of olefins produce β-hydroxyalkyl complexes in water.‡Reaction of (TSPP)RhIII with 2-substituted olefins. Ethene and larger terminal alkene hydrocarbons (CH2=CHR) react regioselectively with (TSPP)RhIII in D2O to form β-hydroxy alkyl rhodium complexes ([(TSPP)Rh–CH2CH(OD)R(D2O)]4−) where rhodium is attached to the terminal primary CH2 unit (eqn (3)).21| | [(TSPP)Rh–OD(D2O)]4− + CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR ⇌ [(TSPP)Rh–CH2CH(OD)R(D2O)]4− CHR ⇌ [(TSPP)Rh–CH2CH(OD)R(D2O)]4− | (3) |
The β-hydroxyalkyl complexes are easily identified by the porphyrin ring current induced high-field 1H NMR resonances of the organic group bonded to the rhodium center. The 1H NMR resonances for the diastereotopic α-CH2 group in (TSPP)Rh–CH2CH(OD)CH2CH2CH3 in D2O are centered at −5.93 and −5.74 ppm. Isolating ((TSPP)Rh–CH2CH(OH)CH2CH2CH3) formed in water and dissolving in anhydrous DMSO-d6 permitted observing the doublet hydroxyl proton resonance ((TSPP)RhCH2CH(OH)CH2CH2CH3) at 0.01 ppm (d, 3JHH = 5.6 Hz). Reactions of (TSPP)RhIII with a series of terminal alkenes in a borate buffer with pH 9.0 produce β-hydroxyalkyl complexes [(TSPP)Rh–CH2CH(OD)R(D2O)]4− (4) that reach equilibrium in time periods of several minutes to hours depending inversely on the olefin solubility (Table 1). Highly soluble alkenes (Table 1, entries 4–6) cleanly react to completion within minutes but poorly soluble alkenes (Table 1, entries 1–3) react much slower which allows time to produce some β-carbonyl organometallic derivatives ([(TSPP)Rh–CH2C(O)R)(D2O)]4−) by β-C–H elimination of 4.22
Table 1 Formation of β-hydroxyalkyl rhodium porphyrin complexes in borate buffer aqueous solution (pH = 9.0) at 298 Ka
Reaction of (TSPP)RhIII with 2,2-disubstituted alkenes in water. Reaction of (TSPP)RhIII with a 2,2-disubstituted alkene (2-methyl-1-butene) also forms a β-hydroxyalkyl complex [(TSPP)Rh–CH2C(OD)(CH3)(CH2CH3)(D2O)]4− (5) (eqn (4)).| | | [(TSPP)Rh–OD(D2O)]4− + CH2C(CH3)CH2CH3⇌ [(TSPP)Rh–CH2C(OD)(CH3)(CH2CH3)(D2O)]4− | (4) |
The 1H–1H and 103Rh–1H coupling patterns that are partially obscured by line broadening in water became clearly resolved when 5 was isolated from water and redissolved in CD3OD (Fig. 1(b)). The characteristic AB pattern for the α-CH2 unit (δ(ppm): −5.55 (HA), −5.48 (HB), JAB = 9.9 Hz, JRhH = 2.9 Hz) in 5 in CD3OD is substantially improved from the broad resonance observed in D2O at −5.72 ppm (Fig. 1(a)). The hydroxyl proton resonance for (TSPP)Rh–CH2C(OH)(CH3)(CH2CH3) was also identified from the 1H NMR spectrum (s, −0.16 ppm) in anhydrous DMSO-d6 by preparation of the sample in H2O and transferring into DMSO-d6 (Fig. 1(c)).
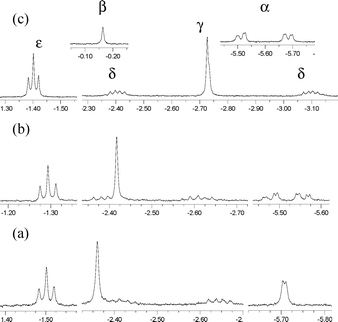 |
| | Fig. 1 High-field 1H NMR (400 MHz) for (TSPP)Rh–CH2αC(OHβ)(CH3γ)(CH2δCH3ε) (a) in D2O, (b) in CD3OD, and (c) in DMSO-d6. | |
Kinetics of alkene reaction with (TSPP)RhIII in water
Evaluation of rate and equilibrium constants (kf, kr, Keq). Formations of (TSPP)Rh–CH2CH(OD)R as a function of time from reactions of (TSPP)RhIII species with pentene and hexene in D2O at 298 K using a pH = 9.0 buffer are illustrated in Fig. 2.![Formation of [(TSPP)Rh–CH2CH(OD)R(D2O)]4− from [(TSPP)RhIII(OD)(D2O)]4− with pentene and hexene at 298 K with pH = 9.0 buffer. The solid line is the nonlinear least-square best fit to the equation d[(TSPP)Rh–CH2CH(OD)R]/dt = β−α[(TSPP)Rh–CH2CH(OD)R], β = kfc(RhT)c0/(([D+]/K1) + (K2/[D+]) + 1), α = (β/c(RhT)) + kr using OriginPro 7.5 software. The initial concentration of (TSPP)RhIII, c(RhT), equals 1.9 × 10−3 M. The saturated solubility of pentene in water, c0, is 2.1 × 10−3 M, and hexane is 1.9 ×10−3 M.23 (A) Reaction of pentene gives α = (5.5 ± 0.1) × 10−4, β = (8.5 ± 0.1) × 10−7, and kf = (2.3 ± 0.1) × 10−1 L mol−1 s−1, kr = (10.0 ± 1.0) × 10−5 s−1, and Keq = (2.3 ± 0.1) × 103 are derived. (B) Reaction of hexane gives α = (2.2 ± 0.1) × 10−4, β = (2.7 ± 0.1) × 10−7 and kf = (8.0 ± 0.1) × 10−2 L mol−1 s−1, kr = (7.8 ± 1.0) × 10−5 s−1, and Keq = (1.0 ± 0.1) × 103 are derived.](/image/article/2010/DT/b912219b/b912219b-f2.gif) |
| | Fig. 2 Formation of [(TSPP)Rh–CH2CH(OD)R(D2O)]4− from [(TSPP)RhIII(OD)(D2O)]4− with pentene and hexene at 298 K with pH = 9.0 buffer. The solid line is the nonlinear least-square best fit to the equation d[(TSPP)Rh–CH2CH(OD)R]/dt = β−α[(TSPP)Rh–CH2CH(OD)R], β = kfc(RhT)c0/(([D+]/K1) + (K2/[D+]) + 1), α = (β/c(RhT)) + kr using OriginPro 7.5 software. The initial concentration of (TSPP)RhIII, c(RhT), equals 1.9 × 10−3 M. The saturated solubility of pentene in water, c0, is 2.1 × 10−3 M, and hexane is 1.9 ×10−3 M.23 (A) Reaction of pentene gives α = (5.5 ± 0.1) × 10−4, β = (8.5 ± 0.1) × 10−7, and kf = (2.3 ± 0.1) × 10−1 L mol−1 s−1, kr = (10.0 ± 1.0) × 10−5 s−1, and Keq = (2.3 ± 0.1) × 103 are derived. (B) Reaction of hexane gives α = (2.2 ± 0.1) × 10−4, β = (2.7 ± 0.1) × 10−7 and kf = (8.0 ± 0.1) × 10−2 L mol−1 s−1, kr = (7.8 ± 1.0) × 10−5 s−1, and Keq = (1.0 ± 0.1) × 103 are derived. | |
Expressions were derived for several plausible mechanisms. The kinetics for this reaction are potentially complicated by having three different aquo/hydroxo complexes, but the monohydroxo complex is the dominant species (>93%) at pH = 9.0 so that effectively all of the β-hydroxyalkyl products were formed through the monohydroxo complex ([(TSPP)RhIII(D2O)(OD)]4− (2)). Thus, the rate of formation of [(TSPP)Rh–CH2CH(OD)R(D2O)]4− is derived as d[Rh–CH2CH(OD)R]/dt = kf[2][alkene] −kr[Rh–CH2CH(OD)R]. The best fit from nonlinear least squares curve fitting is obtained for the mechanism where effectively all of the product forms from reaction of 2 with alkenes (see Experimental section for detailed derivation). The forward rate constant for the pentene reaction at 298 K (kf = (2.3 ± 0.1) × 10−1 L mol−1 s−1) and the reverse reaction rate constant (kr = (10.0 ± 1.0) × 10−5 s−1), along with the corresponding values for the hexene reaction (298 K) (kf = (8.0 ± 0.1) × 10−2 L mol−1 s−1, kr = (7.8 ± 1.0) × 10−5 s−1) are obtained from the kinetic simulation (Fig. 2). The equilibrium constant for pentene Keq (Keq = kf/kr = (2.3 ± 0.1) × 103) that is derived from the kinetic analysis is close to the value obtained from direct equilibrium thermodynamic measurement by integration of the 1H NMR spectrum (Keq = (4.1 ± 0.6) × 103).
Reaction (3) occurs through formation of π complexes ((TSPP)Rh(CH2CHR)) by alkene substitution for a coordinated water molecule followed by nucleophilic attack on the olefin π complex and a proton transfer.
The regioselectivity for the initial nucleophilic attack step is controlled by the sterically bulky porphyrin ligand which favors placing rhodium on the less hindered terminal primary carbon as the kinetic product. In the case of unactivated alkenes the kinetic and thermodynamic products have rhodium on the primary –CH2 ((TSPP)Rh–CH2CH(OD)R), but olefins with electron withdrawing groups (R = phenyl, CO2Na) initially form (TSPP)Rh–CH2CH(OD)R and then rearrange to (TSPP)Rh–CH(R)CH2OD as the thermodynamic products.
Electron withdrawing groups better stabilize the negative charge and provide an electronic energy term that favors placing the metal on the same carbon as the electron withdrawing group which for the phenyl and CO2Na derivatives more than compensates for the unfavorable steric effect.21,24
An octaethylporphyrin rhodium ethene π complex ((OEP)Rh(CH2CH2))+ has been directly observed in toluene by 1H NMR at 243 K. The β-hydroxyalkyl complex of rhodium octaethylporphyrin ((OEP)Rh–CH2CH2OH) was formed in hydrocarbon media through hydroxide nucleophilic addition to ((OEP)Rh(CH2CH2))+.25 Olefin π complexes of (TSPP)RhIII in water at 298 K have not yet been observed by 1H NMR even when the conditions for π complex formation are optimized by using high concentrations of water-soluble olefins and slightly acidic aqueous media where the diaquo complex 1 dominates.
Thermal dissociation of β-hydroxyalkyl rhodium porphyrin complexes
β-Hydroxyalkyl rhodium porphyrin complexes are intermediate organometallic complexes for both β-C–H elimination reactions that produce ketones in pH 9.0 aqueous solution and intramolecular C–O elimination to form epoxides quantitatively in KOH/DMSO (Scheme 1). |
| | Scheme 1 Thermal dissociation pathways of β-hydroxyalkyl rhodium porphyrin complexes. | |
β-Hydrogen elimination of β-hydroxyalkyl rhodium porphyrin complexes in water. β-Hydrogen elimination reactions are among the most important fundamental organometallic transformations in a variety of transition metal alkyl and alkoxide complexes. The hydrogen migration or elimination usually occurs in coordinately unsaturated complexes through use of a vacant cis coordination site. However, β-hydrogen elimination processes for alkyl rhodium porphyrin complexes must utilize a different mechanism because all of the cis coordination sites are occupied by the porphyrin pyrrole nitrogen donors.26 The β-hydroxyalkyl complexes formed by reaction (3) in water spontaneously transform to (TSPP)Rh–H and ketones in the absence of air by an effective β-hydrogen elimination (eqn (5), Table 2).22| | | [(TSPP)Rh–CH2CH(OD)R(D2O)]4−⇌ [(TSPP)Rh(H)(D2O)]4− + CH2DC(O)R | (5) |
Table 2 Formation of ketones through β-hydrogen elimination of β-hydroxyalkyl rhodium porphyrin complexes in pH = 9.0 buffera
Thermal reaction of (TSPP)Rh–CH2CH(OD)(CH2)2CH3 in D2O results in the formation of (TSPP)Rh–H and a mono-deuterated 2-pentanone (CH2(D)C(O)CH2CH2CH3) where deuterium is selectively incorporated into the methyl group. The selective deuteration indicates that β-hydrogen elimination produces an enol containing an OD unit (Scheme 2). This mechanistic feature is different from the Pd(II) Wacker oxidation where deuterium from D2O does not occur in the oxidation product. Isomerization of β-hydroxyalkyl to the α-hydroxy isomer that occurs prior to the product forming step in the Wacker reaction cannot occur for the rhodium porphyrin because the cis coordination sites are blocked.
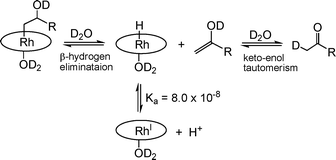 |
| | Scheme 2 Formation of ketones through (i) thermal β-hydrogen elimination of 4 and (ii) keto–enol tautomerism in water. | |
The observed fast reactions of rhodium porphyrin hydrides with olefins in water compared to benzene is ascribed to water supporting ionic reaction pathways.27 In aqueous solution the hydride complex [(TSPP)RhIII(D)(D2O)]4− functions as a weak acid and partially dissociates into D+ and [(TSPP)RhI(D2O)]5−. The rhodium(I) porphyrin complex in water is proposed to activate olefins which subsequently protonate to form rhodium alkyl complexes. The β-C–H elimination or migration process is the microscopic reverse of the addition of the rhodium hydride to olefins in water which is proposed to occur by dissociation of Rh–H into H+ and Rh(I)- and subsequent stepwise addition.27 The complex (TSPP)Rh–CH2C(OD)(CH3)(CH2CH3) 5 lacks a β-hydrogen atom and thus is not capable of producing carbonyl compounds.
Aerobic oxidation of (TSPP)Rh–H to (TSPP)RhIII in water.
The [(TSPP)Rh(H)(D2O)]4− species formed in reaction (5) in a rapid dissociation equilibrium with [(TSPP)RhI(D2O)]5− and H+ undergo an immediate color change to a clear dark red solution when exposed to air. The distinctive pyrrole hydrogen resonance associated with (TSPP)RhIII species is observed by 1H NMR. The sequence of reactions with dioxygen to convert (TSPP)Rh–H/(TSPP)RhI to (TSPP)RhIII is depicted by Scheme 3. |
| | Scheme 3 Aerobic oxidations of (TSPP)Rh–H/(TSPP)RhI to (TSPP)RhIII in water. | |
Rapid air oxidation of Rh(I) to Rh(III) completes the cycle which provides the potential for catalytic oxidation of olefins.
Formation of β-carbonyl alkyl rhodium porphyrin complexes in water
Aqueous buffer solutions of (TSPP)RhIII species react with ketones and aldehydes to produce β-carbonyl derivatives [(TSPP)Rh–CH2C(O)R(D2O)]4− (6), which has a precedent in reaction of [(OEP)RhIII]+ with ketones.28 This electrophilic C–H activation by the cationic metal center is thought to proceed through a stepwise mechanism similar to that of reaction (3), in which the enol intermediate reacts with (TSPP)RhIII to give (TSPP)Rh–CH2C(OD)2R, followed by the dehydration step of the unstable gem-diol complex.Reaction of the acetaldehyde methyl C–H unit occurs with high regioselectivity to form (TSPP)Rh–CH2CHO (eqn (6)). No evidence is found for reaction of the weaker aldehydic C–H bond, which is analogous to reactions of (por)RhII complexes with CH3CHO in benzene.29
| | | [(TSPP)Rh–OD(D2O)]4− + CH3CHO ⇌ [(TSPP)Rh–CH2CHO(D2O)]4− + HOD | (6) |
The compound formed from reaction of (TSPP)RhIII with acetone (eqn (7)) has an identical 1H NMR spectrum with the complex formed from reaction of (TSPP)RhI in water with ClCH2C(O)CH3 that produces (TSPP)Rh–CH2C(O)CH3.
| | | [(TSPP)Rh–OD(D2O)]4− + CH3C(O)CH3⇌ [(TSPP)Rh–CH2C(O)CH3(D2O)]4− + HOD | (7) |
Addition of 300 equiv. of 2-pentanone to an aqueous solution of (TSPP)RhIII in pH 9.0 D2O buffer resulted in an equilibrium distribution of (TSPP)Rh–CH2C(O)CH2CH2CH3 and (TSPP)RhIII which permitted evaluation of the equilibrium constant (Keq(298 K) = 33). The relatively slow reaction rates of ketones compared to alkenes may result from the very low enol concentration. The enolization equilibrium constant KE for acetone30 is (4.69 ± 0.19) × 10−9 and the resulting low concentration of enol place limits on the rate of β-carbonyl formation. 2-Pentanone can enolize in two ways resulting in regio-isomeric enols, CH2C(OD)CH2CH2CH3 (A) and CH3C(OD)CHCH2CH3 (B), but the reaction of (TSPP)RhIII with 2-pentanone exclusively produces (TSPP)Rh–CH2C(O)CH2CH2CH3. The origin of this result may be the thermodynamic preference for primary alkyl rhodium complexes.
The ketone products react with the (TSPP)Rh(III) oxidation catalyst resulting in a product inhibited catalytic oxidation of olefins which limits the numbers of turnovers.
Formation of epoxides from β-hydroxyalkyl rhodium porphyrin complexes
The β-hydroxyalkyl rhodium porphyrin complexes, (TSPP)Rh–CH2CH(OD)(CH2)nCH3 (n = 2–4) (eqn (8)) undergo immediate (<5 min) epoxide forming elimination reactions in KOH/DMSO-d6 (cKOH = 3.3 mg mL−1) at room temperature to give quantitative formation of (TSPP)Rh–D which is rapidly deprotonated to form (TSPP)RhI and 1,2-epoxyalkanes as observed by both 1H NMR and GC-MS (Table 3).| |  | (8) |
Table 3 Formation of 1,2-epoxyalkanes through epoxide-forming elimination of β-hydroxyalkyl rhodium porphyrin complexes in KOH/DMSO
| Entry | Substrate | t | Product |
|---|
| The reactions were performed in KOH/DMSO (cKOH = 3.3 mg mL−1) and the product epoxides were formed quantitatively as measured by 1H NMR spectroscopy. The resulting 1,2-epoxyalkanes were extracted into Et2O and examined by GC-MS. |
|---|
| 1 | 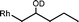 | <5 min | 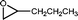 |
| 2 |  | <5 min |  |
| 3 |  | <5 min |  |
| 4 | 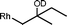 | <5 min |  |
A brownish solution characteristic of (TSPP)RhI is observed to form immediately after (TSPP)Rh–CH2CH(OD)(CH2)nCH3 is dissolved in KOH/DMSO-d6. C–O reductive elimination from the d6 metal center usually occurs through an SN2 route and a direct C–O elimination pathway.31 Groves reported a facile C–O reductive elimination from β-hydroxyalkyl complexes of rhodium tetraphenyl porphyrin in KOtBu/C6D6 by a proposed SN2 pathway.19
The origin of the selective formation of ketone in water and epoxide in KOH/DMSO may be that the nucleophilicity of the alkoxy group is dramatically reduced in water due to the solvation. β-Hydroxyalkyl rhodium porphyrin complexes thus proceed through the alternate usually higher energy β-H elimination pathway which produces ketones. The favored formation of three-membered oxygen heterocycles through intramolecular C–O bond formation is driven by the strong nucleophilicity of the alkoxy group formed in strongly basic conditions of the (KOH/DMSO) medium.
Conclusions
(TSPP)RhIII reacts with terminal alkenes to form β-hydroxy alkyl rhodium porphyrin complexes with high conversion in pH 9.0 aqueous buffer solution at room temperature. Regioselectivity of the reaction is controlled by the sterically bulky porphyrin ligand and the less-substituted alkyl rhodium complexes are formed as the kinetic products. The equilibrium constant of reaction of (TSPP)RhIII with pentene was evaluated as K = (2.3 ± 0.1) × 103 from kinetic simulation. The β-hydroxyalkyl rhodium porphyrin complexes underwent the β-hydrogen elimination reaction to produce ketones in pH 9.0 aqueous solution and near quantitative intramolecular formation of epoxides in KOH/DMSO. Product inhibited catalytic oxidation of olefins to ketones by (TSPP)RhIII is accomplished by using dioxygen as the oxidant.Experimental
Typical procedure for preparation of (TSPP)Rh–CH2CH(OD)R in water
Alkenes (0.01 mmol) and 1 (1.1 mg, 0.001 mmol) were dissolved in 0.4 mL borate buffer D2O solution (pH = 9.0) in J. Young valve adapted NMR tubes at room temperature. The progress of the reaction was monitored by 1H NMR spectroscopy.Typical procedure for β-hydrogen elimination of (TSPP)Rh–CH2CH(OD)R in water
The (TSPP)Rh–CH2CH(OD)R complexes were prepared according to the procedure given above, which exclusively converted (TSPP)RhIII into (TSPP)Rh–CH2CH(OD)R. The excesses of alkenes and solvent D2O were pumped out. Fresh D2O was added into the NMR tube and subjected to three freeze–pump–thaw cycles. The initial 1H NMR spectrum was recorded to show the formation of Rh–CH2CH(OD)R and a clean range from 0 to 4 ppm. The sample (TSPP)Rh–CH2CH(OD)R was heated in a water-bath at 60 °C (or 80 °C) for a period of hours, and the progress of the reaction was monitored by 1H NMR spectroscopy. When the reactions reached completion after all (TSPP)Rh–CH2CH(OD)R complexes were converted to ketones and (TSPP)Rh–D (which was rapidly deprotonated to form (TSPP)RhI), the product ketones were extracted by CDCl3. A parallel sample of (TSPP)Rh–CH2CH(OH)R dissolved in H2O was also heated under the same reaction conditions, and extracted by CDCl3. Both 1H NMR and GC-MS were used to characterize the product ketones.Kinetic simulation for reaction of (TSPP)RhIII with pentene
| |  | (9) |
| |  | (10) |
| |  | (11) |
We then have:
| [1] + [2] + [3] + [4] = c(RhT) |
| c0 = [CH2CHCH2CH2CH3] |
The concentration of
2 is related to
4,
As stated above:
where
so
| [4]t = (β/α)[1 − exp(−αt)] |
β = (8.5 ± 0.1) × 10
−7,
α = (5.5 ± 0.1) × 10
−4 are obtained from simulation, and
k3 = (2.3 ± 0.1) × 10
−1 L mol
−1 s
−1,
k-3 = (10.0 ± 1.0) × 10
−5 s
−1, and
K3 = (2.3 ± 0.1) × 10
3 are derived when
c(RhT) = 1.9 × 10
−3 M and
c0 = [CH
2CHCH
2CH
2CH
3] = 2.1 × 10
−3 M are used.
Acknowledgements
This work was supported by a starter grant from Peking University, NSFC (grants 20841002 and 20801002), and the U.S. Department of Energy, Division of Chemical Sciences, Office of Science DE-FG02–09ER16000.Notes and references
- K. F. McDaniel, in Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Elsevier Science Ltd., Oxford, UK, 1995, vol. 12, ch. 7 Search PubMed.
- L. S. Hegedus, Palladium in Organic Synthesis, in Organometallics in Synthesis: a Manual, ed. M. Schlosser, Wiley-Interscience, Chichester, UK, 2nd edn, 1998 Search PubMed.
- A. R. Chianese, S. J. Lee and M. R. Gagné, Angew. Chem., Int. Ed., 2007, 46, 4042–4059 CrossRef CAS.
- C. Hahn, Chem.–Eur. J., 2004, 10, 5888–5899 CrossRef CAS.
-
(a) B. de Bruin, P. H. M. Budzelaar and A. W. Gal, Angew. Chem., Int. Ed., 2004, 43, 4142–4157 CrossRef CAS;
(b) R. S. Pryadun and J. D. Atwood, Organometallics, 2007, 26, 4830–4834 CrossRef CAS; D. S. Helfer and J. D. Atwood, Organometallics, 2004, 23, 2412–2420 CrossRef CAS.
-
(a) S. S. Stahl, Science, 2005, 309, 1824 CrossRef CAS;
(b) M. M. Konnick and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 5753–5762 CrossRef CAS;
(c) B. A. Steinhoff and S. S. Stahl, J. Am. Chem. Soc., 2006, 128, 4348–4355 CrossRef CAS;
(d) J. L. Brice, J. E. Harang, V. I. Timokhin, N. R. Anastasi and S. S. Stahl, J. Am. Chem. Soc., 2005, 127, 2868–2869 CrossRef CAS.
-
(a) M. Utsunomiya and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 2702–2703 CrossRef CAS;
(b) J. Takaya and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 5756–5757 CrossRef CAS.
- R. Pryadun, D. Sukumaran, R. Bogadi and J. D. Atwood, J. Am. Chem. Soc., 2004, 126, 12414–12420 CrossRef CAS.
- M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368–3398 CrossRef CAS.
- C. Liu, C. F. Bender, X. Han and R. A. Widenhoefer, Chem. Commun., 2007, 3607–3618 RSC.
- E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318–5365 CrossRef CAS.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS.
-
(a) S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400–3420 CrossRef CAS;
(b) V. I. Timokhin, N. R. Anastasi and S. S. Stahl, J. Am. Chem. Soc., 2003, 125, 12996–12997 CrossRef CAS.
- T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675–703 CrossRef.
-
(a) G.-J. ten Brink, I. W. C. E. Arends, G. Papadogianakis and R. A. Sheldon, Chem. Commun., 1998, 2359–2360 RSC;
(b) G.-J. ten Brink, 2288; I. W. C. E. Arends, G. Papadogianakis and R. A. Sheldon, Appl. Catal. A, 2000, 194–195, 435–442 Search PubMed.
-
(a) B. de Bruin and D. G. H. Hetterscheid, Eur. J. Inorg. Chem., 2007, 211–230 CrossRef CAS;
(b) D. G. H. Hetterscheid, M. Bens and B. de Bruin, Dalton Trans., 2005, 979–984 RSC;
(c) D. G. H. Hetterscheid, J. M. M. Smits and B. de Bruin, Organometallics, 2004, 23, 4236–4246 CrossRef CAS;
(d) B. de Bruin, M. J. Boerakker, J. A. W. Verhagen, R. de Gelder, J. M. M. Smits and A. W. Gal, Chem.–Eur. J., 2000, 6, 298–312 CrossRef CAS;
(e) B. de Bruin, J. A. W. Verhagen, C. H. J. Schouten, A. W. Gal, D. Feichtinger and D. A. Plattner, Chem.–Eur. J., 2001, 7, 416–422 CrossRef CAS.
-
(a) M. Beller, C. Breindl, M. Eichberger, C. G. Hartung, J. Seayad, O. R. Thiel, A. Tillack and H. Trauthwein, Synlett, 2002, 1579–1594 CrossRef CAS;
(b) K. D. Hesp and M. Stradiotto, Org. Lett., 2009, 11, 1449–1452 CrossRef CAS.
- Z. Liu and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 1570–1571 CrossRef CAS.
-
(a) M. S. Sanford and J. T. Groves, Angew. Chem., Int. Ed., 2004, 43, 588–590 CrossRef CAS;
(b) Y. Z. Han, M. S. Sanford, M. D. England and J. T. Groves, Chem. Commun., 2006, 549–551 RSC.
- X. Fu and B. B. Wayland, J. Am. Chem. Soc., 2004, 126, 2623–2631 CrossRef CAS; X. Fu, L. Basickes and B. B. Wayland, Chem. Commun., 2003, 520–521 RSC.
- X. Fu, S. Li and B. B. Wayland, J. Am. Chem. Soc., 2006, 128, 8947–8954 CrossRef CAS.
- J. Zhang, S. Li, X. Fu and B. B. Wayland, Dalton Trans., 2009, 3661–3663 RSC.
- M. H. Abraham and J. Le, J. Pharm. Sci., 1999, 88, 868–880 CrossRef CAS.
- S. Li, S. Sarkar and B. B. Wayland, Inorg. Chem., 2009, 48, 8550–8558 CrossRef CAS.
- B. B. Wayland, S. L. VanVoorhees and K. J. Del Rossi, J. Am. Chem. Soc., 1987, 109, 6513–6515 CrossRef CAS.
-
(a) J. P. Collman, A. S. Chien, T. A. Eberspacher and J. I. Brauman, J. Am. Chem. Soc., 1998, 120, 425–426 CrossRef CAS;
(b) I. H. Grate and G. N. Schrauzer, J. Am. Chem. Soc., 1979, 101, 4601–4611 CrossRef CAS;
(c) G. N. Schrauzer and I. H. Grate, J. Am. Chem. Soc., 1981, 103, 541–546 CrossRef CAS;
(d) K. W. Mak and K. S. Chan, J. Am. Chem. Soc., 1998, 120, 9686–9687 CrossRef CAS;
(e) K. W. Mak, F. Xue, T. C. W. Mak and K. S. Chan, J. Chem. Soc., Dalton Trans., 1999, 3333–3334 RSC;
(f) S. K. Yeung and K. S. Chan, Organometallics, 2005, 24, 2561–2563 CrossRef CAS.
- X. Fu and B. B. Wayland, J. Am. Chem. Soc., 2005, 127, 16460–16467 CrossRef CAS.
- Y. Aoyama, Y. Tanaka, T. Yoshida, H. Toi and H. Ogoshi, J. Organomet. Chem., 1987, 329, 251 CrossRef CAS.
- K. J. Del Rossi, X. Zhang and B. B. Wayland, J. Organomet. Chem., 1995, 504, 47 CrossRef CAS.
- Y. Chiang, A. J. Kresge and N. P. Schepp, J. Am. Chem. Soc., 1989, 111, 3977–3980 CrossRef CAS.
- E. J. Alexanian and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 15627–15635 CrossRef CAS; J. R. Khusnutdinova, L. L. Newman, P. Y. Zavalij, Y.-F. Lam and A. N. Vedernikov, J. Am. Chem. Soc., 2008, 130, 2174–2175 CrossRef CAS; J. R. Khusnutdinova, P. Y. Zavalij and A. N. Vedernikov, Organometallics, 2007, 26, 2402–2413 CrossRef CAS; G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2006, 128, 7179–7181 CrossRef CAS; L. V. Desai and M. S. Sanford, Angew. Chem., Int. Ed., 2007, 46, 5737–5740 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.