 Open Access Article
Open Access ArticleMicrofluidic lab-on-a-chip platforms: requirements, characteristics and applications†
Daniel
Mark‡
b,
Stefan
Haeberle‡
ab,
Günter
Roth‡
ab,
Felix
von Stetten‡
ab and
Roland
Zengerle‡
*abc
aLaboratory for MEMS Applications, Department of Microsystems Engineering (IMTEK), University of Freiburg, Georges-Koehler-Allee 106, 79110 Freiburg, Germany. E-mail: zengerle@imtek.de; Fax: +49 761 203 7539; Tel: +49 761 203 7477
bHSG-IMIT—Institut für Mikro- und Informationstechnik, Wilhelm-Schickard-Straße 10, 78052 Villingen-Schwenningen, Germany
cCentre for Biological Signalling Studies (bioss), Albert-Ludwigs-University of Freiburg, Germany
First published on 25th January 2010
Abstract
This critical review summarizes developments in microfluidic platforms that enable the miniaturization, integration, automation and parallelization of (bio-)chemical assays (see S. Haeberle and R. Zengerle, Lab Chip, 2007, 7, 1094–1110, for an earlier review). In contrast to isolated application-specific solutions, a microfluidic platform provides a set of fluidic unit operations, which are designed for easy combination within a well-defined fabrication technology. This allows the easy, fast, and cost-efficient implementation of different application-specific (bio-)chemical processes. In our review we focus on recent developments from the last decade (2000s). We start with a brief introduction into technical advances, major market segments and promising applications. We continue with a detailed characterization of different microfluidic platforms, comprising a short definition, the functional principle, microfluidic unit operations, application examples as well as strengths and limitations of every platform. The microfluidic platforms in focus are lateral flow tests, linear actuated devices, pressure driven laminar flow, microfluidic large scale integration, segmented flow microfluidics, centrifugal microfluidics, electrokinetics, electrowetting, surface acoustic waves, and dedicated systems for massively parallel analysis. This review concludes with the attempt to provide a selection scheme for microfluidic platforms which is based on their characteristics according to key requirements of different applications and market segments. Applied selection criteria comprise portability, costs of instrument and disposability, sample throughput, number of parameters per sample, reagent consumption, precision, diversity of microfluidic unit operations and the flexibility in programming different liquid handling protocols (295 references).
 Daniel Mark | Mr Daniel Mark studied physics at the University of Ulm, Germany and the University of Oregon, USA, receiving an MSc degree and German diploma in 2006/2007. In 2007, he started his work as an R&D engineer and PhD candidate at the Institute of Microsystems Technology (IMTEK) of the University of Freiburg, focussing on lab-on-a-chip applications for medical diagnostics. In 2008, he became group leader of the centrifugal microfluidics team of the joint lab-on-a-chip research division of IMTEK and the Hahn Schickard Society. His research experience includes microfluidic design, prototyping, and validation of biomedical applications. |
 Stefan Haeberle | Dr Stefan Haeberle received his PhD at the Laboratory for MEMS Applications at the Department of Microsystems Engineering (IMTEK) at the University of Freiburg, Germany in 2009. He received his diploma degree in microsystem engineering in 2004 from the University of Freiburg. His research concentrates on the development of lab-on-a-chip systems based on the pressure driven and centrifugal microfluidic platform. He recently accepted a position at a global consulting firm. |
 Günter Roth | Dr Günter Roth studied interdisciplinary physics and biochemistry in parallel at the Eberhard-Karls-University in Tübingen, Germany. He received the German diploma in physics 2001 for a microstructure to separate cell lysate and in biochemistry 2002 for establishing an micro-ELISA with one micron spatial resolution. At the EMC microcollections GmbH, Tübingen, Germany he developed two different high-throughput screening platforms within his PhD thesis. In 2007, he was post-doc in the Institute for Cell Biology, Tübingen, Germany and finally joined the Laboratory for MEMS Applications at IMTEK, University of Freiburg, as group leader for lab-on-a-chip assay development in July 2008. |
 Felix von Stetten | Dr Felix von Stetten studied Agricultural Engineering and Dairy Sciences at the Technical University of Munich, Germany. After additional studies in Biotechnology and a research period in food microbiology he received his PhD in microbiology, also from the Technical University of Munich in 1999. Then he spent three years in the diagnostic industry and was involved in the development of methods for sample preparation, real-time PCR and DNA-arrays. Afterwards he joined the Laboratory for MEMS Applications at IMTEK, University of Freiburg, where he became involved in biofuel cell- and lab-on-a-chip-research. Today Felix von Stetten heads the joint research division for lab-on-a-chip of IMTEK and HSG-IMIT. |
 Roland Zengerle | Prof. Dr Roland Zengerle received his diploma in physics from the Technical University of Munich in 1990, and a PhD from the “Universität der Bundeswehr München” based on the development of micropumps in 1994. Since 1999 he has been full professor at the Department of Microsystems Engineering (IMTEK) at the University of Freiburg, Germany. Today Dr Zengerle in addition is a director at the Institut für Mikro- und Informationstechnik of the Hahn-Schickard-Gesellschaft (HSG-IMIT) and vice director of the Centre for Biological Signalling Studies (bioss). The research of Dr Zengerle is focused on microfluidics and nanofluidics. He acts also as European editor of the journal “Microfluidics and Nanofluidics”. |
Introduction
Almost 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 papers have been published over the last 10 years on the topic of microfluidics1 and the annual numbers of new publications are still increasing continuously. According to the ISI Web of Science they currently receive around 40000 citations per year (see Fig. 1). Additionally, over 1000 patents referring to microfluidics have been issued in the USA alone.2 Consequently, microfluidics is established very well in academia and industry as a toolbox for the development of new methods and products in life sciences. However, the number of commercial products based on microfluidics is, with few exceptions, still quite low. The question is: will microfluidics remain a toy for academic and industrial research or will it finally make the transition to an end-user product?
000 papers have been published over the last 10 years on the topic of microfluidics1 and the annual numbers of new publications are still increasing continuously. According to the ISI Web of Science they currently receive around 40000 citations per year (see Fig. 1). Additionally, over 1000 patents referring to microfluidics have been issued in the USA alone.2 Consequently, microfluidics is established very well in academia and industry as a toolbox for the development of new methods and products in life sciences. However, the number of commercial products based on microfluidics is, with few exceptions, still quite low. The question is: will microfluidics remain a toy for academic and industrial research or will it finally make the transition to an end-user product?
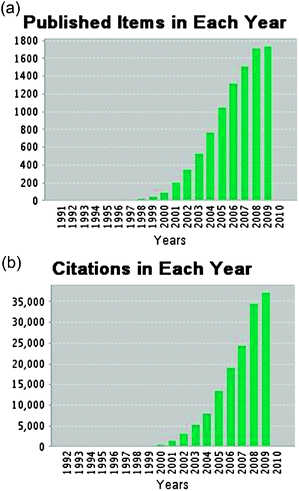 | ||
| Fig. 1 Growth of publications (a) and citations (b) of articles related to microfluidics.1 The data from 2009 are incomplete due to the editorial deadline of this review (November, 24, 2009) but already show a further increase in publications and citations. | ||
Looking into the past, the first microfluidic technology was developed in the early 1950s when efforts to dispense small amounts of liquids in the nanolitre and picolitre range were made, providing the basis for today's ink-jet technology.3 In terms of fluid propulsion within microchannels with sub-millimetre cross sections, the year 1979 set a milestone when a miniaturized gas chromatograph (GC) was realized by Terry et al. on a silicon (Si) wafer.4 The first high-pressure liquid chromatography (HPLC) column microfluidic device, fabricated using Si-Pyrex technology, was published in 1990 by Manz et al.5 By the end of the 1980s and the beginning of the 1990s, several microfluidic structures, such as microvalves6 and micropumps7,8 had been realized by silicon micromachining, providing the basis for automation of complex liquid handling protocols by microfluidic integration.9,10 This was the advent of the newly emerging field of “micro total analysis systems” (μTAS11), also called “lab-on-a-chip”.12
At the same time, much simpler yet very successful microfluidic analysis systems based on capillary liquid transport in wettable fleeces emerged: First very simple “dipsticks” for e.g. pH measurement based on a single fleece paved the way for more complex “test strips” that have been sold as “lateral-flow tests” since the late 80s.13 Examples that are still on the market today are test strips for pregnancy,14 drug abuse,15–17 cardiac markers18 and also upcoming bio-warfare protection.19 Among the devices that completely automated a biochemical analysis by microfluidic integration into one miniature piece of hardware, the test strips became the first devices that obtained a remarkable market share with billions of units sold per year. Yet they remain one of the few microfluidic systems which are sold in high numbers.
Until today, in many cases, the revenue in the field of lab-on-a-chip is created on a business-to-business, rather than a business-to-consumer basis,20 as the vast majority of research in the field only approaches the stage of demonstrations and is not followed up by the development of products for end-users. Among the hurdles for market entry are high initial investments and running fabrication costs.21 Regardless of the 10000 available publications, offering solutions for almost every problem that might occur, the development of a lab-on-a-chip product is still a risky adventure. Quite often the existing microfluidic building blocks are not compatible to or combinable with each other. In addition, in some cases the fabrication technologies do not match or are too expensive. Therefore implementing an application specific assay on a chip is still a very complex and cumbersome task bearing technical risks and with it also financial risks.
Instead of the development of individual and isolated lab-on-a-chip solutions, the constraint of using building blocks to form well-defined microfluidic platforms enables the implementation of biochemical assays in a much better, foreseeable and less risky manner. A microfluidic platform comprises an easily combinable set of microfluidic unit-operations that allows assay miniaturization within a consistent fabrication technology. Hence, the intention of this review is to provide an overview and classification of existing microfluidic platforms that enable the miniaturization, integration, automation and parallelization of (bio-)chemical assays in an easy, consistent and therefore less risky manner. This classification also enables us to categorize the huge amount of literature available in the field of microfluidics into solutions that are compatible to each other and therefore can be combined within a given microfluidic platform.
According to their dominating main liquid propulsion principle, we subdivide microfluidic platforms into 5 groups, namely: capillary, pressure driven, centrifugal, electrokinetic and acoustic systems, as depicted in Fig. 2. Each listed platform within these groups will be discussed. As a guide, we provide a characterization of the respective platforms in Table 1. After providing a short general introduction to the unique properties, requirements, and applications for microfluidic platforms, this review focuses on a detailed discussion of the microfluidic platforms listed in Fig. 2. For each platform, the characterization and the general principle is presented first. After that the microfluidic unit operations as well as application examples are briefly discussed. Finally, each platform is characterized by providing an overview of its strengths and limitations. We conclude by an attempt to provide a selection scheme for microfluidic platforms which is based on platform characteristics and application requirements.
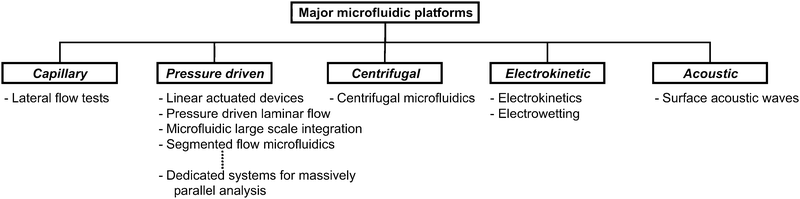 | ||
| Fig. 2 Microfluidic platforms classified according to main liquid propulsion principle. | ||
| Microfluidic platform | Characterization |
|---|---|
| Definition of a microfluidic platform | A microfluidic platform provides a set of fluidic unit operations, which are designed for easy combination within a well-defined fabrication technology. A microfluidic platform paves a generic and consistent way for miniaturization, integration, automation and parallelization of (bio-)chemical processes. |
| Lateral flow tests | In lateral flow tests, also known as test strips (e.g. pregnancy test strip), the liquids are driven by capillary forces. Liquid movement is controlled by the wettability and feature size of the porous or microstructured substrate. All required chemicals are pre-stored within the strip. The readout of a test is typically done optically and is quite often implemented as color change of the detection area that can be seen by the naked eye. |
| Linear actuated devices | Linear actuated devices control liquid movement by mechanical displacement of liquid e.g. by a plunger. Liquid control is mostly limited to a one-dimensional liquid flow in a linear fashion without branches or alternative liquid pathways. Typically liquid calibrants and reaction buffers are pre-stored in pouches. |
| Pressure driven laminar flow | A pressure driven laminar flow platform is characterized by liquid transport mechanisms based on pressure gradients. Typically this leads to hydrodynamically stable laminar flow profiles in microchannels. There is a broad range of different implementations in terms of using external or internal pressure sources such as using syringes, pumps or micropumps, gas expansion principles, pneumatic displacement of membranes, etc. The samples and reagents are processed by injecting them into the chip inlets either batch-wise or in a continuous mode. |
| Microfluidic large scale integration | Microfluidic large scale integration describes a microfluidic channel circuitry with chip-integrated microvalves based on flexible membranes between a liquid-guiding layer and a pneumatic control-channel layer. The microvalves are closed or open corresponding to the pneumatic pressure applied to the control-channels. Just by combining several microvalves more complex units like micropumps, mixers, multiplexers, etc. can be built up with hundreds of units on one single chip. |
| Segmented flow microfluidics | Segmented flow microfluidics describes the principle of using small liquid plugs and/or droplets immersed in a second immiscible continuous phase (gas or liquid) as stable micro-confinements within closed microfluidic channels. Those micro-confinements are in the picolitre to microlitre volume range. They can be transported by pressure gradients and can be merged, split, sorted, and processed without any dispersion in microfluidic channels. |
| Centrifugal microfluidics | In centrifugal microfluidics all processes are controlled by the frequency protocol of a rotating microstructured substrate. The relevant forces for liquid transport are centrifugal force, Euler force, Coriolis force and capillary force. Assays are implemented as a sequence of liquid operations arranged from radially inward positions to radially outward positions. Microfluidic unit operations include metering, switching, aliquoting, etc. |
| Electrokinetics | In electrokinetics platforms microfluidic unit operations are controlled by electric fields acting on electric charges, or electric field gradients acting on electric dipoles. Depending on buffers and/or sample, several electrokinetic effects such as electroosmosis, electrophoresis, dielectrophoresis, and polarization superimpose each other. Electroosmosis can be used to transport the whole liquid bulk while the other effects can be used to separate different types of molecules or particles within the bulk liquid. |
| Electrowetting | Electrowetting platforms use droplets immersed in a second immiscible continuous phase (gas or liquid) as stable micro-confinements. The droplets reside on a hydrophobic surface that contains a one- or two-dimensional array of individually addressable electrodes. The voltage between a droplet and the electrode underneath the droplet defines its wetting behavior. By changing voltages between neighboring electrodes, droplets can be generated, transported, split, merged, and processed. These unit operations are freely programmable for each individual droplet by the end-user enabling online control of an assay. |
| Surface acoustic waves | The surface acoustic waves platform uses droplets residing on a hydrophobic surface in a gaseous environment (air). The microfluidic unit operations are mainly controlled by acoustic shock waves travelling on the surface of the solid support. The shock waves are generated by an arrangement of surrounding sonotrodes, defining the droplet manipulation area. Most of the unit operations such as droplet generation, transport, mixing, etc. are freely programmable. |
| Dedicated systems for massively parallel analysis | Within the category of dedicated systems for massively parallel analysis we discuss specific platforms that do not comply with our definition of a generic microfluidic platform. The characteristics of those platforms are not given by the implementation of the fluidic functions but by the specific way to process up to millions of assays in parallel. Prominent examples are platforms used for gene expression and sequencing such as microarrays, bead-based assays and pyro-sequencing in picowell-plates. |
This review does not claim completeness. It contains examples of microfluidic platforms which were selected as fitting to our platform definition. The review should, however, provide the reader with some orientation in the field and the ability to select platforms with appropriate characteristics on the basis of application-specific requirements.
The framework for microfluidic platforms: unique properties, requirements and applications
Microfluidics as an enabling technology: from classical liquid handling to single-cell handling
A number of classical, macroscopic liquid handling systems for performing analytical and diagnostic assays have been in use for many decades. Examples are petri dishes, culture bottles and microtitre plates (also called microplates). Petri dishes were first described in 188722 and culture bottles23 have been in use since around 1850. Since roughly 60 years ago, they have been manufactured as plastic disposables. In comparison, microtiter plates are quite “modern,” having first been described in 1951.24 Based on these standards, highly automated liquid handling solutions have been developed within the last few decades (“pipetting robots”) and are the current “gold standard” for automated sample processing in pharma and diagnostics. They offer a huge potential for many applications since they are very flexible as well as freely programmable. Microfluidic platforms have to compete against these established systems by offering new opportunities. Expectations often quoted in this context are:25• Portability/wearability
• Higher sensitivity
• Lower cost per test
• Shorter time-to-result
• Less laboratory space consumption
Additionally, scaling effects lead to new phenomena and permit entirely new applications that are not accessible to classical liquid handling platforms, such as:
• Well-defined, laminar flow
• Controllable diffusion enabling defined concentration gradients on the length scales of single-cells
• Surface forces dominate over gravitational forces
• Liquid compartments of the size of a single cell or smaller
• High-speed serial processing (at single cell level)
• High degree of parallelization (up to around 106)
In the following, the effects and phenomena leading to the above-mentioned expectations and the potential for new applications will be outlined briefly.
It is obvious that the amount of reagent consumption can be decreased significantly by scaling down the assay volume. Additionally, by reducing the footprint of each individual test, a higher degree of parallelization can be achieved in a limited laboratory space. A prime example for microfluidic tests with minimal reagent consumption are parallel reactions in hundreds of thousands of individual wells with picolitre-volumes,26 which took genome sequencing to a new level27 hardly achievable by classical liquid handling platforms.
With decreasing length scales, surface phenomena (e.g.capillary forces, surface charges, etc.) become increasingly dominant over volume phenomena. This permits purely passive liquid actuation based on capillary forces used in the popular lateral flow assays also know as capillary test strips. Another effect is the onset of laminar flow at low Reynolds numbers in small channels. This enables the creation of well-defined and stable liquid–liquid interfaces down to cellular dimensions. Therefore, large concentration gradients can be applied and the effects monitored at the single cell level28 (Fig. 3). In summary, laminar flow conditions and controlled diffusion enable temporally and spatially highly resolved reactions with little reagent consumption.
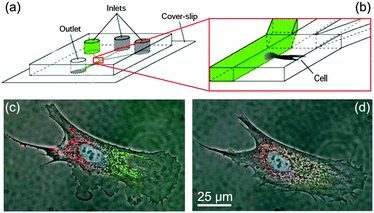 | ||
| Fig. 3 Concept of differential manipulation in a single bovine capillary endothelial cell using multiple laminar flows. (a, b), Chip layout: 300 μm × 50 μm channels are used to create laminar interfaces between liquids from different inlets. (c) Fluorescence image of a cell locally exposed to red and green fluorophores in a laminar flow. (d) Migration of fluorophores over time (scale bars, 25 μm). This shows the high potential for accurate spatial control and separation of liquids achievable in microfluidic laminar flows. Adapted by permission from Macmillan Publishers Ltd: Nature,28 copyright 2001. | ||
A different paradigm using the possibility of controlling interfaces in microfluidic applications is the concept of droplet-based microfluidics, also called “digital microfluidics”.29 The on-demand generation of liquid micro-cavities either in air or a second immiscible liquid enables the manipulation of small quantities of reagents down to single cells with high throughput.30 Control and manipulation of such droplets can be achieved by another favorable aspect of the high surface-to-volume ratio in microfluidics: the possibility to control the liquid flow by electrically induced forces or electrowetting.31 Having the huge background of theoretical and practical knowledge in electronics, this is obviously a desirable property. Additional helpful properties of small assay volumes are fast thermal relaxation and low power consumption for liquid manipulation and thermal control. This can speed up assays that require thermocycling, such as PCR, which was realized in numerous microfluidic applications.32
This short summary shows that there is the potential for many novel applications and improvements over the state-of-the-art within the above-mentioned criteria of sensitivity, cost, time, and size. However, despite a myriad of publications about microfluidic components, principles and applications, only a limited number of successful products with a relevant market share have emerged from this field so far. In the next chapter, we will outline hurdles and present emerging paradigm changes that will influence future research in microfluidics.
The need for the microfluidic platform approach
Definition of a microfluidic platform: A microfluidic platform provides a set of fluidic unit operations, which are designed for easy combination within a well-defined fabrication technology. A microfluidic platform paves a generic and consistent way for miniaturization, integration, automation and parallelization of (bio-)chemical processes.
In the last two decades, thousands of researchers spent a huge amount of time to develop micropumps,33–36 microvalves,37 micromixers,38,39 and microfluidic liquid handling devices in general. However, a consistent fabrication and interfacing technology as one prerequisite for the efficient development of lab-on-a-chip systems is very often still missing. This missing link can only be closed by establishing a microfluidic platform approach which allows the fast and easy implementation of (bio-)chemical protocols based on common building blocks. The idea follows the tremendous impact of platforms in the application-specific integrated circuit (ASIC) industry in microelectronics, where validated elements and processes enabled faster design and cheaper fabrication of electronic circuitries.
Conveying this to the microfluidic platform approach, a set of validated microfluidic elements is required, each able to perform a certain basic fluid handling step or unit operation. Such basic unit operations are building blocks of laboratory protocols and comprise fluid transport, fluid metering, fluid mixing, valving, separation or concentration of molecules or particles (see Table 2) and others. Every microfluidic platform should offer an adequate number of microfluidic unit operations that can be easily combined and thereby enable easy implementation of application-specific assays within that given platform.
| Microfluidic unit operations | Fabrication technology |
|---|---|
| • Fluid transport | • Validated manufacturing technology for the whole set of fluidic unit operations (prototyping and mass fabrication) |
| • Fluid metering | |
| • Fluid valving | |
| • Fluid mixing | |
| • Separation | |
| • Accumulation/amplification | • Seamless integration of different elements |
| • Reagent storage & release | … preferable in a monolithic way |
| • Incubation | … or by a well defined easy packaging technique |
| • … |
This concept, however, does not imply that every microfluidic platform needs to provide a complete set of all the unit operations listed in Table 2. It is much more important that the different elements are connectable, ideally in a monolithically integrated way or at least by a well defined, ready-to-use interconnection and packaging process. Therefore at least one validated fabrication technology is required to realize complete microfluidic solutions from the individual elements within a microfluidic platform.
Market requirements and platform selection criteria
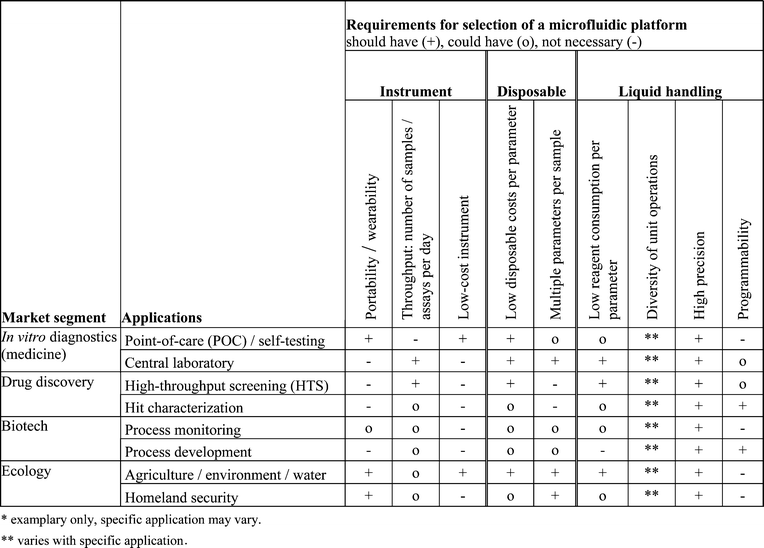
The requirements on microfluidic platforms differ greatly between different market segments. Following a roadmap on microfluidics for life sciences,40 the four key market segments for microfluidic lab-on-a-chip applications are, according to their market size: in vitro diagnostics, drug discovery, biotechnology, and ecology.The largest market segment, in vitro diagnostics, can be subdivided into point-of-care testing (e.g. for self-testing in diabetes monitoring or cardiac marker testing in emergency medicine) and central laboratory-based testing (e.g. core laboratory in a hospital). Especially the self- and point-of-care testing segments offer huge potential for microfluidics, since portability and/or wearability is an important requirement.
Drug discovery in the pharmaceutical industry is the second largest segment. Here, enormous effort is undertaken to identify new promising drug candidates in so called high-throughput screening (HTS) or massively parallel analysis.41 After screening promising candidates, so-called hits have to be validated and characterized (hit characterization). In this context cell-based assays have received increasing interest over recent years.42,43 These assays often require the handling of single cells, which becomes possible using microfluidic approaches. This market segment requires high sample throughput and low costs per test.
The third segment is the biotech market with fermentation-based production (e.g. for biopharmaceuticals or food). This industry shows a great demand for on-line process monitoring and analyses in the field of process development. Here, low sample volumes and flexibility (programmability) are important factors.
Ecology is another market segment, comprising the field of agricultural- and water-analysis, either as on-site spot tests or as continuous monitoring. Included are also applications related to homeland security, e.g. the detection of agents that pose biological threats. This market benefits from portable systems with preferably multi-parameter capabilities.
These diverse fields of applications are associated with a number of analytical and diagnostic tasks. This outlines the field for the microfluidic technology, which has to measure itself against the state-of-the-art in performance and costs. Table 3 gives an overview on some important requirements of the different market segments and application examples, with respect to the following selection criteria:
• Portability/wearability: miniaturized, hand-held device with low energy consumption
• Throughput: number of samples/assays per day
• Cost of instrument: investment costs of the instrument (“reader”)
• Cost of disposables: defining the costs per assay (together with reagent consumption)
• Number of parameters per sample: number of different parameters to be analyzed per sample
• Low reagent consumption: amount of sample and/or reagents required per assay
• Diversity of unit operations: the variety/completeness of laboratory operations that can be realized
• Precision: the volume and time resolution that is possible
• Programmability: the flexibility to adapt liquid handling protocols without fabricating a new chip
These criteria will be discussed for each of the platforms described in this review.
Biochemical applications for microfluidic platforms
Here, a short overview of the fields of applications that are typically addressed by microfluidic platforms is presented.A first field of application is biotransformation, the breakdown and generation of molecules and products by the help of enzymes, bacteria, or eukaryotic cell cultures. This comprises fermentation, the break down and re-assembly of molecules (e.g. fermentation of sugar to alcohol), and (bio)synthesis the build-up of complex molecules (e.g. antibiotics, insulin, interferon, steroids). Especially in the field of process development challenges are to handle a large number of different liquids under controlled conditions such as temperature or pH, in combination with precise liquid control down to nL or even pL volumes. Some examples of microfluidic liquid handling platforms are given for fermentation in micro bioreactors,44–51 the biosynthesis of radiopharmaceuticals,52 and antibody screening, phage- and ribosome-display technologies.53,54
Another major field of application is analytics. The analysed molecule (analyte) can be from a variety of biomolecules, including proteins and nucleic acids. Here, the main requirements are effective mixing strategies and highly precise liquid metering and liquid handling which are needed to get accurate quantitative results. Also, automation and portability/wearability combined with a large set of unit operations for the implementation of complex analytical protocols are required.
As an emerging field, cellular assays are the most challenging format, since the cells have to be constantly kept in an adequate surrounding to maintain their viability and activity (control of pH, O2, CO2, nutrition, etc.). Cellular tests are useful to assess the effect of new pharmaceutical entities at different dosing concentrations on toxicity, mutagenicity, bioavailability and unwanted side effects. The most exciting prospect is the establishment of assays with single-cell analyses.55,56 Requirements on cellular assays include high-throughput solutions as well as a low reagent consumption per test.
After this short overview, the next chapter will summarize the liquid handling challenges that arise from the different liquids associated with these fields of applications.
Requirements on microfluidic platforms related to liquids with biochemical content
Performing microfluidics with pure water cannot be compared to the challenge of developing a microfluidic platform for handling of liquids with biochemical content. Here, a large variety of changing liquid properties needs to be considered, ranging from surface tension, non-Newtonian viscosities and the contact angle on a certain surface. In addition, when handling biological samples, such as blood, an inter-sample variation, e.g. due to physiological differences between patients, has to be managed by the microfluidic system. In the following, a short summary of typical sample materials and their interactions with the microfluidic substrate is provided. Also, strategies to prevent unfavorable interactions are outlined.In general, microfluidic substrates should be inert against the expected sample and assay reagents which might comprise organic or inorganic solvents or extreme pH values.57 Likewise, the sample must not be affected by the microfluidic substrate in any way that could influence the analytical result. For example, nucleic acids are critical molecules because of their negative charge and tendency to adhere to charged surfaces such as metal oxides. Similar problems occur with proteins or peptides which exist in a variety of electrical charges, molecular sizes, and physical properties. In addition to possible adsorption onto the surfaces, the catalytic activity of enzymatic proteins can be reduced by interaction with the substrate.58–61 A general counter-measure against the interaction of biomolecules and microfluidic substrates is to block the substrates with another suitable biomolecule which is added in excess. For instance, bovine serum albumin (BSA) adsorbs to nearly any surface thus passivating it.62,63 Another significant challenge in microfluidic production technology is to maintain the activity of proteins during processes such as thermal bonding64,65 or UV curing steps. In addition, the long-term stability of pre-stored dry reagents is required, hence materials with low vapor transition rates have to be selected.
Experience shows that this set of challenges needs to be considered at the very beginning of a fluidic design, since the listed problems can jeopardize the functionality of the whole system if addressed too late.
Lateral flow tests
Characterization of lateral flow tests
In lateral flow tests, also known as test strips (e.g. pregnancy test strip), the liquids are driven by capillary forces. Liquid movement is controlled by the wettability and feature size of the porous or microstructured substrate. All required chemicals are pre-stored within the strip. The readout of a test is typically done optically and is quite often implemented as a color change in the detection area that can be seen by the naked eye.General principle
The first immunoassay performed in a capillary driven system was reported in 1978.66 Based on this technique, the commonly known “over-the-counter pregnancy test” was introduced into the market in the middle of the ‘80s. Today, this microfluidic platform is commonly designated as a “lateral flow test (LAT)”.13 Other terms are “test strip”, “immunochromatographic strip”, “immunocapillary tests” or “sol particle immunoassay (SPIA)”.67 Astonishingly, hardly any publications from a microfluidic point of view or in terms of material classification exist, and apparently many “company secrets” are kept unpublished.68The “standard LAT” consists of an inlet port and a detection window (Fig. 4(a)). The core comprises several wettable materials providing all biochemicals for the test and enough capillary capacity to wick the sample through the whole strip. The sample is introduced into the device through the inlet into a sample pad (Fig. 4(b)), which holds back contaminations and dust. Through capillary action, the sample is transported into the conjugate pad, where antibodies conjugated onto a signal-generating particle are rehydrated and bind to the antigens in the sample (Fig. 4(c)). This binding reaction continues as the sample flows in the incubation and detection pad. On the test line a second type of antibody catches the particles coated with antigens, while a third type of antibody catches particles which did not bind to an analyte on the control line. The control line shows a successfully processed test while the detection line shows the presence or absence of a specific analyte (Fig. 4(d)). Typically the result becomes visible after 2 to 15 min.
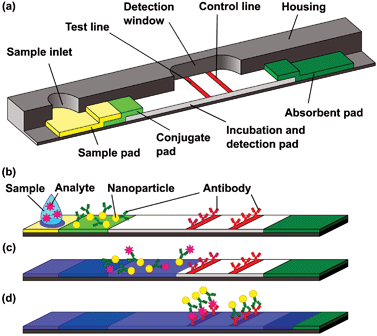 | ||
| Fig. 4 Schematic design of a lateral flow test (according to ref. 68), (a) Sample pad (sample inlet and filtering), conjugate pad (reactive agents and detection molecules), incubation and detection zone with test and control lines (analyte detection and functionality test) and final absorbent pad (liquid actuation). (b) Start of assay by adding liquid sample. (c) Antibodies conjugated to colored nanoparticles bind the antigen. (d) Particles with antigens bind to test line (positive result), particles w/o antigens bind to the control line (proof of validity). | ||
Over the last decades, LAT transformed from a simply constructed device into a more and more sophisticated high-tech platform with internal calibrations and quantitative readout by a hand-held reader (Fig. 5).69
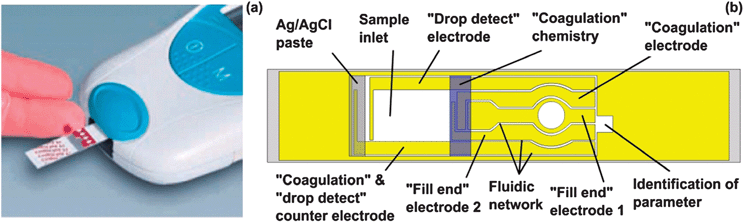 | ||
| Fig. 5 LAT for blood coagulation with hand-held readout according to Cosmi et al.69,73 (image (a) courtesy of Roche Diagnostics). (a) Loading of blood. (b) The blood flows from the inlet into the fluidic network rehydrating the coagulation chemistry. The “drop detect” electrodes detect whether blood is applied and measure the incubation times. Several capillaries are filled and the filling is monitored with according electrodes. A Ag/AgCl electrode is used as standard electrode for calibration and analysis. Finally the analyte gets quantified by optical or electrochemical detection. | ||
Unit operations
The different pads in the test strip represent different functions such as loading, reagent pre-storage, reaction, detection, absorption and liquid actuation. The characteristic unit operation of LATs is the passive liquid transportvia capillary forces, acting in the capillaries of a fleece, a microstructured surface, or a single capillary. The absorption volume of an absorption pad defines how much sample is wicked through the strip and provides metering of the sample.68 The sample pad usually consists of cellulose or cross-linked silica and is used for filtering of particles and cells as well as separating the analyte from undesired or interfering molecules, which is absorbed in the pad.70 The conjugation pad is made of cross-linked silica and is used as dry-reagent storage for antibodies specific to the antigen conjugated to the signal generating particle. The conjugates are typically colored or fluorescent nanoparticles with sizes up to 800 nm, which flow without obstruction through the fleeces together with the sample. Most often colloidal gold19 or latex71 and more rarely carbon, selenium, quantum dots, or liposomes72 are the choice of nanoparticles.The length, material (mainly nitro-cellulose) and pore-size (50 nm to 12 μm, depending on the applied nanoparticles) of the detection and incubation pad define the incubation time.68 The detection and enrichment of the conjugates is achieved on the antibody-bearing lines. Analyte detection is performed on the test line and proof of assay validity on the control line. The readout is typically done by naked eye for absence (1 colored line) or presence (2 colored lines) of a minimum analyte amount. A readout with a reader enables quantitative analyte detection.69,73 For multi-analyte detection68 or semi-quantitative setups74 several test lines are applied.
Within the last few years, new LAT designs have been developed in combination with the device-based readout in hand-held systems. Here a complex capillary channel network provides the liquid actuation (Fig. 5). Antibodies conjugated to nanoparticles or special enzymes are pre-stored at the inlet. The incubation time is defined by the filling time of the capillary network. Typically, readout is done quantitatively by fluorescence or electrochemical detection. The time-to-result is usually several seconds. Blood glucose or coagulation monitoring are the most common applications for such quantitative readouts.69 To accommodate aging, batch-to-batch variations and sample differences, and also to achieve higher precision and yield of the assay, several internal controls and calibrations are automatically performed during analysis by the readout device.
Application examples
Lateral flow tests were among the first successfully commercialized microfluidic products. A huge amount of assays have been developed on the capillary test strip platform during the past 30 years.75 Today, they serve a wide field of applications, including health biomarkers (pregnancy,13,76 heart attack,70 blood glucose,77 metabolic disorders78), small molecules (drug abuse,16 toxins,79 antibiotics80), infectious agents (anthrax,81salmonella,82 viruses83), immunodiagnostics,84 RNA applications,81 and even whole bacteria.85 Some of the more recent designs and publications even show the detection of DNA83 without the need of amplification by PCR, which would open yet another vast field of new applications. The first trials for massively parallel screening in combination with microarrays were made in lateral flow tests.70,81Strengths and limitations
The fact that 6 billion glucose test strips were sold in 200786 already indicates that the LAT may be seen as a gold-standard microfluidic platform in terms of cost, handling simplicity, robustness, market presence and the number of implemented lab-on-a-chip applications.68 The amount of sample and reagent consumption are moderate, and the concept is mainly used for qualitative or semi-quantitative assays. Especially the complete disposable test carriers with direct visual readout, easy handling, and a time-to-result between seconds and several minutes are predestined for untrained users.The simplicity of the test strip is also its major drawback. Assay protocols within capillary driven systems follow a fixed process scheme with a limited number of unit operations, imprinted in the microfluidic channel design itself. Highly precise liquid handling and metering is also extremely challenging.68 The dependency of the purely capillary liquid actuation on the sample properties can also be a major problem, leading to false positive or negative results14 or decreased precision. New designs allow applications with quantitative analysis, but require a readout device (mainly hand-held).69,73 High-throughput or screening applications are possible, but quite difficult to implement.
In total, the lateral flow test is a well established platform with a large but limited field of applications and consequently a benchmark for the home-care and in vitro diagnostics (IVD) sector in terms of cost per assay and simplicity.
Linear actuated devices
Characterization of linear actuated devices
Linear actuated devices control liquid movement by mechanical displacement of liquid e.g. by a plunger. Liquid control is mostly limited to a one-dimensional liquid flow in a linear fashion without branches or alternative liquid pathways. Typically liquid calibrants and reaction buffers are pre-stored in pouches.General principle
One of the first examples of a linear actuated device was the i-STAT® for quantitative bedside testing, introduced in the early 1990s by Abbott Point of Care Inc., NJ, USA. It relied on active liquid actuation by displacement.87 Compared to lateral flow tests, this principle was one step ahead in result quantification and possible applications, but also in complexity of the processing device and disposable test carrier.The characteristic actuation principle of the linear actuated platform is the mechanical linear propulsion of liquids with no branching. Normally, the liquid actuation is performed by a plunger which presses on a flexible pouch, displacing its content. Another common attribute is the pre-storage of all required reagents (liquid and dry) on the disposable test carrier (cartridge). Systems based on this platform thus offer fully integrated sample-to-result processing in a relatively short time.
Unit operations
Basically, the linear actuated platform relies on only two unit operations: liquid transport and reagent storage. Liquid transport is achieved by mechanical displacement (e.g. with a plunger). By pressing on flexible compartments of the disposable, the liquid can be transported between reservoirs.87 Alternatively, a weakly bonded connection to an adjacent reservoir can be disrupted, or the connection to a neighbouring cavity selectively blocked.88 Liquid reagent storage can easily be implemented by integrating pouches into the cartridge. Mixing can also be realized on the linear actuated platform by moving liquids between neighbouring reservoirs.88Application examples
One example of a linear actuated device is of course the previously mentioned i-STAT® analyzer from Abbott Point-of-Care.89 Using different disposable cartridges, several blood parameters (blood gases, electrolytes, coagulation, cardiac markers, and hematology) can be determined with the same portable hand-held analyzer for automated sample processing and readout (Fig. 6(a)). Since only the disposable polymer cartridge is contaminated with the blood sample and thus has to be disposed after performing the diagnostic assay, the analyzer device itself is reusable. Typical response times of the system are in the order of a few minutes.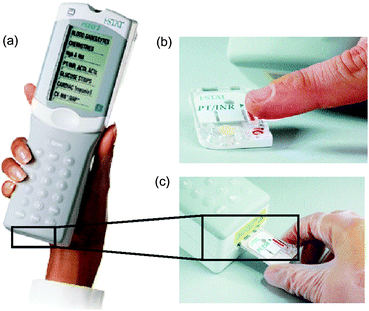 | ||
| Fig. 6 Images and handling procedure of the i-STAT® analyzer. (a) Photograph depicting the portable i-STAT® analyzer for clinical blood tests.89 (b) Depending on the blood parameters to be measured, a certain disposable cartridge is filled with blood by capillary forces from the finger tip and (c) afterwards loaded into the analyzer for assay processing and readout (images courtesy of Abbott Point of Care Inc., NJ, USA). | ||
The system features an integrated calibration solution that is pre-stored in the disposable. The analysis process takes only a few steps: As depicted in Fig. 6, the blood sample (a few drops) is filled into the cartridge by capillary forces (b), and placed into the analyzer (c). First, the calibrant solution is released and provides the baseline for an array of thin-film electrodes integrated in the disposable. Then the sample is pushed into the measuring chamber and displaces the calibrant. Thereby, the blood parameters which can be determined by the sensor array of the specific disposable are measured and presented at the integrated display of the hand-held analyzer. Several studies showed good agreement between laboratory results and this POC-system.87,90,91
A second example is the lab-in-a-tube (Liat™) analyzer from IQuum.92 This bench-top device with disposable test tubes contains all necessary reagents for amplification-based nucleic acid tests. It integrates sample preparation, amplification and detection and is a fully integrated sample-to-result platform with response times between 30 and 60 min. Handling of the platform requires only a few steps: The sample (e.g. 10 μL of whole blood) is collected in the collection tube that is integrated into the disposable, the barcode on the disposable is scanned, and the tube is then inserted into the analyzer. The disposable features compartmentalized chambers in a tube which contain different reagents and can be connected via peelable seals (Fig. 7). Liquid control is performed by actuators that compress the compartments, displacing the liquid into adjacent chambers.88 Sample preparation includes a nucleic acid purification step: magnetic beads serve as solid nucleic acid binding phase and are controlled by a built-in magnet. For nucleic acid amplification, compartments can be heated and the liquid is transferred between two different temperature zones thus cycling the sample. The system is capable of real-time fluorescence readout.
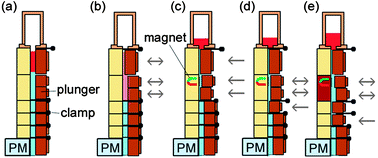 | ||
| Fig. 7 Functional principle and processing steps in a nucleic acid test in the lab-in-a-tube analyzer according to Chen et al.88 The disposable contains pouches with reagents (light blue) which are actuated by plungers while clamps open and close fluidic connections to adjacent pouches. (a) Sample is inserted (red). (b) Sample is mixed with pre-stored chemicals containing magnetic capture-beads. (c) Unwanted sample components are moved to a waste reservoir while the capture-beads are held in place by a magnet. (d, e) Further processing steps allow sequential release of additional (washing) buffers and heating steps (red block) for lysis and thermocycling demands. The system allows optical readout by a photometer (PM). | ||
Strengths and limitations
The presented commercially available examples show that automation and time-reduction by microfluidic systems with active processing devices can indeed be achieved in a market-relevant context. The potential of the linear actuated device platform certainly lies in its simplicity and the ability for long-term liquid reagent storage. The presented application examples are portable and show a high degree of assay integration, requiring no external sample pre- or post-processing steps. Typical liquid (sample) volumes handled on the platform are in the range of 10–100 μL, which is adequate for point-of-care diagnostic applications (capillary blood from finger tip). While disposables can generally be mass-produced, these can become somewhat expensive due to the integration of sensors (i-STAT®) and liquid reagents (i-STAT® and Liat™). Time-to-result varies between minutes and approximately one hour, depending on the assay.The advantage of full integration with pre-stored reagents comes at the price of an imprinted protocol that cannot be changed for a specific test carrier. The number of unit operations is somewhat limited, in particular separation, switching, and aliquoting as well as precise metering are difficult to realize. This hinders the implementation of more complex assays and laboratory protocols in linear actuated systems, such as integrated genotyping with a plurality of genetic markers or multiparameter assays.
Pressure driven laminar flow
Characterization of pressure driven laminar flow
A pressure driven laminar flow platform is characterized by liquid transport mechanisms based on pressure gradients. Typically this leads to hydrodynamically stable laminar flow profiles in microchannels. There are a broad range of different implementations in terms of using external or internal pressure sources such as using syringes, pumps or micropumps, gas expansion principles, pneumatic displacement of membranes, etc. The samples and reagents are processed by injecting them into the chip inlets either batch-wise or in a continuous mode.General principle
As mentioned earlier, liquid flow in microchannels is typically strictly laminar over a wide range of flow rates and channel dimensions. Pressure driven laminar flow offers several opportunities for assay implementation:• Predictable velocity profiles
• Controllable diffusion mixing
• Stable phase arrangements, e.g. in co-flowing streams
These advantages have been utilized for several lab-on-a-chip applications in the past. Probably the oldest example is the so-called “hydrodynamic focusing” technology,93 used to align cells in continuous flow for analysis and sorting in flow cytometry.94,95 Today, many technologies still use laminar flow effects for particle counting96 or separation.97–101 However, pressure driven laminar flow can also be utilized to implement other (bio-)chemical assays for lab-on-a-chip applications as described within this section. In particular, nucleic acid-based diagnostic systems received a great deal of interest in the last decade, since the first introduction of a combined microfluidic PCR and capillary electrophoresis in 1996 by Woolley et al.102
Unit operations
The basic unit operation on the pressure driven laminar flow platform is the contacting of at least two liquid streams at a microfluidic channel junction (see Fig. 8). This leads to controlled diffusional mixing at the phase interface, e.g. for initiation of a (bio-)chemical reaction.103 It can also be applied for the lateral focusing of micro-objects like particles or cells in the channel.93 The required “flow focusing” channel network consists of one central and two symmetric side channels, connected at a junction to form a common outlet channel. By varying the ratio of the flow rates, the lateral width of the central streamline within the common outlet channel can be adjusted very accurately. Consequently, micro-objects suspended in the liquid flowing through the central channel are focused and aligned to this well-defined streamline position. If the available duration for a (bio-)chemical reaction needs to be limited, the contacted liquid streams can again be separated further downstream as shown in ref. 103.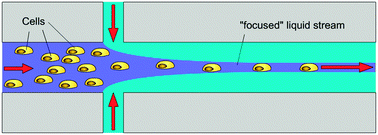 | ||
| Fig. 8 Contacting on the laminar flow platform. Three different liquid streams are symmetrically contacted at an intersection point. This microfluidic structure is also referred to as a “flow focusing structure”.93 | ||
For the separation of micro-objects like living cells or micro-beads from a liquid stream, several technologies have been presented relying either on geometrical barriers,103 or magnetic forces.104,105Sorting of micro-objects, i.e. the selective separation based on size or any other feature, was implemented using magnetic forces,106,107 acoustic principles,108 dielectrophoresis,109 or hydrodynamic principles97–99,110 on the pressure driven laminar flow platform. The common principle of all these technologies is a force acting selectively on the suspended micro-objects (particles or cells), while the liquid stream stays more or less unaffected.
A great number of valving principles exist on the pressure driven laminar flow platform, summarized in a review by Oh and Ahn.37 Active as well as passive solutions have been presented. However, no standards have emerged so far, so the choice and implementation of valves remains a difficulty on this platform. A possible approach is to transfer the valving functionality off-chip,111 thus decreasing the complexity and cost of the disposable.
Application examples
One recently established technology on the pressure driven laminar flow platform is so called “phase transfer magnetophoresis (PTM)”.104 Magnetic microparticles flowing through a microfluidic channel network are attracted by a rotating off-chip permanent magnet, and can consequently be transferred between different co-flowing liquid streams. As a first application, DNA purification with magnetic beads was successfully demonstrated with a yield of approximately 25%104 (first prototype). Thus, this system provides continuous DNA-extraction capability which could serve as an automated sample preparation step for flow-through PCR, in e.g. bioprocess monitoring (of fermentation) applications.Other microfluidic applications based on the manipulation of magnetic microparticles with external permanent magnets have been shown. One example is the free-flow magnetophoresis,106,107 which can be utilized to sort magnetic microparticles by size.
A large number of microfluidically automated components for batch-wise nucleic acid diagnostics based on pressure driven laminar flow chips have been published and summed up in several reviews.32,112,113 However, a totally integrated system remains a challenge, since the integration of sample preparation proved difficult,113 although it seems to be in reach, as the next two examples show.
Easley et al. showed integrated DNA purification, PCR, electrophoretic separation and detection of pathogens in less than 30 min.114 The assay was performed on a pressure driven four layer glass/PDMS chip with elastomeric valves. Temperature cycling for PCR was achieved by IR radiation. Only the sample lysis step was not integrated in the microfluidic chip. Detection of Bacillus anthracis from infected mice and Bordetella pertussis from a clinical sample was successfully demonstrated.
An integrated μTAS system for the detection of bacteria including lysis, DNA purification, PCR and fluorescence readout has also been published recently.111 A microfluidic plastic chip with integrated porous polymer monoliths and silica particles for lysis and nucleic acid isolation was used for detection (Fig. 9). A custom-made base device provided liquid actuation and off-chip valving by stopping liquid flow from the exits of the chip, utilizing the incompressibility of liquids. Detection of 1.25 × 106 cells of Bacillus subtilis was demonstrated with all assay steps performed on-chip.
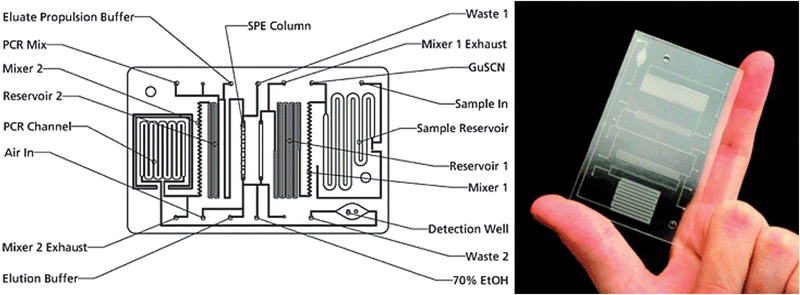 | ||
| Fig. 9 Chip for integrated detection of bacteria including lysis, DNA isolation and PCR published by Sauer-Budge et al.111 | ||
Strengths and limitations
One strength of the platform lies in its potential for continuous processing of samples. Continuous sample processing is of utmost importance for online monitoring of clinical parameters, process control in fermentation, water quality control or cell sorting. Typically one or a few parameters are monitored. The application examples showed one system capable of continuous DNA extraction as well as other implementations that integrated complex batch-wise protocols such as nucleic acid analysis. The platform is in principle compatible with polymer mass-production technologies such as injection molding, enabling inexpensive disposable microfluidic chips.A difficulty of the platform is the necessity to connect the pressure source to the (disposable) chip, which decreases the portability and requires additional manual steps. Another challenge is the Taylor dispersion115 of streamwise dispersed samples which can make it hard to accurately track analyte concentrations. Unit operations on the platform are optimized for mixing and separation processes and somewhat limited in other aspects such as aliquoting.
Microfluidic large scale integration
Characterization of microfluidic large scale integration
Microfluidic large scale integration describes a microfluidic channel circuitry with chip-integrated microvalves based on flexible membranes between a liquid-guiding layer and a pneumatic control-channel layer. The microvalves are closed or open corresponding to the pneumatic pressure applied to the control-channels. Just by combining several microvalves more complex units like micropumps, mixers, multiplexers, etc. can be built up with hundreds of units on one single chip.General principle
The microfluidic large scale integration (LSI) platform arose in 1993.116 At the same time, a novel fabrication technology for microfluidic channels, called soft lithography made its appearance. Soft lithography is based on the use of elastomeric stamps, molds and conformable photomasks to fabricate and replicate microstructures.117 Using this technology, the monolithic fabrication of all necessary fluidic components within one single elastomer material (polydimethylsiloxane, PDMS) became possible, similar to the silicon-based technology in microelectronics. PDMS, also known as silicone elastomer, is an inexpensive material offering several advantages compared to silicon or glass. It is a cheap, rubber-like elastomer with good optical transparency and biocompatibility. A detailed review on the use of PDMS for different fields of applications can be found in ref. 118.The strength of the technology became obvious, when Stephen Quake's group expanded the technology towards the multilayer soft-lithography process, MSL.119 With this technology, several layers of PDMS can be hermetically bonded on top of each other resulting in a monolithic, multilayer PDMS structure. This enables the fabrication of microfluidic chips with densely integrated microvalves, pumps and other functional elements. Today, this technology is pushed forward by the company Fluidigm Corp., CA, USA.
Unit operations
Based on the high elasticity of PDMS, the elementary microfluidic unit operation is a valve which is typically made of a planar glass substrate and two layers of PDMS on top of each other. One of the two elastomer layers contains the fluidic ducts while the other elastomer layer features pneumatic control channels. To realize a microfluidic valve, a pneumatic control channel crosses a fluidic duct as depicted in Fig. 10(a). A pressure p applied to the control channel squeezes the elastomer into the lower layer, where it blocks the liquid flow. Because of the small size of this valve, on the order of 100 × 100 μm2, a single integrated fluidic circuit can accommodate thousands of valves. Comparable to developments in microelectronics, this approach is called “microfluidic large scale integration” (LSI).120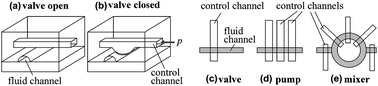 | ||
| Fig. 10 Realization of the main unit operations on the multilayer PDMS-based LSI platform.121 The NanoFlex™ valve (a) can be closed (b) by applying a pressure p to the control channel. Therewith, microfluidic valves (c), peristaltic pumps (d) and mixing structures (e) can be designed. | ||
The valve technology called NanoFlex™ (Fluidigm) is the core technology of the complete platform. For example, by placing two such valves at the two arms of a T-shaped channel a fluidic switch for the routing of liquid flows between several adjacent channels can be realized. Liquid transport within the fluid channels can be accomplished by external pumps while the PDMS multilayer device merely works passively as integrated valves, or an integrated pumping mechanism can be achieved by combining several micro-valves and actuating them in a peristaltic sequence (Fig. 10(d)).
Metering of liquid volumes can be achieved by crossed fluid channels and a set of microvalves. Therefore, the liquid is initially loaded into a certain fluid channel and afterwards segmented into separated liquid compartments by pressurizing the control channel.
Also mixing can be realized using the above described pumping mechanism by the subsequent injection of the liquids into a fluidic loop (Fig. 10(e)) through the left inlet (right outlet valve is closed). Afterwards, the inlet and outlet valves are closed and the three control channels on the orbit of the mixing loop are displaced with a peristaltic actuation scheme leading to a circulation of the mixture within the loop.122 Thereby the liquids are mixed and can be flushed out of the mixer by a washing liquid afterwards. Using this mixing scheme, the increase of reaction kinetics by nearly two orders of magnitude has been demonstrated in surface binding assays.123
However, the key feature to tap the full potential of the large scale integration approach is the multiplexing technology allowing for the control of N fluid channels with only 2 log2N control channels. Based on this principle, a microfluidic storage device with 1000 independent compartments of approximately 250 pL volume and 3574 microvalves has been demonstrated.120
Application examples
One application example on the microfluidic LSI platform is the extraction of nucleic acids (NA) from a small amount of cells124,125 for cell-based assays. For the extraction of NA from a cell suspension, the cell membrane has to be destroyed first (chemical lysis of the cell). Afterwards, the NA are specifically separated from the residual cell components using a solid phase extraction method based on a NA affinity column (paramagnetic beads). This extraction protocol is completely implemented on the microfluidic platform using the basic unit operations for valving, metering, mixing and switching of liquids. Measurable amounts of mRNA were extracted in an automated fashion from as little as a single mammalian cell and recovered from the chip.124 Based on this technology, the development of a nucleic acid processor for complete single cell analysis is under way.126–128Also many other applications have been implemented on the LSI platform over the last few years: protein crystallization,129 immunoassays,130 automated culturing of cells131 or multicellular organisms132 and DNA synthesizing.133
From a commercial perspective, Fluidigm Corp. has launched three different products based on the large scale integration platform within the last years: the BioMarkTM technology for molecular biology (e.g. TaqMan® assay), the TOPAZ® system for protein crystallography, and the Fluidigm® EP1 system for genetic analysis. The EP1 system in particular, bears great potential for high-throughput screening applications such as sequencing.134 multiparallel PCR,135 single-cell analysis,136 siRNA-137 or antibody-screening,138 kinase-139 or expression-profiling.140
Strengths and limitations
The microfluidic LSI platform certainly has the potential to become one of the most versatile microfluidic platforms especially for high-throughput applications. It is a flexible and configurable technology which stands out by its suitability for large scale integration. The PDMS fabrication technology is comparably cheap and robust, and thus suitable to fabricate disposables. Reconfigured layouts can be assembled from a small set of validated unit operations and design iteration periods for new chips are in the order of days. Some of the system functions are hardware defined by the fluidic circuitry but others like process sequences can easily be programmed externally.Limitations of the platform are related to the material properties of PDMS: for example, chemicals which the elastomer is not inert to cannot be processed, and elevated temperatures such as in micro-reaction technology are not feasible. Also for the implementation of applications in the field of point-of-care diagnostics, where a hand-held device is often required, the LSI platform seems not to be beneficial at the moment. Thereto external pressure sources and valves would have to be downsized to a smaller footprint, which is of course technically feasible, but the costs would be higher in comparison to other platform concepts. However, as a first step towards downsizing the liquid control equipment, the use of a Braille system was successfully demonstrated.141
Segmented flow microfluidics
Characterization of segmented flow microfluidics
Segmented flow microfluidics describes the principle of using small liquid plugs and/or droplets immersed in a second immiscible continuous phase (gas or liquid) as stable micro-confinements within closed microfluidic channels. Those micro-confinements are in the picolitre to microlitre volume range. They can be transported by pressure gradients and can be merged, split, sorted, and processed without any dispersion in microfluidic channels.General principle
The segmented flow microfluidic platform relies on a multiphase fluid flow through microchannels. Generally, the applied technologies can be divided into the following categories:• 2-phase gas–liquid
• 2-phase liquid–liquid
• 3-phase liquid–liquid
In principal, droplets of a dispersed liquid phase are immersed in a second continuous gas (2-phase gas–liquid) or liquid (2-phase liquid–liquid) phase within a microchannel. Thereby, the inner liquid droplets are separated by the continuous carrier liquid along the channel. If the size of the inner phase exceeds the cross sectional dimensions of the channel, the droplets are squeezed to form non-spherical segments, also called “plugs”. Following this flow scheme, the platform is called segmented flow microfluidics.
In some applications, the stability of the phase-arrangement is increased by additional surfactants as the third phase, stabilizing the plug interface (3-phase liquid–liquid).142 An external pressure is applied for the transport of the plugs. A comprehensive general discussion of the platform can also be found in recent review papers.29,143,144
Unit operations
The most elementary unit operation on the segmented flow platform is the initial generation of the droplets (see Table 4). This step can also be considered a metering, since the liquid volumes involved in the subsequent reaction within the droplet are defined during the droplet formation process. Generally, two different microfluidic structures have been reported for a controlled and continuous generation of droplets: the flow focusing structure as depicted in Fig. 8145,146 and the T-shaped junction,147,148 respectively. The size of the droplet is influenced by the strength of the shear forces at the channel junction (higher shear forces lead to smaller droplets) for both droplet formation mechanisms.To use droplets inside channels as reaction confinements, the different reactants have to be loaded into the droplet. Therefore, a method to combine 3 different sample liquid streams by a sheath flow arrangement with subsequent injection as a common droplet into the carrier fluid has been shown by the group of Rustem F. Ismagilov at the University of Chicago, IL, USA149 (see Fig. 11). Different concentrations and ratios of two reagent sub-streams plus a dilution buffer merge into one droplet and perform a so called on-chip dilution.150 The mixing ratios can be adjusted by the volume flow ratio of the three streams.
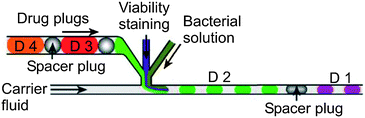 | ||
| Fig. 11 Droplet-based drug screening. The plugs containing the drugs (D1 to D4) get mixed with a bacterial solution and a viability dye. In the case of potent drugs the bacteria die and the droplet shows no staining. Image adapted from Boedicker et al.167 | ||
Using a combination of two opposing T-junctions connected to the same channel, the formation of droplets of alternating composition has been demonstrated.151 Using a similar technique, the injection of an additional reactant into a liquid plug moving through the channel at an additional downstream T-junction has been demonstrated.152 Not only liquid chemical reagents but also other components like cells have been loaded into droplets.153
The merging of different sized droplets showing different velocities to single droplets has been demonstrated successfully.149 In the same work, the controlled splitting of droplets at a channel branching point has been shown. Using a similar method, the formation of droplet emulsions with controlled volume fractions and drop sizes has been realized.154
Mixing inside the droplets can be accelerated by a recirculating flow due to shear forces induced by the motion along the stationary channel wall.155 This effect is even more pronounced if two liquids of differing viscosities are mixed within the droplet.156 Based on the recirculating flow, a mixing scheme for the segmented flow platform has been proposed using serpentine microchannels.157 Within each channel curvature the orientation between the phase pattern in the droplet and the direction of motion is changed so that the inner recirculation leads to stretching and folding of the phases. Under favorable conditions, sub-millisecond mixing can be achieved and has been employed for multi-step synthesis of nanoparticles.152 A detailed and theoretical description of this mixing effect is given in ref. 158.
Besides the mixing within liquid droplets dispersed into another liquid carrier phase, mixing within the carrier phase can also be accelerated by a segmented flow. The injection of gas-bubbles into a continuous liquid stream forming a segmented gas–liquid flow has been described by Klavs Jensen and his group at MIT.159,160 The gas bubbles are introduced into the liquid flow and initiate recirculation flows within the liquid segments in between due to the motion along the channel wall. The gas bubbles can be completely separated from the liquid stream using a planar capillary separator after the reaction is finished. Using that technology, the synthesis of colloidal silica particles has been demonstrated.161 Another microfluidic mixing scheme based on a gas–liquid segmented flow uses an additional repeated separation and re-combining of the channel.162
The incubation time of the reagents combined inside a droplet at the injection position can easily be calculated at a certain point of observation from the travelling distance of the droplet divided by the droplet velocity. Thus, the incubation time can be temporally monitored by simply scanning along the channel from the injection point to positions farther downstream. This is a unique feature of the platform and enables the investigation of chemical reaction kinetics on the order of only a few milliseconds.150 On the other hand, also stable incubation times on the order of a week have been demonstrated.163 This is enabled by separating the droplet compartments with a carrier fluid that prevents evaporation and diffusion. Using this approach, several 60 nL liquid droplets containing one or a few cells were generated within a microfluidic chip and afterwards flushed into a Teflon capillary tube for cultivation. The cell densities were still as high as in conventional systems after 144 h of growth within the droplets.
Additional unit operations based on charged droplets and electric fields have been added to the segmented flow platform by David A. Weitz and co-workers.164 Using dielectrophoresis, the sorting of single droplets out of a droplet train (switching) at rates up to 4 kHz has been shown.165 The segmented flow technology augmented with electric field-based unit operations is currently commercialized by the company Raindance Technologies, MA, USA.
Application examples
Table 4 gives an overview of the microfluidic unit operations and applications that have been already implemented on the segmented flow platform. They all take advantage of the enclosed reaction confinement within the droplets, either for analytical applications (cell analysis, single organism analysis, DNA assays, drug screening, protein crystallization) or chemical synthesis.Protein crystallization, for example, is realized on the segmented flow platform by forming droplets out of three liquids, namely the protein solution, a buffer and the precipitant within oil as the carrier phase.174,180 The precipitant concentration inside the droplet is adjusted via the buffer and precipitant flow rates, respectively. Therewith, different concentrations are generated and transferred into a glass capillary for later X-ray analysis.175 The effect of mixing on the nucleation of protein crystallization has been investigated by combining the described crystallization structure with a serpentine mixing channel.179 Fast mixing has been found to be favorable for the formation of well-crystallized proteins within the droplets.178
Recently, a chip for rapid detection and drug susceptibility screening of bacteria has also been presented167 as one example of a high-throughput screening application. The channel design is depicted in Fig. 11. Plugs of the bacterial solution, a fluorescent viability indicator, and the drugs to be screened are injected into the carrier fluid. The different drug solutions (antibiotics: vancomycin (VCM), levofloxixin (LVF), ampicillin (AMP), cefoxitin (CFX), oxicillin (OXA), and erythromycin (ERT)) are separated by an air spacer plug within the drug trial channel. Plugs containing VCM were used as baseline, because VCM inhibited this Staphylococcus aureus strain in macro-scale experiments. No plugs containing VCM or LVF had a fluorescence increase greater than three times the baseline, indicating that MRSA was sensitive to these antibiotics.
Strengths and limitations
The main advantages of the segmented flow microfluidic platform are the small volume liquid segments (controllable with high precision in the nanolitre range), acting as reaction confinements. This leads to little reagent consumption as well as a high number of different experiments that can be performed within a short period of time, which makes the platform a promising candidate for high-throughput screening applications, e.g. in the pharmaceutical industry. The quasi-batch-mode operation scheme within nanolitre to microlitre-sized droplets is beneficial since it represents a consistent further development of classic assay protocols in e.g. well plates. The large number of existing unit operations enables the effective manipulation of the liquid segments. Furthermore, the completely enclosed liquid droplets allow the incubation and storage of liquid assay results over a long period of time without evaporation.However, a limitation of the platform is that handling of small overall sample volumes is not possible due to the volume consumption during the run-in phase of the flow within the microchannels. This and the manual connection to external pumps renders the platform less suitable for point-of-care applications. Another drawback is the need for surfactants that are required for high stability of the plugs. They sometimes interfere with the (bio-)chemical reaction within the plugs and thus can limit the number of possible applications on the platform.
Centrifugal microfluidics
Characterization of centrifugal microfluidics
In centrifugal microfluidics all processes are controlled by the frequency protocol of a rotating microstructured substrate. Relevant forces for liquid transport are centrifugal force, Euler force, Coriolis force and capillary force. Assays are implemented as a sequence of liquid operations arranged from radially inward positions to radially outward positions. Microfluidic unit operations include metering, switching, aliquoting, etc.General principle
The approach of using centrifugal forces to automate sample processing dates back to the end of the 1960s.181 At that time, centrifugal analyzers were first used to transfer and mix a series of samples and reagents in the volume range from 1 μL to 110 μL into several cuvettes, followed by spectrometric monitoring of reactions and real-time data processing. Controlling microfluidic networks by just one rotary axis has an obvious charm to it, since no connections to the macro-world, such as pumps, are required. Moreover, the required centrifugal base devices can be simple and therefore robust. Rotational frequencies can be controlled very well and a radially constant centrifugal pseudo-force guarantees pulse-free liquid flow. Scientific work and applications based on centrifugal microfluidics have continuously been published since these early beginnings, although most attention to the topic arose again in the last two decades, as summarized in several reviews.121,182–184 However, the concept is still somewhat exotic compared to the large number of pressure driven systems existing today, possibly attributed to the difficulty of monitoring liquid flow under rotation and the dependency of liquid flow on microchannel surface quality.185 This results in high initial investment in monitoring equipment and prototyping lines. Nevertheless, considerable advances towards integrated systems have been made in the last few decades.In the beginning of the 1990s, the company Abaxis186 developed the portable clinical chemistry analyzer.187 This system consists of a plastic disposable rotating cartridge for processing of the specimen, preloading of dried reagents on the cartridge, and an analyzer instrument for actuation and readout.
A next generation of centrifugal devices emerged from the technical capabilities offered by microfabrication and microfluidic technologies.188–191 Length scales of the fluidic structures in the range of a few hundred micrometres allow parallel processing of up to a hundred units assembled on a single disk. This enables high throughput by highly parallel and automated liquid handling. In addition, assay volumes can be reduced to less than 1 μL. Particular fields such as drug screening,189 where precious samples are analyzed, benefit from these low assay volumes.
Today, many basic unit operations for liquid control on the centrifugal microfluidic platform are known and new ones are continuously being developed, enabling a number of applications in the fields of point-of-care testing, research, and security.
Unit operations
Liquid transport is initiated by the centrifugal force, fω, directed outwards in the radial direction. The centrifugal force can be scaled over a wide range by the frequency of rotation ω. Together with a tunable flow resistance of the fluidic channels, small flow rates in the order of nL s−1 as well as high throughput continuous flows up to 1 mL s−1192 can be generated. Therefore, scaling of flow rates over 6 orders of magnitude independent of the chemical composition, ionic strength, conductivity or pH value of the liquid can be accomplished, opening a wide field of possible applications. Also, liquid transport at rest can be achieved by capillary forces, depending on the channel geometry and the wetting properties of the liquid.Liquid valves can be realized by several different microfluidic structures on the centrifugal platform. In general, they can be purely passive, as depicted in Fig. 12, or require an active component outside the microfluidic substrate. First, the passive valves will be summarized: A very simple valve arises at the sudden expansion of a microfluidic channel, e.g. into a bigger reservoir: the geometric capillary valve (Fig. 12(a)). The valving mechanism of this capillary valve is based on the energy barrier for the proceeding of the meniscus, which is pinned at the sharp corner. This barrier can be overcome under rotation due to the centrifugal pressure load of the overlying liquid plug.189,193,194 For a given liquid plug position, length, liquid surface tension and contact angle, the valve is influenced by only the frequency of rotation, and a critical burst frequency ωc can be attributed to every valve structure. Another possibility to stop the liquid flow within a channel is the local hydrophobic coating of the channel walls (hydrophobic valve) (Fig. 12(b)).183,195–197 This valve is opened as soon as the rotational frequency exceeds the critical burst frequency ωc for this geometry and surface properties. A third method (Fig. 12(c)) utilizes the stopping effect of compressed air in an unvented receiving chamber. This centrifugo-pneumatic valve stops liquid up to much higher pressures than capillary valves for small receiving chamber volumes (≤40 μL). The air counter-pressure in the unvented receiving chamber can be overcome at high centrifugal frequencies, at which the liquid–air interface becomes unstable and enables a phase exchange, permitting liquid flow.198,199 Another method is based on a hydrophilic S-shaped siphon channel (hydrophilic siphon valve), wherein the two liquid–gas interfaces are leveraged at high frequencies of rotation183 (Fig. 12(d)). Below a critical frequency ωc however, the right-hand meniscus proceeds beyond the bend, thus allowing the centrifugal force to drain the complete liquid from the siphon.
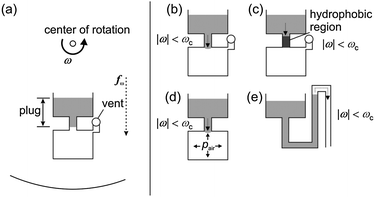 | ||
| Fig. 12 Passive centrifugal microfluidic valves. (a) Positioning of valves relative to center of rotation and centrifugal force, (b) geometric capillary valve,189 (c) hydrophobic valve,195 (d) centrifugo-pneumatic valve198 and (e) hydrophilic siphon valve.183 | ||
One example of an active valve is an irradiation-triggered “sacrificial” valve published by Samsung Advanced Institute of Technology (Laser Irradiated Ferrowax Microvalve, LIFM).200 A ferrowax plug is used to close channels off during the fabrication of the microfluidic network. A laser source in the processing device can be utilized to melt the ferrowax plug and thus allow liquid passage (normally-closed valve). A modification of this technique also allows closing channels off by illuminating a ferrowax reservoir that expands into a channel and seals it (normally-open valve). An advantage of this valve is that it allows liquid control depending solely on the moment of the laser actuation, so it does not depend on the rotational speed or liquid properties. This comes at the cost of a more complex production process and base device. An alternative approach for the active control of liquid flows on the centrifugal platform is followed by the company Spin-X technologies, Switzerland. A laser beam individually opens fluidic interconnects between different channel layers on a plastic substrate (Virtual Laser Valve, VLV). This enables online control of the liquid handling process on the rotating module for adjusting metered volumes and incubation times within a wide range. Due to this, the Spin-X platform works with a standardized fluidic cartridge that is not custom made for each specific application, but can be programmed online during a running process.
Combining one of the above-mentioned valve principles at the radially outward end of a chamber with an overflow channel at the radially inward end results in a metering structure.201 The metered liquid portion is directly set by the volume capacity of the chamber. With highly precise micro-fabrication technologies, small coefficients of variations (CV, standard deviation divided by mean value), e.g. a CV < 5% for a volume of 300 nL202 and also metered volumes of as little as 5 nL have been achieved.196 By arranging several metering structures interconnected via an appropriate distribution channel, simple aliquoting structures can be realized.198,203 These structures split a sample into several defined volumes, enabling the conduction of several assays from the same sample in parallel.
Different mixing schemes have been proposed on the centrifugal platform. Considering mixing of continuous liquid flows within a radially directed rotating channel, the perpendicular Coriolis force automatically generates a transverse liquid flow.192 A continuous centrifugal micromixer, utilizing the Coriolis stirring effect, showed an increasing mixing quality towards very high volume throughputs of up to 1 mL s−1 per channel192 (Coriolis mixer). Besides the mixing of continuous liquid flows, also the homogenization of discrete and small liquid volumes located in chambers is of importance especially when analyzing small sample volumes (batch-mode mixing), since homogenous mixing obviously speeds up diffusion-limited chemical and biological reactions due to the close proximity between analytes. One possibility to enhance the mixing is the active agitation of the liquid within a mixing chamber by inertia related shear forces (Euler force), induced by a fast change of the sense of rotation (shake-mode-mixing)201 or change of rotational frequency (unidirectional shake-mode-mixing).204 Shake-mode mixing leads to reduced mixing times on the order of several seconds compared to several minutes for pure diffusion-based mixing. A further downscaling of mixing times below one second using magnetic microparticles, located in the mixing chamber, has also been demonstrated.205 Accelerated mixing can also be achieved by an interplay of capillary and intermittent centrifugal forces.206
For routing (switching) of liquids, a switch utilizing the transversal Coriolis force to guide liquid flows between two outlets at the bifurcation of an inverse Y-shaped channel207 or at a nozzle leading into a chamber208 has been presented. Depending on the sense of rotation, the Coriolis force is either directed to the left or to the right, guiding the liquid stream into one of two downstream reservoirs at the bifurcation. Another method for liquid routing based on different wetting properties of the connected channels has been reported by Gyros AB, Sweden.209 The liquid stream is initially guided towards a radial channel, exhibiting a hydrophobic patch at the beginning. Therefore, the liquid is deflected into a branching non-hydrophobic channel next to the radial one. For high frequencies of rotation, the approaching liquid possesses enough energy to overcome the hydrophobic patch and is therefore routed into the radial channel.210 A further possibility to switch liquid flows is to utilize an “air cushion” between an initial first liquid entering a downstream chamber and a subsequent liquid. The centrifugally generated pressure of the first liquid is transmitted via the air cushion to the subsequent liquid and forces it via an alternative route into a chamber placed to the side of the main channel.211
The separation of plasma from a whole blood sample is the prevalent first step within a complete analytical protocol for the analysis of whole blood. Since blood plasma has lower density compared to the white and red blood cells it can be found in the upper phase after sedimentation in the artificial gravity field under rotation. The spatial separation of the obtained plasma from the cellular pellet can be achieved via a capillary channel that branches from the sedimentation chamber at a radial position where only plasma is expected.187 Another method uses pre-separation of the cellular and plasma phase during the sample flow through an azimuthally aligned channel of 300 μm radial width.197 The obtained plasma fraction is thereafter split from the cellular components by a decanting process. Another concept enables plasma separation of varying blood sample volumes in a continuous process. The sedimentation occurs in an azimuthally curved channel due to centrifugal and Coriolis forces, enabling up to 99% separation efficiency between two outlets for a diluted sample with 6% hematocrit.212 An overview of centrifugal microfluidic unit operations and related applications can be found in Table 5.
| Microfluidic unit operations | Reference |
|---|---|
| Capillary valving | 183; 189; 191; 193; 194; 213–220 |
| Hydrophobic valving | 183; 195–197 |
| Siphon valving | 183; 186; 187; 204; 221; 222 |
| Laser-triggered valving | 200; 223–225 |
| Centrifugo-pneumatic valving | 198; 211 |
| Metering | 183; 187; 191; 195–197; 200–202; 221; 222; 224 |
| Aliquoting | 181; 183; 186; 187; 195; 198; 226 |
| Mixing | 181; 183; 186; 187; 191; 192; 200–202; 204; 205; 217; 221; 222; 224; 226–229 |
| Coriolis switching | 183; 201; 207; 211; 212; 230 |
| Reagent storage | 217; 231 |
| Applications | Reference |
|---|---|
| Integrated plasma separation | 183; 197; 201; 212; 221–224; 232 |
| Cell lysis and/or DNA extraction | 224; 230; 233 |
| Protein-based assays | 181; 189; 195; 201; 213; 217; 219; 221–223; 226; 234 |
| Nucleic acid-based assays | 213; 218; 235 |
| Clinical chemistry assays | 186; 187; 201; 202; 214–216; 222; 229; 236 |
| Chromatography | 237 |
| Protein crystallization | 196 |
Application examples
Table 5 shows some applications that have been realized on the centrifugal microfluidic platform. At the top of the applications section, sample preparation modules (plasma separation, DNA extraction) are shown. This is followed by assays based on the detection of proteins, nucleic acids and small molecules (clinical chemistry). Two additional applications are presented at the end of the table, demonstrating chromatography and protein crystallization. Some instructive examples are discussed in more detail below.Madou et al. from the University of California, Irvine showed a series of capillary valves to perform enzyme-linked immunosorbent assays (ELISAs) on the centrifugal platform.219 The different assay liquids are held back in reservoirs connected to the reaction chamber via valves of different burst frequency. The capillary valves are opened subsequently by increasing the frequency of rotation. It was shown that in terms of detection range the centrifugally conducted assay has the same performance as the conventional method on a 96-well plate, but with less reagent consumption and shorter assay time.
Gyros AB, Sweden209 use a flow-through sandwich immunoassay at the nanolitre scale to quantify proteins within their Gyrolab™ Workstation. A column of pre-packed and streptavidin-coated microparticles is integrated into each one of 112 identical assay units on the microfluidic disk. Each unit has an individual sample inlet and a volume definition chamber that leads to an overflow channel. Defined volumes (200 nL) of samples and reagents can be applied to the pre-packed particle column. The laser induced fluorescent (LIF) detector is incorporated into the Gyrolab™ Workstation. Using this technology, multiple immunoassays have been carried out to determine the imprecision of the assay result. The day-to-day (total) imprecisions (CV) of the immunoassays on the microfluidic disk are below 20%.195 The assays are carried out within 50 min with sample volumes of 200 nL. In comparison, the traditional ELISA performed in a 96-well plate typically takes several hours and requires sample volumes of several hundred microlitres.
A fully integrated colorimetric assay for determination of alcohol concentrations in human whole blood has been shown on the centrifugal Bio-Disk platform.202 After loading the reagents into the reagents reservoir, a droplet of untreated human blood taken from a finger tip is loaded into the inlet port of the microstructure. By mixing the blood sample with the reagents, an enzymatic reaction is initiated, changing the color of the mixture depending on the alcohol concentration. After sedimentation of the residual blood cells, the absorbance is monitored in a real-time manner via a laser beam that is reflected into the disk plane on integrated V-grooves.229 Using this automated assay and readout protocol the concentration of alcohol in human whole blood was determined within only 150 s. The results were comparable to common point-of-care tests and required a minute blood volume of just 500 nL.
Also a protein crystallization assay has been demonstrated on the centrifugal microfluidic platform.196 First, a defined volume of the protein solution is dispensed into the protein inlet and transported into the crystallization chamber. Afterwards, the pre-loaded precipitant is metered under rotation and transferred into the crystallization chamber as soon as a hydrophobic valve breaks. In the last step, the pre-loaded oil is released at yet a higher frequency and placed on top of the liquid stack within the crystallization chamber, to prevent evaporation. The successful crystallization of proteinase K and catalase was demonstrated.
Samsung Advanced Institute of Technology showed a fully integrated immunoassay for Hepatitis B and other antibodies, starting from 150 μL whole blood on a centrifugal base device including a laser for controlling ferrowax valves and a readout-unit.223 A limit of detection comparable to a conventional ELISA and an assay time of 30 min were reported. On the same platform, enrichment of pathogens and subsequent DNA extraction was also shown (Fig. 13).224 The microfluidic structure features an integrated magnet that controls the position of coated magnetic particles which are used to capture target pathogens and lyse them by laser irradiation. With a total extraction time of 12 min, down to 10 copies/μL DNA concentration in a spiked blood sample of 100 μL could be specifically extracted and detected in a subsequent external PCR. Reagents are loaded by the operator prior to the process.
 | ||
| Fig. 13 Centrifugal microfluidic structure for pathogen-specific cell capture, lysis and DNA purification published by Cho et al.224 The microfluidic network comprises structures for plasma separation, mixing, and laser-triggered valves. For manipulation of the magnetic capture-beads, a movable magnet is integrated into the cartridge. | ||
Strengths and limitations
Two major advantages of the centrifugal microfluidic platform are the modular setup of the system with disposable and easily exchangeable plastic cartridges and the many existing unit operations, which allow highly precise liquid handling. The fabrication costs of the disposables are governed by the specific implementation of unit operations. Necessary global or local surface modification or the integration of active (ferrowax) valves, post-replication treatment, assembly and reagent pre-storage steps can increase the cost of the disposables. Mostly, they are made out of plastic and thus suitable for mass-production. The presented unit operations allow the automation of complex assay protocols. The cost for the base instrument depends heavily on readout and temperature control modules. The motor required for liquid control is generally required to be able to achieve very stable and defined rotational speed and acceleration, also adding to the costs. However, compared to (several) high-precision syringe pumps, this solution is generally cheaper and allows a higher degree of integration. Due to the rotational symmetry of the disks, optionally some degree of parallelization can be achieved. Also, the rotational symmetry is beneficial for fast readout and temperature uniformity between cavities at the same radial position.However, as soon as any additional actuation or sensing function is required on the module during rotation and if a contact free interfacing is not applicable, things become challenging from a technical point of view. Especially interfacing to electric readout modules on the disk is difficult, since the rotating setup does not allow for wire connections between the disposable and the base instrument. The platform also lacks flexibility compared to others that allow online programming of fluidic networks within one piece of hardware that fits all, since most of the logic functions as well as their critical frequencies are permanently imprinted into the channel network. However, the Virtual Laser Valve technology is an exception in this respect and allows online programming in a centrifugal system. Space restrictions are also an issue, since the required footprint (disk surface) increases quadratically with the number of connected unit operations (radial length). The low centrifugal forces near the center of rotation and the difficulty of transporting liquids radially inward are other challenges in the fluidic design process. Also, completely portable solutions are currently still only a vision.
Electrokinetics
Characterization of electrokinetics
In electrokinetics platforms microfluidic unit operations are controlled by electric fields acting on electric charges, or electric field gradients acting on electric dipoles. Depending on buffers and/or sample, several electrokinetic effects such as electroosmosis, electrophoresis, dielectrophoresis, and polarization superimpose each other. Electroosmosis can be used to transport the whole liquid bulk while the other effects can be used to separate different types of molecules or particles within the bulk liquid.General principle
One of the first applications for electrokinetics was the analysis of chemical compounds via electrophoretic separation within capillaries in 1967,238 long before the term “microfluidics” emerged. In the beginning, glass capillaries made from drawn glass tubes were used, whereas today well defined microchannels are established and commonly used. The actuation principle of the electrokinetic platform relies on the movement of liquid in an induced electric double layer and charged particles (ions) in an electric field applied along a microfluidic channel. The simple setup of electrokinetic systems consisting of microfluidic channels and electrodes without moving parts explains the early advent of electrokinetic platforms for microfluidic lab-on-a-chip applications.Unit operations
In a microfluidic channel, a charged solid surface induces an opposite net charge in the adjacent liquid layer (electric double layer). As soon as an electric potential is applied along the channel, the positively charged liquid molecules are attracted by electrostatic forces and thus move towards a corresponding electrode (Fig. 14(a)). Due to viscous coupling, the bulk liquid is dragged along by the moving layer and liquid actuation with a planar velocity profile is generated (electroosmotic flow (EOF)239). The velocity profile is constant and dispersion only occurs by molecular diffusion. This motion is superimposed by the movement of ions and charged molecules, which are attracted or repelled by the electrodes depending on their charge (Fig. 14(b)). The velocity of the molecule depends on its charge and hydrodynamic radius and enables the distinction between different molecular entities. This effect is used for separation of charged molecules and is called electrophoresis.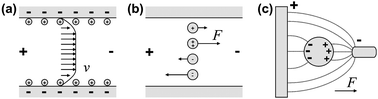 | ||
| Fig. 14 Basic electrokinetic effects (according to Atkins239). (a): electroosmotic flow (EOF), (b): electrophoresis (EP), (c) dielectrophoresis (DEP). | ||
Based on the electroosmotic flow, metering of volumes down to the picolitre range can be achieved. While the sample liquid is injected and crosses an intersection point of two perpendicular channels, the electrodes and therefore the flow along the main channel is switched off. Then, the electrodes in the side channel are activated. This displaces a small plug at the intersection into the side channel, resulting in metering of a sample volume depending on the geometry of the intersection area. The mixing of two co-flowing streams was shown on the electrokinetic platform by applying an AC voltage.238 A 20-fold reduction in mixing time compared to molecular diffusion has been reported. Also complete biological assays comprising cell lysis, mixing, and DNA amplification have been presented.240
A modification to electrophoresis is free-flow electrophoresis, which enables the continuous separation of a mixture according to charge with subsequent collection of the sample band of interest.241 For this, an transverse electric field is applied in pressure driven flow within a broad and flat microchamber. While passing this extraction chamber, the species contained in the sample flow are deflected depending on their charge and thus exit the chamber through one of several outlets.
Another electrokinetic effect is based on polarization of particles within an oscillating electrical field or field gradient (dielectrophoresis), as depicted in Fig. 14(c). Dielectrophoresis is applied in many fields, e.g. for the controlled separation and trapping of submicron bioparticles,242 for the fusion and transport of cells,243 gene transfection244 or the separation of metallic from semiconducting carbon nanotubes.12,245,246 Other applications are cell sorting247,248 and apoptosis of cells.249,250
Application examples
Capillary electrophoresis systems were the first micro total analysis systems and emerged as single chip solutions from the analytical chemistry field in the 1990s.251 Several companies utilize microfluidic capillary electrophoretic chips for chemical analysis, with capillaries of typically 10 to 100 μm diameter.252Today, Caliper Life Sciences, MA, USA252 and Agilent Technologies, CA, USA253 offer microfluidic chips for DNA and protein analysis. Liquid propulsion is provided via electroosmosis and combined with capillary electrophoretic separation. The sample is electroosmotically transported and metered inside the chip, then separated via capillary electrophoresis and analysed by fluorescence detection. (Fig. 15). The whole assay is performed within minutes, instead of hours or days.
 | ||
| Fig. 15 Microfluidic realization of capillary electrophoresis analysis on the electrokinetic platform (adapted from ref. 121) (© Agilent Technologies, Inc. 2007. Reproduced with permission, courtesy of Agilent Technologies, Inc.). After the sample has been transported to the junction area (a) it is metered by the activated horizontal flow and injected into the separation channel (b). Therein, the sample components are electrophoretically separated (c) and readout by their fluorescence signal (d). The complete microfluidic CE-chip is depicted in the center. | ||
The first combinations of microfluidic integrated electrophoresis with microarrays were published in 1998 by Nanogen Inc., CA, USA.254 This approach resulted in a 20-fold faster hybridization and more specific binding of DNA onto the microarray. This was the first step in the direction of a platform for massively parallel analysis.
Strengths and limitations
Electroosmotic actuation of liquids enables pulse-free pumping without any moving parts. Liquid manipulation at high precision can be achieved by the existing unit operations. In addition, electroosmotic flow does not lead to Taylor dispersion115 as in pressure driven systems and thus enables high efficiency separations. The seamless integration with electrophoresis, an established technology in use for 100 years,255 is another obvious strength. In microfluidic systems, applications can benefit from faster heat dissipation, better resolution, and faster separation. Miniaturization of electrophoretic analysis enables the automation and parallelization of tests with small dead volumes, thus reducing the required amount of sample.A technical problem in capillary electrophoresis systems is the changing pH-gradient due to electrolysis or electrophoresis itself. Also streaming currents which counteract the external electric field or gas bubbles as a result of electrolysis at the electrodes are problematic. Also a massively parallel setup is problematic due to the heat generated by the electrophoresis itself. In addition, the realization of hand-held devices is challenging due to the necessity of high voltages in combination with high energy consumption. Overall, miniaturized electrophoresis is established as a fast and efficient method for the separation and analysis of bio-molecules.
Electrowetting
Characterization of electrowetting
Electrowetting platforms use droplets immersed in a second immiscible continuous phase (gas or liquid) as stable micro-confinements. The droplets reside on a hydrophobic surface that contains a one- or two-dimensional array of individually addressable electrodes. The voltage between a droplet and the electrode underneath the droplet defines its wetting behavior. By changing voltages between neighboring electrodes, droplets can be generated, transported, split, merged, and processed. These unit operations are freely programmable for each individual droplet by the end-user enabling online control of an assay.General principle
The electrowetting effect was first described by Lippmann in 1875.256 Interest in this effect was spurred again in the 1990s, when researchers started placing thin insulating layers on the metallic electrodes to separate it from the often conductive liquids in order to eliminate electrolysis.257 The basic electrowetting effect is depicted in Fig. 16(a). The wettability of a solid surface increases due to polarization and electric fields as soon as a voltage is applied between the electrode and the liquid droplet above (separated by the dielectric insulating layer).257 This so-called “electrowetting-on-dielectric” (EWOD)258 effect is therefore a tool to control the contact angle of liquids on surfaces.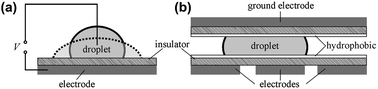 | ||
| Fig. 16 The electrowetting effect (according to Mugele and Baret257). (a) If a voltage V is applied between a liquid and an electrode separated by an insulating layer, the contact angle of the liquid–solid interface is decreased and the droplet “flattens”. (b) Hydrophobic surfaces enhance the effect of electrowetting. For “electrowetting-on-dielectrics” (EWOD) several individual addressable control electrodes (here on the bottom) and a large counter-electrode are used. The droplet is pulled to the charged electrodes. | ||
This invention paved the way for the application of the electrowetting effect as a liquid propulsion principle for lab-on-a-chip systems.259,260 To utilize the EWOD technology for programmable liquid actuation, a liquid droplet is placed between two electrodes covered with insulating, preferably hydrophobic, dielectric layers (Fig. 16(b)). The liquid droplet is steered by the electrode array on one side and by a large planar ground electrode on the opposite side. Activating selected electrodes allows programming of a path which the droplet follows. The droplet needs to be large enough to cover parts of at least four addressable electrodes at all times, allowing two-dimensional movement. If a voltage is applied to one of the control electrodes covered by the droplet, it moves onto the activated electrode pad. Successive activation of one electrode after the other will drag the droplet along a defined path. This freedom to program the liquid movement enables the implementation of different assays on the same chip.
The universal applicability of moving droplets by EWOD was shown with several media such as ionic liquids, aqueous surfactant solutions,261 and also biological fluids like whole blood, serum, plasma, urine, saliva, sweat, and tear fluid.262
Unit operations
The droplet formation, i.e. initial metering, is the elementary unit operation of the platform. Metered droplets can be produced from an on-chip reservoir in three steps.262 First, a liquid column is extruded from the reservoir by activating a series of adjacent electrodes. Second, once the column overlaps the electrode on which the droplet is to be formed, all the remaining electrodes are turned off, forming a neck in the column. The reservoir electrode is then activated during the third and last step, pulling back the liquid and breaking the neck, leaving a droplet behind on the metering electrode. Using this droplet metering structure, droplets down to 20 nL volume can be generated with a standard deviation of less than 2%.262 A similar technology can be used for the splitting of a droplet into several smaller droplets.31 Since the droplet volume is of great importance for the accuracy of all assays, additional volume control mechanisms such as on-chip capacitance volume control263 or the use of numerical methods for the design of EWOD metering structures264 have been proposed. Once the droplets are formed, their actuation is accomplished by the EWOD effect as described above. Also the merging of droplets can be achieved easily with the use of three electrodes. Two droplets are individually guided to electrodes separated from each other by a third one. Deactivating these two electrodes and activating the third separation electrode pulls the droplets together.265 The most basic type of mixing within droplets on the EWOD platform is an oscillation, forwards and backwards, between at least two electrodes. Another mixing scheme is the repetitive movement of the droplet on a rectangular path. The shortest mixing time for two 1.3 μL droplets in linear oscillation on 4 electrodes was about 4.6 s.266 In another work, the mixing times of 1.4 μL droplets could be further reduced to less than 3 s using two-dimensional arrays.267Application examples
Applications based on EWOD are in the development phase and quite close to market products. For example, an enzymatic colorimetric assay for (point-of-care) diagnostic applications has been successfully implemented, and glucose concentration in several biological liquids (serum, plasma, urine, and saliva) was determined with comparable results to standard methods.262 The microfluidic chip layout for the colorimetric glucose assay is depicted in Fig. 17. It features reservoirs, injection structures (metering) and a network of electrodes for droplet transport, splitting and detection. | ||
| Fig. 17 Electrowetting platform (EWOD). Implementation of a colorimetric glucose assay in a single chip. Four reservoirs with injection elements are connected to an electrode circuitry, where the droplets are mixed, split and transported to detection sites for readout (adapted from Srinivasan et al.262). | ||
Also the use of an EWOD system for the automated sample preparation of peptides and proteins for matrix-assisted laser desorption-ionization mass spectrometry (MALDI-MS) was reported. In that work, standard MALDI-MS reagents, analytes, concentrations, and recipes have been demonstrated to be compatible with the EWOD technology, and mass spectra comparable to those collected by conventional methods were obtained.268 Also a PCR assay has been realized on the platform by temperature cycling of a droplet at rest.269 Additional information about the EWOD platform can be found in a comprehensive review.270
Strengths and limitations
The strengths of the platform are the very small liquid volumes in the nanolitre range that can be handled with high precision, and the freedom to program the droplet movement. This cuts down sample and reagent consumption and allows a maximum of flexibility for the implementation of different assay protocols. The simple setup without any moving parts can be fabricated using standard lithographic processes. The programmable control of small droplets has its particular potential in assay optimization, since it allows the protocol to be varied over a certain range on the same chip.However, although the sample and reagent consumption is low, portable systems for e.g. point-of-care applications have not yet been demonstrated due to the bulky electronic instrumentation required to operate the platform. Another drawback is the influence of the liquid properties on the droplet transport behaviour, i.e. different patient materials will show different wetting abilities and thus lead to differences in volume or movement speed. Also the long-term stability of the hydrophobic surface coatings and the contamination risk is problematic, since every droplet can potentially contaminate the surface and thus lead to false results and also change the contact angle for the successor droplets. Another issue is the possible electrolysis caused by the electric fields themselves. Strategies for high-throughput applications have not been demonstrated to date.
In summary, the EWOD technique bears great potential to manipulate many single droplets in parallel. While first applications have been shown, the EWOD concept is still at a stage of development, shortly before entering the IVD markets.270
Surface acoustic waves
Characterization of surface acoustic waves
The surface acoustic waves platform uses droplets residing on a hydrophobic surface in a gaseous environment (air). The microfluidic unit operations are mainly controlled by acoustic shock waves travelling on the surface of the solid support. The shock waves are generated by an arrangement of surrounding sonotrodes, defining the droplet manipulation area. Most of the unit operations such as droplet generation, transport, mixing, etc. are freely programmable.General principle
An alternative to the electrowetting-based transportation of droplets on a plane surface has been proposed by the group of Achim Wixforth at the University of Augsburg, Germany.271 The approach is based on surface acoustic waves (SAW), which are mechanical waves with amplitudes of typically only a few nanometres. The surface acoustic waves are generated by a piezoelectric transducer chip (e.g. quartz) fabricated by placing interdigital electrodes (interdigital transducer, IDT) on top of a piezoelectric layer. Liquid droplets situated on the hydrophobic surface of the chip can be moved by the SAWs if the acoustic pressure exerted on the liquid droplet is high enough (Fig. 18).272 The actuation of small amounts of liquids with viscosities extending over a large range (from 1 to 1000 mPa s) has been shown.273 This approach is also sometimes referred to as “flat fluidics”, because no cover or slit is required as in the EWOD approach.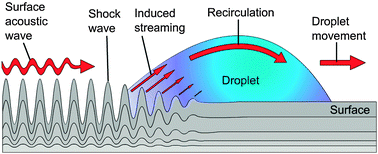 | ||
| Fig. 18 Surface acoustic wave (SAW) (according to Tan et al.274). The shock waves induce a stream on the solid–liquid interface and lead finally to a movement of the droplet (amplitude of acoustic wave not to scale). | ||
Unit operations
Metering is accomplished by moving a liquid droplet over a small hydrophilic “metering spot” via surface acoustic waves, leaving behind a small metered liquid portion due to the interplay between the surface tension force (keeping the droplet on the spot) and the acoustic force (pushing the droplet forward). Since those two forces scale differently over the droplet size, the splitting of the initial droplet into two droplets (one sitting on the metering spot and the other propagating forward) occurs. The smaller droplet is not transported since it stays unaffected by the acoustic wave. Also aliquoting has been shown by moving the initial droplet over a hydrophobic/hydrophilic checkerboard pattern.271Mixing is an intrinsic unit operation of the SAW platform. A droplet which is placed on the substrate and is influenced by a SAW shows internal liquid circulation due to the vibrating forces of the wave. This internal circulation leads to mixing.271
Application examples
A PCR protocol has been implemented on the SAW platform, based on 200 nL droplets and an additional heating element placed underneath the substrate surface for temperature cycling while the droplet is at rest.275 However, since the nanolitre-sized droplet possesses a high surface-to-volume ratio, the liquid volume would decrease rapidly due to evaporation at the elevated temperatures required for the PCR reaction. Therefore, the aqueous liquid droplet is covered with a droplet of immiscible mineral oil with a smaller contact angle. This droplet-in-droplet configuration can still be moved via surface acoustic waves on the substrate surface. The concentration of DNA could be monitored by online fluorescent measurement providing a sensitivity of 0.1 ng.275Strengths and limitations
As in the EWOD platform, the SAW platform also allows the handling of small nanolitre-sized liquid volumes in droplets on planar surfaces. The transport mechanism using surface acoustic waves though is more flexible since it depends only on the viscosity and surface tension of the liquid.However, the programmability is in turn limited since the position of the interdigital electrodes and especially the hydrophobic/hydrophilic areas determine the possible liquid handling processes. Another disadvantage is the long-term stability and the complexity of these hydrophobic and hydrophilic surface coatings, and thus costs of the disposable chip as well as the instrument.
Dedicated systems for massively parallel analysis
Characterization of massively parallel analysis
Within the category of dedicated systems for massively parallel analysis we discuss specific platforms that do not comply with our definition of a generic microfluidic platform. The characteristics of those platforms is not given by the implementation of the fluidic functions but by the specific way to process up to millions of assays in parallel. Prominent examples are platforms used for gene expression and sequencing such as microarrays, bead-based assays and pyro-sequencing in picowell-plates.General principle
In this chapter, solutions for highly parallel assay processing are presented. These are not per se microfluidic platforms by our definition, since they do not offer a set of easily combined unit operations and are quite inflexible in terms of assay layout. They are nevertheless presented here, since the small reaction volumes per assay and partly the liquid control systems are based on microfluidic platforms. The significant market for repetitive analyses, which allows high development costs for proprietary, optimized systems, does not necessarily require a platform approach, but can benefit from microfluidic production technologies and liquid handling systems.The massively parallel assay systems are a result of the increasing demand of the pharmaceutical industry for repetitive assays276,277 to cover the following objectives:
• Screening of chemical libraries with millions of compounds278
• Screening of known drugs against new targets, different cell lines or patient material279,280
• Multiparameter analysis of cell signaling and single cell analysis281
• All -omic analyses such as genomics, transcriptomics, proteomics, glucomics, metabolomics…282
With every newly discovered receptor or protein, all known drugs, pre-drugs, and chemical compounds should be tested for interaction by means of binding, activity change, or enzymatic activity. Also the analysis of gene activity or gene sequencing requires new and massively parallel testing in numbers of hundred thousands to billions. These tests consume a lot of time, material, effort, and money, but could lead to precious results (e.g. in case of a new blockbuster drug).283 The challenging task to monitor millions of different binding reactions is partially solved by microarrays284 (mainly in the case of DNA and RNA) or bead-based assays in combination with picowell plates.
Microarrays 284 are matrices with spots of different chemical compounds on a surface (Fig. 19(a)). The number of spots ranges from a few dozen to up to several millions. The microarray is incubated with the sample and each spot interacts with the sample in parallel, leading to as many parallel assays as there are spots on the microarray. Typically a microarray is read out by fluorescence and used for nucleic acid or protein analysis.
 | ||
| Fig. 19 Images of the different systems for massively parallel screening. (a) Microarray284 after binding, providing two different fluorophores in red and green. Unchanged genes remain yellow. Up- or down-regulated genes appear in red or green. (b) 3 μm silica spheres, as an example for bead-based assays,278,287 deposited on the front end of glass fibers. (c) Empty wells of a picowell plate.285,286 In each well single cells or beads are deposited, incubated and analyzed. | ||
Picowell plates 285,286 consist of millions of small wells (<50 μm in diameter) (Fig. 19(c)). In each well, either one chemical compound or one single cell is deposited. After the deposition, the picowell plate acts as a “microarray” with each position bearing a unique chemical compound or cell. Afterwards, all assays are performed similar to a microarray.
In bead-based assays278,287 small solid phase spheres (Fig. 19(b)) or particles are used. Each bead bears one unique chemical compound. Such a bead library can consist of billions of different beads. For screening, the beads are mixed and incubated with the sample and consecutively with the assay buffers, performing one assay on each bead in parallel. The readout is commonly fluorescence based and the positive beads are sorted out and analysed one by one in series. Typically this technique is used for binding assays or DNA analysis.
The pioneers of each field who introduced this system to the market are: Microarrays by Affymetrix, CA, USA,288 bead-based arrays by Luminex Corp., TX, USA289,290 and Illumina Inc., CA, USA,291,292 and picowell plates by 454 Life Sciences, CT, USA.286
Microfluidic components and applications
Here, the microfluidic actuation principles that are utilized in massively parallel analysis are outlined briefly. This is followed by some commercial application examples. Due to the similar principle, microarrays and picowell plates are presented together, followed by bead-based assays.Micorarrays/picowell plates
For micorarrays/picowell plates, liquid actuation and metering can be achieved by different actuation principles. Mainly capillary filling of a cartridge,288 or pressure driven systems are used.283,284 In other cases, the liquid actuation is achieved by centrifugal systems, electrophoresis, surface acoustic waves, electrowetting, and several other principles. Incubation and mixing is realized by diffusion and in some cases enhanced by sonication, surface acoustic waves, or electric fields. Washing is achieved by displacing the sample with the consecutive liquid. The classical (parallel) readout of binding or interaction between the molecules is performed by fluorescence (Fig. 19(a) and (c)).288 An interesting feature is that some of the picowell plates are made from glass fiber bundles and thus present a perfect interface between the light generating bead and the detector, often a CCD camera.286,291,292Today, the company Affymetrix offers microarrays with >2000000 unique compounds. The fluidic system is quite simple. The sample is manually loaded with a pipette into the chip, and capillary forces transfer the sample to the incubation chamber. Incubation and mixing is enhanced by a moving air bubble actuated by slow rotation.
The company 454 Life Sciences offers picowell plate systems for the performance of massively parallel gene sequencing.286 Beads containing roughly 10 million identical DNA copies are loaded into the picowell plate with a pressure driven system, where each bead sediments into one cavity. Different biomolecules are washed over the wells, interacting with the beads inside. In the case of a positive reaction, a quantitative enzymatic reaction, the pyro-sequencing,293 results in the emission of light. This system allows for parallel sequencing of 106 beads in a single run.
Bead-based assays
For bead-based assays, liquid actuation and metering is most often pressure driven or performed with a pipetting robot in a microtiter plate. Mixing can be performed by any kind of mixing process according to the different actuation principles (diffusion, sonication, SAW, shaking, electrokinetic, electrophoretic, pressure driven pumping through microchannels, etc.). The beads are separated from the liquid by centrifugation or with the help of magnetic fields and can then be transferred into another liquid. Typically, detection and readout are enabled with a fluorescent marker. The beads are then analyzed either sequentially or in parallel. For sequential analysis the beads are transferred into a capillary and cross several laser beams and detectors one after the other. In that case, the beads bear a coding to identify them.289,290 For the massively parallel analysis the beads are transferred onto a planar surface or into a picowell plate (Fig. 19(b) and (c)).Bead-based assays have been commercialized by Luminex since 1997.289 A microtiter plate is used for incubation and a capillary for bead transfer into the reader. Illumina291,292 expanded this concept radically by the use of 3 μm silica spheres, each bearing a unique DNA strand. The spheres are deposited on one end of a glass fiber connected to a detector. The spheres are incubated with a DNA sample, and in the case of a binding event, the according sphere emits a light signal into the glass fiber. The current system allows handling of millions of unique compounds.294
Strengths and limitations
Today, many manual steps and skilled personnel are required for the described systems and a “real” microfluidic platform is still not reached. However, microarrays, picowell plates and bead-based assays are a very useful combination of solid phase and liquid handling for massively parallel assays in the number of millions. The material consumption per assay is quite low and the reaction time quite fast. The time-to-result is longer compared to a single assay, but several orders of magnitude faster compared to serially performing the same number of assays.A significant limitation of these systems is the reliability, reproducibility, and identification of artefacts. Therefore a positive binding event in these systems is always counterchecked in a microtiter plate experiment to verify the binding event. The whole system itself cannot be designed as hand-held and is quite expensive (several 10000 € per run for sequencing), but is inexpensive in terms of cost-per-assay and material consumption (less than a cent per sequenced base).295
Criteria for the selection of a microfluidic platform
After the previous discussion of the platform approach and the presentation of some prominent examples for microfluidic platforms, this section will attempt to summarize the strengths and limitations of each platform presented in Fig. 2. This should provide the reader with some guidance to select platforms based on the selection criteria presented in Table 3. The given platform characteristics are based on the reviewed literature and the experience of the authors, taking into consideration properties such as the material of the disposable, necessary processing equipment, production technologies, published variety of unit operations, published data concerning precision, throughput, or multiparameter testing. Beneficial platforms can be selected by identifying imperative requirements of a certain application, e.g. portability, low reagent consumption and high precision for point-of-care diagnostics, which are then compared to the characteristics of the available platforms. The platform characteristics are compiled to the best of our knowledge in Table 6. Nevertheless the performance of certain platforms according to some of the criteria is still debatable and could easily change in the future with upcoming innovations in this fast developing field. Nevertheless the strong position of classical liquid handling technologies using pipetting robots can clearly be seen.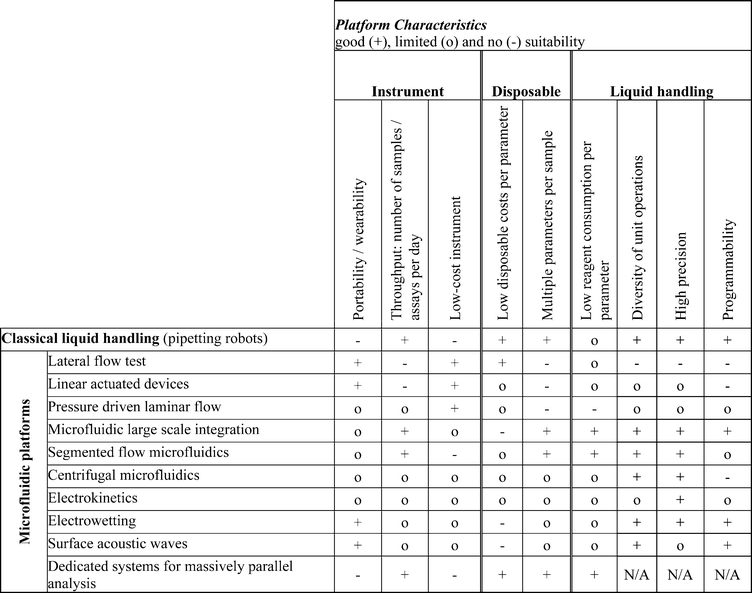
It is obvious that some of the microfluidic platform approaches are dedicated to certain fields of application. For example, the classical liquid handling technology enables high sample throughput and is programmable with high flexibility, but the main drawback is the lack of portability/wearability and the high equipment costs for complex automated workstations. These properties limit its use to large laboratories. The lateral flow test platform fulfils the requirements for point-of-care diagnostic applications quite well (moderate reagent consumption, good portability, and additionally low costs). However, as soon as the diagnostic assay requires higher precision or exceeds a certain level of complexity (e.g. if an exact metering of the sample volume or sample aliquoting is required), new approaches like linear actuated devices and centrifugal microfluidics become advantageous for point-of-care applications. They enable more sophisticated liquid handling functions, which is for instance required for nucleic acid-based tests.
The pressure driven laminar flow platform is especially interesting for online monitoring applications, since it enables continuous flows compared to the merely “batch-wise” operation of most of the other microfluidic platforms (i.e. handling discrete liquid volumes).
Some of the platforms can also be considered as “multi-application” platforms, which is of special interest in the field of research instrumentation. Here, portability is of less importance, and the number of multiple parameters per sample as well as programmability (potentially also during an assay run) gains impact. The microfluidic large scale integration and the droplet-based electrowetting and surface acoustic waves platforms are such versatile examples.
For high-throughput screening applications, on the contrary, a high number of assays need to be performed within an acceptable period of time at a minimum consumption of reagents per test. Consequently flexibility is less important, and throughput and costs are the main issues. Thus, approaches like segmented flow and dedicated systems for massively parallel analysis are interesting candidates for these applications.
An increasing number of application examples benefits from the transfer of unit operations and fabrication technologies between research groups by literature, collaboration or commercial supply (e.g. foundries). This shows the advance of the platform approach in the research community. We strongly believe that this trend of platform-based development will continue and speed up the variety of assay implementations in the field of microfluidics. If research time and development costs of microfluidic applications can be reduced significantly by this approach, and the spectrum of applications increases correspondingly, this could finally lead to the commercial breakthrough of microfluidic products.
Acknowledgements
We would like to thank our colleagues Peter Koltay, Junichi Miwa and Sven Kerzenmacher for their helpful suggestions and assistance during the preparation of this manuscript. We would also like to thank our colleagues Nicolai Wangler and Jan Lienemann (Lab for Simulation, IMTEK) for their support during the composition of the graphical abstract.Notes and references
- ISI Web of Science, search for topic “microfluidic”, http://www.isiknowledge.com, accessed 2009.
- United States Patent and Trademark office, search issued patents for “microfluidic” in title or abstract, http://patft.uspto.gov, accessed 2009.
- H. P. Le, J. Imaging Sci. Technol., 1998, 42, 49–62 CAS.
- S. C. Terry, J. H. Jerman and J. B. Angell, IEEE Trans. Electron Devices, 1979, 26, 1880–1886 CrossRef.
- A. Manz, Y. Miyahara, J. Miura, Y. Watanabe, H. Miyagi and K. Sato, Sens. Actuators, B, 1990, 1, 249–255 CrossRef.
- S. Shoji, M. Esashi and T. Matsuo, Sens. Actuators, 1988, 14, 101–107 CrossRef CAS.
- H. T. G. Van Lintel, F. C. M. Vandepol and S. Bouwstra, Sens. Actuators, 1988, 15, 153–167 CrossRef.
- R. Zengerle, J. Ulrich, S. Kluge, M. Richter and A. Richter, Sens. Actuators, A, 1995, 50, 81–86 CrossRef.
- E. Verpoorte, A. Manz, H. Ludi, A. E. Bruno, F. Maystre, B. Krattiger, H. M. Widmer, B. H. Vanderschoot and N. F. Derooij, Sens. Actuators, B, 1992, 6, 66–70 CrossRef.
- A. van den Berg and T. S. J. Lammerink, Top. Curr. Chem., 1998, 194, 21–49 CAS.
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, 1, 244–248 CrossRef.
- D. J. Harrison, A. Manz, Z. H. Fan, H. Ludi and H. M. Widmer, Anal. Chem., 1992, 64, 1926–1932 CrossRef.
- T. Chard, Hum. Reprod., 1992, 7, 701–710 CAS.
- J. M. Hicks and M. Iosefsohn, N. Engl. J. Med., 1989, 320, 320–321 CAS.
- D. J. Litman, R. H. Lee, H. J. Jeong, H. K. Tom, S. N. Stiso, N. C. Sizto and E. F. Ullman, Clin. Chem., 1983, 29, 1598–1603 CAS.
- L. Wilhelm, S. Jenckel and R. Junker, Laboratoriumsmedizin, 2008, 32, 168–174 CrossRef CAS.
- R. Pacifici, M. Farre, S. Pichini, J. Ortuno, P. N. Roset, P. Zuccaro, J. Segura and R. de la Torre, J. Anal. Toxicol., 2001, 25, 144–146 CAS.
- A. H. B. Wu, J. Clin. Ligand Assay, 1999, 22, 32–37 Search PubMed.
- R.-H. Shyu, H.-F. Shyu and S.-S. Tang, Toxicon, 2002, 40, 255–258 CrossRef CAS.
- H. Becker, Lab Chip, 2009, 9, 1659–1660 RSC.
- H. Becker, Lab Chip, 2009, 9, 2759–2762 RSC.
- R. J. Petri, Centralblatt für Bacteriologie und Parasitenkunde, 1887, 1, 279–280 Search PubMed.
- J. O. Corliss, Protist, 2001, 152, 69–85 CrossRef CAS.
- J. Hüser, R. Mannhold, H. Kubinyi and G. Folkers, in High-Throughput Screening in Drug Discovery (Methods and Principles in Medicinal Chemistry), Wiley-VCH Verlagsgesellschaft mbH & Co. KGaA, Weinheim, Germany, 1st edn, 2006, vol. 1 Search PubMed.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS.
- J. H. Leamon, W. L. Lee, K. R. Tartaro, J. R. Lanza, G. J. Sarkis, A. D. deWinter, J. Berka and K. L. Lohman, Electrophoresis, 2003, 24, 3769–3777 CrossRef.
- M. Margulies, M. Egholm, W. E. Altman, S. Attiya, J. S. Bader, L. A. Bemben, J. Berka, M. S. Braverman, Y. J. Chen, Z. T. Chen, S. B. Dewell, L. Du, J. M. Fierro, X. V. Gomes, B. C. Godwin, W. He, S. Helgesen, C. H. Ho, G. P. Irzyk, S. C. Jando, M. L. I. Alenquer, T. P. Jarvie, K. B. Jirage, J. B. Kim, J. R. Knight, J. R. Lanza, J. H. Leamon, S. M. Lefkowitz, M. Lei, J. Li, K. L. Lohman, H. Lu, V. B. Makhijani, K. E. Mcdade, M. P. McKenna, E. W. Myers, E. Nickerson, J. R. Nobile, R. Plant, B. P. Puc, M. T. Ronan, G. T. Roth, G. J. Sarkis, J. F. Simons, J. W. Simpson, M. Srinivasan, K. R. Tartaro, A. Tomasz, K. A. Vogt, G. A. Volkmer, S. H. Wang, Y. Wang, M. P. Weiner, P. G. Yu, R. F. Begley and J. M. Rothberg, Nature, 2005, 437, 376–380.
- S. Takayama, E. Ostuni, P. Leduc, K. Naruse, D. E. Ingber and G. M. Whitesides, Nature, 2001, 411, 1016 CrossRef CAS.
- S. Y. Teh, R. Lin, L. H. Hung and A. P. Lee, Lab Chip, 2008, 8, 198–220 RSC.
- A. Huebner, M. Srisa-Art, D. Holt, C. Abell, F. Hollfelder, A. J. deMello and J. B. Edel, Chem. Commun., 2007, 1218–1220 RSC.
- S. K. Cho, H. J. Moon and C. J. Kim, J. Microelectromech. Syst., 2003, 12, 70–80 CrossRef.
- C. S. Zhang and D. Xing, Nucleic Acids Res., 2007, 35, 4223–4237 CrossRef CAS.
- S. Shoji and M. Esashi, J. Micromech. Microeng., 1994, 4, 157–171 CrossRef CAS.
- D. J. Laser and J. G. Santiago, J. Micromech. Microeng., 2004, 14, R35–R64 CrossRef.
- P. Woias, Sens. Actuators, B, 2005, 105, 28–38 CrossRef.
- P. Gravesen, J. Braneberg and O. S. Jensen, J. Micromech. Microeng., 1993, 3, 168–182 CrossRef CAS.
- K. W. Oh and C. H. Ahn, J. Micromech. Microeng., 2006, 16, R13–R39 CrossRef.
- N. T. Nguyen and Z. G. Wu, J. Micromech. Microeng., 2005, 15, R1–R16 CrossRef.
- V. Hessel, H. Lowe and F. Schonfeld, Chem. Eng. Sci., 2005, 60, 2479–2501 CrossRef CAS.
- J. Ducrée and R. Zengerle, in FlowMap-Microfluidics Roadmap for the Life Sciences, Books on Demand GmbH, Norderstedt, Germany, 2004 Search PubMed.
- M. Banks, A. Cacace, J. O'Connel and J. Houston, in Drug Discovery Handbook, ed. S. C. Gad, John Wiley & Sons, Inc., Cary, North Carolina, 1st edn, 2005, ch. 13, vol. 1, pp. 559–602 Search PubMed.
- G. E. Croston, Trends Biotechnol., 2002, 20, 110–115 CrossRef CAS.
- S. A. Sundberg, Curr. Opin. Biotechnol., 2000, 11, 47–53 CrossRef CAS.
- H. L. T. Lee, P. Boccazzi, R. J. Ram and A. J. Sinskey, Lab Chip, 2006, 6, 1229–1235 RSC.
- S. T. Yang, X. D. Zhang and Y. Wen, Curr. Opin. Drug Discovery Dev., 2008, 11, 111–127 CAS.
- Y. Wen and S. T. Yang, Expert Opin. Drug Discovery, 2008, 3, 1237–1253 Search PubMed.
- V. Hessel and H. Lowe, Chem. Eng. Technol., 2003, 26, 13–24 CrossRef CAS.
- H. Löwe and W. Ehrfeld, Electrochim. Acta, 1999, 44, 3679–3689 CrossRef CAS.
- W. Ehrfeld, H. Lowe, V. Hessel and T. Richter, Chem. Ing. Tech., 1997, 69, 931–934 CrossRef CAS.
- D. M. Roberge, L. Ducry, N. Bieler, P. Cretton and B. Zimmermann, Chem. Eng. Technol., 2005, 28, 318–323 CrossRef CAS.
- H. Pennemann, V. Hessel and H. Lowe, Chem. Eng. Sci., 2004, 59, 4789–4794 CrossRef CAS.
- A. M. Elizarov, Lab Chip, 2009, 9, 1326–1333 RSC.
- Y. L. Liu, J. D. Adams, K. Turner, F. V. Cochran, S. S. Gambhir and H. T. Soh, Lab Chip, 2009, 9, 1033–1036 RSC.
- P. H. Bessette, X. Y. Hu, H. T. Soh and P. S. Daugherty, Anal. Chem., 2007, 79, 2174–2178 CrossRef CAS.
- L. M. Borland, S. Kottegoda, K. S. Phillips and N. L. Allbritton, Annu. Rev. Anal. Chem., 2008, 1, 191–227 Search PubMed.
- T. C. Chao and A. Ros, J. R. Soc. Interface, 2008, 5, S139–S150 CrossRef CAS.
- H. Becker and L. E. Locascio, Talanta, 2002, 56, 267–287 CrossRef CAS.
- T. B. Christensen, C. M. Pedersen, K. G. Grondhal, T. G. Jensen, A. Sekulovic, D. D. Bang and A. Wolff, J. Micromech. Microeng., 2007, 17, 1527–1532 CrossRef CAS.
- F. Reynolds, J. Pitha, P. M. Pitha and D. Grundberg, Biochemistry, 1972, 11, 3261–3266 CrossRef CAS.
- S. Landi, H. R. Held and M. C. Tseng, Appl. Microbiol., 1970, 20, 696–703 Search PubMed.
- L. Gunasekara, W. M. Schoel, S. Schurch and M. W. Amrein, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 433–444 CrossRef CAS.
- H. Schonheyder and P. Andersen, J. Immunol. Methods, 1984, 72, 251–259 CrossRef CAS.
- A. G. Papavassiligy and D. Bohmann, Nucleic Acids Res., 1992, 20, 4365–4366 CrossRef.
- J. Steigert, S. Haeberle, T. Brenner, C. Muller, C. P. Steinert, P. Koltay, N. Gottschlich, H. Reinecke, J. Ruhe, R. Zengerle and J. Ducrée, J. Micromech. Microeng., 2007, 17, 333–341 CrossRef CAS.
- C. W. Tsao and D. L. Devoe, Microfluid. Nanofluid., 2009, 6, 1–16 CrossRef CAS.
- C. Glad and A. O. Grubb, Anal. Biochem., 1978, 85, 180–187 CrossRef CAS.
- J. H. W. Leuvering, P. J. H. M. Thal, M. V. D. Waart and A. H. W. M. Schuurs, Fresenius' Z. Anal. Chem., 1980, 301, 132 CrossRef CAS.
- G. A. Posthuma-Trumpie, J. Korf and A. van Amerongen, Anal. Bioanal. Chem., 2009, 393, 569–582 CrossRef CAS.
- B. Cosmi, G. Palareti, M. Moia, M. Carpenedo, V. Pengo, A. Biasiolo, P. Rampazzo, G. Morstabilini and S. Testa, Thromb. Res., 2000, 100, 279–286 CrossRef CAS.
- T. J. Clark, P. H. McPherson and K. F. Buechler, Point of Care, 2002, 1, 42–46 Search PubMed.
- S. Birnbaum, C. Uden, C. G. M. Magnusson and S. Nilsson, Anal. Biochem., 1992, 206, 168–171.
- H. W. Wen, W. Borejsza-Wysocki, T. DeCory and R. Durst, Anal. Bioanal. Chem., 2005, 382, 1217–1226 CrossRef CAS.
- Evaluation of the CoaguChek XS System, International Evaluation Workshop, Heidelberg, Germany, 2009.
- W. Leung, C. P. Chan, T. H. Rainer, M. Ip, G. W. H. Cautherley and R. Renneberg, J. Immunol. Methods, 2008, 336, 30–36 CrossRef CAS.
- A. Heller and B. Feldman, Chem. Rev., 2008, 108, 2482–2505 CrossRef CAS.
- J. Daviaud, D. Fournet, C. Ballongue, G. Guillem, A. Leblanc, C. Casellas and B. Pan, N. Engl. J. Med., 1989, 320, 320–321 CAS.
- P. Bohme, M. Floriot, M. A. Sirveaux, D. Durain, O. Ziegler, P. Drouin and B. Guerci, Diabetes Care, 2003, 26, 1170–1175 CrossRef CAS.
- S. C. Lou, C. Patel, S. F. Ching and J. Gordon, Clin. Chem., 1993, 39, 619–624 CAS.
- R. Krska and A. Molinelli, Anal. Bioanal. Chem., 2009, 393, 67–71 CrossRef CAS.
- H. L. Xie, W. Ma, L. Q. Liu, W. Chen, C. F. Peng, C. L. Xu and L. B. Wang, Anal. Chim. Acta, 2009, 634, 129–133 CrossRef CAS.
- D. J. Carter and R. B. Cary, Nucleic Acids Res., 2007, 35, e74 CrossRef.
- J. A. A. Ho, S. C. Zeng, W. H. Tseng, Y. J. Lin and C. H. Chen, Anal. Bioanal. Chem., 2008, 391, 479–485 CrossRef CAS.
- K. A. Edwards and A. J. Baeumner, Methods Mol. Biol., 2009, 185–215 CAS.
- L. Gervais and E. Delamarche, Lab Chip, 2009, 9, 3330–3337 RSC.
- P. Yager, T. Edwards, E. Fu, K. Helton, K. Nelson, M. R. Tam and B. H. Weigl, Nature, 2006, 442, 412–418 CrossRef CAS.
- J. Hu, Biosens. Bioelectron., 2009, 24, 1083–1089 CrossRef CAS.
- K. A. Erickson and P. Wilding, Clin. Chem., 1993, 39, 283–287 CAS.
- S. Chen, G. Selecman and B. Lemieux, IVD Technology, 2004, 7, 51 Search PubMed.
- Abbott Point-of-Care, USA, http://www.abbottpointofcare.com, accessed 2006.
- B. S. Karon, R. D. Mcbane, R. Chaudhry, L. K. Beyer and P. J. Santrach, Am. J. Clin. Pathol., 2008, 130, 88–92 CrossRef.
- E. Jacobs, E. Vadasdi, L. Sarkozi and N. Colman, Clin. Chem., 1993, 39, 1069–1074 CAS.
- IQuum, Inc., http://www.iquum.com, accessed 2009.
- L. Spielman and S. L. Goren, J. Colloid Interface Sci., 1968, 26, 175–182 CrossRef.
- G. Valet, J. Biol. Regul. Homeost. Agents, 2003, 17, 213–222 Search PubMed.
- D. Huh, W. Gu, Y. Kamotani, J. B. Grotberg and S. Takayama, Physiol. Meas., 2005, 26, R73–R98 CrossRef.
- X. D. Wu, C. H. Chon, Y. N. Wang, Y. J. Kang and D. Q. Li, Lab Chip, 2008, 8, 1943–1949 RSC.
- M. Yamada and M. Seki, Anal. Chem., 2006, 78, 1357–1362 CrossRef CAS.
- M. Yamada and M. Seki, Lab Chip, 2005, 5, 1233–1239 RSC.
- M. Yamada, M. Nakashima and M. Seki, Anal. Chem., 2004, 76, 5465–5471 CrossRef CAS.
- S. Chang and Y. H. Cho, Lab Chip, 2008, 8, 1930–1936 RSC.
- A. A. S. Bhagat, S. S. Kuntaegowdanahalli and I. Papautsky, Lab Chip, 2008, 8, 1906–1914 RSC.
- A. T. Woolley, D. Hadley, P. Landre, A. J. de Mello, R. A. Mathies and M. A. Northrup, Anal. Chem., 1996, 68, 4081–4086 CrossRef CAS.
- K. Sato, A. Hibara, M. Tokeshi, H. Hisamoto and T. Kitamori, Anal. Sci., 2003, 19, 15–22 CAS.
- M. Karle, J. Miwa, G. Roth, R. Zengerle and F. von Stetten, Proceedings of the 22nd IEEE International Conference on Micro Electro Mechanical Systems, Sorrento, 2009 Search PubMed.
- J. H. Kang and J. K. Park, Small, 2007, 3, 1784–1791 CrossRef CAS.
- N. Pamme and A. Manz, Anal. Chem., 2004, 76, 7250–7256 CrossRef CAS.
- N. Pamme and C. Wilhelm, Lab Chip, 2006, 6, 974–980 RSC.
- T. Laurell, F. Petersson and A. Nilsson, Chem. Soc. Rev., 2007, 36, 492–506 RSC.
- U. Kim, C. W. Shu, K. Y. Dane, P. S. Daugherty, J. Y. J. Wang and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20708–20712 CrossRef CAS.
- J. Takagi, M. Yamada, M. Yasuda and M. Seki, Lab Chip, 2005, 5, 778–784 RSC.
- A. F. Sauer-Budge, P. Mirer, A. Chatterjee, C. M. Klapperich, D. Chargin and A. Sharon, Lab Chip, 2009, 9, 2803–2810 RSC.
- P. A. Auroux, Y. Koc, A. deMello, A. Manz and P. J. R. Day, Lab Chip, 2004, 4, 534–546 RSC.
- L. Chen, A. Manz and P. J. R. Day, Lab Chip, 2007, 7, 1413–1423 RSC.
- C. J. Easley, J. M. Karlinsey, J. M. Bienvenue, L. A. Legendre, M. G. Roper, S. H. Feldman, M. A. Hughes, E. L. Hewlett, T. J. Merkel, J. P. Ferrance and J. P. Landers, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19272–19277 CrossRef CAS.
- G. Taylor, Proc. R. Soc. London, Ser. A, 1953, 219, 186–203 Search PubMed.
- A. Kumar and G. M. Whitesides, Appl. Phys. Lett., 1993, 63, 2002–2004 CrossRef.
- J. A. Rogers and R. G. Nuzzo, Mater. Today, 2005, 8, 50–56 CrossRef CAS.
- S. K. Sia and G. M. Whitesides, Electrophoresis, 2003, 24, 3563–3576 CrossRef CAS.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS.
- S. Haeberle and R. Zengerle, Lab Chip, 2007, 7, 1094–1110 RSC.
- S. R. Quake and A. Scherer, Science, 2000, 290, 1536–1540 CrossRef CAS.
- H. P. Chou, M. A. Unger and S. R. Quake, Biomed. Microdevices, 2001, 3, 323–330 CrossRef.
- J. W. Hong, V. Studer, G. Hang, W. F. Anderson and S. R. Quake, Nat. Biotechnol., 2004, 22, 435–439 CrossRef CAS.
- J. W. Hong and S. R. Quake, Nat. Biotechnol., 2003, 21, 1179–1183 CrossRef CAS.
- J. S. Marcus, W. F. Anderson and S. R. Quake, Anal. Chem., 2006, 78, 3084–3089 CrossRef CAS.
- J. S. Marcus, W. F. Anderson and S. R. Quake, Anal. Chem., 2006, 78, 956–958 CrossRef CAS.
- J. Liu, C. Hansen and S. R. Quake, Anal. Chem., 2003, 75, 4718–4723 CrossRef CAS.
- M. J. Anderson, C. L. Hansen and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16746–16751 CrossRef CAS.
- E. P. Kartalov, J. F. Zhong, A. Scherer, S. R. Quake, C. R. Taylor and W. F. Anderson, BioTechniques, 2006, 40, 85–90 CrossRef CAS.
- R. Gómez-Sjöberg, A. A. Leyrat, D. M. Pirone, C. S. Chen and S. R. Quake, Anal. Chem., 2007, 79, 8557–8563 CrossRef.
- J. Melin, A. Lee, K. Foygel, D. E. Leong, S. R. Quake and M. W. M. Yao, Dev. Dyn., 2009, 238, 950–955 CrossRef.
- Y. Y. Huang, P. Castrataro, C. C. Lee and S. R. Quake, Lab Chip, 2007, 7, 24–26 RSC.
- R. A. White, P. C. Blainey, H. C. Fan and S. R. Quake, BMC Genomics, 2009, 10, 116 CrossRef.
- S. Bhat, J. Herrmann, P. Armishaw, P. Corbisier and K. R. Emslie, Anal. Bioanal. Chem., 2009, 394, 457–467 CrossRef CAS.
- A. R. Wheeler, W. R. Throndset, R. J. Whelan, A. M. Leach, R. N. Zare, Y. H. Liao, K. Farrell, I. D. Manger and A. Daridon, Anal. Chem., 2003, 75, 3581–3586 CrossRef CAS.
- J. D. Berndt, T. L. Biechele, R. T. Moon and M. B. Major, Sci. Signal., 2009, 2, t4 Search PubMed.
- J. A. Weinstein, N. Jiang, R. A. White, D. S. Fisher and S. R. Quake, Science, 2009, 324, 807–810 CrossRef CAS.
- V. G. Oehler, J. Qin, R. Ramakrishnan, G. Facer, S. Ananthnarayan, C. Cummings, M. Deininger, N. Shah, F. McCormick, S. Willis, A. Daridon, M. Unger and J. P. Radich, Leukemia, 2009, 23, 396–399 CrossRef CAS.
- J. E. Lee, M. L. Fusco and E. O. Saphire, Nat. Protoc., 2009, 4, 592–604 Search PubMed.
- W. Gu, X. Y. Zhu, N. Futai, B. S. Cho and S. Takayama, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15861–15866 CrossRef CAS.
- C. Holtze, A. C. Rowat, J. J. Agresti, J. B. Hutchison, F. E. Angile, C. H. J. Schmitz, S. Koster, H. Duan, K. J. Humphry, R. A. Scanga, J. S. Johnson, D. Pisignano and D. A. Weitz, Lab Chip, 2008, 8, 1632–1639 RSC.
- C. E. Sims and N. L. Allbritton, Lab Chip, 2007, 7, 423–440 RSC.
- A. Huebner, S. Sharma, M. Srisa-Art, F. Hollfelder, J. B. Edel and A. J. deMello, Lab Chip, 2008, 8, 1244–1254 RSC.
- S. L. Anna, N. Bontoux and H. A. Stone, Appl. Phys. Lett., 2003, 82, 364–366 CrossRef CAS.
- M. Joanicot and A. Ajdari, Science, 2005, 309, 887–888 CrossRef CAS.
- T. Nisisako, T. Torii and T. Higuchi, Lab Chip, 2002, 2, 24–26 RSC.
- D. Malsch, N. Gleichmann, M. Kielpinski, G. Mayer, T. Henkel, D. Mueller, V. van Steijn, C. Kleijn and M. Kreutzer, Microfluid. Nanofluid., 2009 DOI:10.1007/s10404-009-0479-5.
- H. Song, J. D. Tice and R. F. Ismagilov, Angew. Chem., Int. Ed., 2003, 42, 768–772 CrossRef CAS.
- H. Song and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 14613–14619 CrossRef CAS.
- B. Zheng, J. D. Tice and R. F. Ismagilov, Anal. Chem., 2004, 76, 4977–4982 CrossRef CAS.
- I. Shestopalov, J. D. Tice and R. F. Ismagilov, Lab Chip, 2004, 4, 316–321 RSC.
- M. Y. He, J. S. Edgar, G. D. M. Jeffries, R. M. Lorenz, J. P. Shelby and D. T. Chiu, Anal. Chem., 2005, 77, 1539–1544 CrossRef CAS.
- D. R. Link, S. L. Anna, D. A. Weitz and H. A. Stone, Phys. Rev. Lett., 2004, 92, 054503 CrossRef CAS.
- J. D. Tice, H. Song, A. D. Lyon and R. F. Ismagilov, Langmuir, 2003, 19, 9127–9133 CrossRef CAS.
- J. D. Tice, A. D. Lyon and R. F. Ismagilov, Anal. Chim. Acta, 2004, 507, 73–77 CrossRef CAS.
- H. Song, M. R. Bringer, J. D. Tice, C. J. Gerdts and R. F. Ismagilov, Appl. Phys. Lett., 2003, 83, 4664–4666 CrossRef CAS.
- M. R. Bringer, C. J. Gerdts, H. Song, J. D. Tice and R. F. Ismagilov, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 1087–1104 CrossRef CAS.
- A. Günther, S. A. Khan, M. Thalmann, F. Trachsel and K. F. Jensen, Lab Chip, 2004, 4, 278–286 RSC.
- A. Günther, M. Jhunjhunwala, M. Thalmann, M. A. Schmidt and K. F. Jensen, Langmuir, 2005, 21, 1547–1555 CrossRef.
- S. A. Khan, A. Günther, M. A. Schmidt and K. F. Jensen, Langmuir, 2004, 20, 8604–8611 CrossRef CAS.
- P. Garstecki, M. A. Fischbach and G. M. Whitesides, Appl. Phys. Lett., 2005, 86, 244108 CrossRef.
- K. Martin, T. Henkel, V. Baier, A. Grodrian, T. Schon, M. Roth, J. M. Kohler and J. Metze, Lab Chip, 2003, 3, 202–207 RSC.
- D. R. Link, E. Grasland-Mongrain, A. Duri, F. Sarrazin, Z. D. Cheng, G. Cristobal, M. Marquez and D. A. Weitz, Angew. Chem., Int. Ed., 2006, 45, 2556–2560 CrossRef CAS.
- K. Ahn, C. Kerbage, T. P. Hunt, R. M. Westervelt, D. R. Link and D. A. Weitz, Appl. Phys. Lett., 2006, 88, 024104 CrossRef.
- J. F. Edd, D. Di Carlo, K. J. Humphry, S. Koster, D. Irimia, D. A. Weitz and M. Toner, Lab Chip, 2008, 8, 1262–1264 RSC.
- J. Q. Boedicker, L. Li, T. R. Kline and R. F. Ismagilov, Lab Chip, 2008, 8, 1265–1272 RSC.
- W. W. Shi, J. H. Qin, N. N. Ye and B. C. Lin, Lab Chip, 2008, 8, 1432–1435 RSC.
- A. Huebner, L. F. Olguin, D. Bratton, G. Whyte, W. T. S. Huck, A. J. de Mello, J. B. Edel, C. Abell and F. Hollfelder, Anal. Chem., 2008, 80, 3890–3896 CrossRef CAS.
- A. Funfak, A. Brosing, M. Brand and J. M. Kohler, Lab Chip, 2007, 7, 1132–1138 RSC.
- A. Reichert, J. Felbel, M. Kielpinski, M. Urban, B. Steinbrecht and T. Henkel, J. Bionic Eng., 2008, 5, 291–298 Search PubMed.
- Y. Schaerli, R. C. Wootton, T. Robinson, V. Stein, C. Dunsby, M. A. A. Neil, P. M. W. French, A. J. deMello, C. Abell and F. Hollfelder, Anal. Chem., 2009, 81, 302–306 CrossRef CAS.
- M. Srisa-Art, A. J. deMello and J. B. Edel, Anal. Chem., 2007, 79, 6682–6689 CrossRef CAS.
- B. Zheng, J. D. Tice, L. S. Roach and R. F. Ismagilov, Angew. Chem., Int. Ed., 2004, 43, 2508–2511 CrossRef CAS.
- M. K. Yadav, C. J. Gerdts, R. Sanishvili, W. W. Smith, L. S. Roach, R. F. Ismagilov, P. Kuhn and R. C. Stevens, J. Appl. Crystallogr., 2005, 38, 900–905 CrossRef CAS.
- L. S. Roach, H. Song and R. F. Ismagilov, Anal. Chem., 2005, 77, 785–796 CrossRef CAS.
- B. Zheng, J. D. Tice and R. F. Ismagilov, Adv. Mater., 2004, 16, 1365–1368 CrossRef CAS.
- B. Zheng, C. J. Gerdts and R. F. Ismagilov, Curr. Opin. Struct. Biol., 2005, 15, 548–555 CrossRef CAS.
- D. L. Chen, C. J. Gerdts and R. F. Ismagilov, J. Am. Chem. Soc., 2005, 127, 9672–9673 CrossRef CAS.
- B. Zheng, L. S. Roach and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 11170–11171 CrossRef CAS.
- N. G. Anderson, Science, 1969, 166, 317–324 CrossRef CAS.
- M. Madou, J. Zoval, G. Y. Jia, H. Kido, J. Kim and N. Kim, Annu. Rev. Biomed. Eng., 2006, 8, 601–628 CrossRef CAS.
- J. Ducrée, S. Haeberle, S. Lutz, S. Pausch, F. von Stetten and R. Zengerle, J. Micromech. Microeng., 2007, 17, S103–S115 CrossRef CAS.
- D. D. Nolte, Rev. Sci. Instrum., 2009, 80, 101101 CrossRef.
- J. M. Koo and C. Kleinstreuer, J. Micromech. Microeng., 2003, 13, 568–579 CrossRef.
- Abaxis Inc., USA, http://www.abaxis.com, accessed 2006.
- C. T. Schembri, T. L. Burd, A. R. Kopfsill, L. R. Shea and B. Braynin, J. Autom. Chem., 1995, 17, 99–104 CAS.
- M. Madou and G. J. Kellogg, Proc. Soc. Photo-Opt. Instrum. Eng., 1998, 3259, 80–93.
- D. C. Duffy, H. L. Gillis, J. Lin, N. F. Sheppard, Jr. and G. J. Kellogg, Anal. Chem., 1999, 71, 4669–4678 CrossRef CAS.
- G. Ekstrand, C. Holmquist, A. E. Örlefors, B. Hellman, A. Larsson and P. Andersson, Proceedings of the 4th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Enschede, 2000 Search PubMed.
- M. Madou, J. Lee, S. Daunert, S. Lai and C.-H. Shih, Biomed. Microdevices, 2001, 3, 245–254 CrossRef CAS.
- S. Haeberle, T. Brenner, H. P. Schlosser, R. Zengerle and J. Ducrée, Chem. Eng. Technol., 2005, 28, 613–616 CrossRef CAS.
- H. Cho, H. Y. Kim, J. Y. Kang and T. S. Kim, J. Colloid Interface Sci., 2007, 306, 379–385 CrossRef CAS.
- J. M. Chen, P. C. Huang and M. G. Lin, Microfluid. Nanofluid., 2008, 4, 427–437 CrossRef.
- N. Honda, U. Lindberg, P. Andersson, S. Hoffman and H. Takei, Clin. Chem., 2005, 51, 1955–1961 CrossRef CAS.
- C. P. Steinert, J. Mueller-Dieckmann, M. Weiss, M. Roessle, R. Zengerle and P. Koltay, Proceedings of 19th International Conference on Micro Electro Mechanical Systems, Kobe, 2007 Search PubMed.
- S. Haeberle, T. Brenner, R. Zengerle and J. Ducrée, Lab Chip, 2006, 6, 776–781 RSC.
- D. Mark, T. Metz, S. Haeberle, S. Lutz, J. Ducrée, R. Zengerle and F. von Stetten, Lab Chip, 2009, 9, 3599–3603 RSC.
- D. H. Sharp, Phys. D, 1984, 12, 3–18 CrossRef.
- J. M. Park, Y. K. Cho, B. S. Lee, J. G. Lee and C. Ko, Lab Chip, 2007, 7, 557–564 RSC.
- J. Steigert, M. Grumann, T. Brenner, K. Mittenbühler, T. Nann, J. Rühe, I. Moser, S. Haeberle, L. Riegger, J. Riegler, W. Bessler, R. Zengerle and J. Ducrée, JALA, 2005, 10, 331–341 Search PubMed.
- J. Steigert, M. Grumann, T. Brenner, L. Riegger, J. Harter, R. Zengerle and J. Ducrée, Lab Chip, 2006, 6, 1040–1044 RSC.
- J. V. Zoval and M. J. Madou, Proc. IEEE, 2004, 92, 140–153 CrossRef CAS.
- S. Lutz, V. Reitenbach, D. Mark, J. Ducrée, R. Zengerle and F. von Stetten, Proceedings of the 12th International Conference on Miniaturized Systems for Chemistry and Life Sciences, San Diego, 2008 Search PubMed.
- M. Grumann, A. Geipel, L. Riegger, R. Zengerle and J. Ducrée, Lab Chip, 2005, 5, 560–565 RSC.
- US Pat., 7147362, 2006.
- T. Brenner, T. Glatzel, R. Zengerle and J. Ducrée, Lab Chip, 2005, 5, 146–150 RSC.
- S. Haeberle, L. Naegele, R. Zengerle and J. Ducrée, Proceedings of the 10th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Tokyo, 2006 Search PubMed.
- Gyros AB, Sweden, http://www.gyros.com, accessed 2006.
- WO Pat., 2005032999, 2005.
- J. Kim, H. Kido, R. H. Rangel and M. J. Madou, Sens. Actuators, B, 2008, 128, 613–621 CrossRef.
- J. L. Zhang, Q. Q. Guo, M. Liu and J. Yang, J. Micromech. Microeng., 2008, 18, 125025 CrossRef.
- G. J. Kellogg, T. E. Arnold, B. L. Carvalho, D. C. Duffy and N. F. Sheppard, Jr., Proceedings of the 4th International Conference on Miniaturized Systems for Chemistry and Life Sciences, 2000 Search PubMed.
- I. H. A. Badr, R. D. Johnson, M. J. Madou and L. G. Bachas, Anal. Chem., 2002, 74, 5569–5575 CrossRef CAS.
- R. D. Johnson, I. H. Badr, G. Barrett, S. Lai, Y. Lu, M. J. Madou and L. G. Bachas, Anal. Chem., 2001, 73, 3940–3946 CrossRef CAS.
- A. S. Watts, A. A. Urbas, E. Moschou, V. G. Gavalas, J. V. Zoval, M. Madou and L. G. Bachas, Anal. Chem., 2007, 79, 8046–8054 CrossRef CAS.
- L. G. Puckett, E. Dikici, S. Lai, M. Madou, L. G. Bachas and S. Daunert, Anal. Chem., 2004, 76, 7263–7268 CrossRef CAS.
- G. Jia, K. S. Ma, J. Kim, J. V. Zoval, R. Peytavi, M. G. Bergeron and M. J. Madou, Sens. Actuators, B, 2006, 114, 173–181 CrossRef.
- S. Lai, S. Wang, J. Luo, L. J. Lee, S. T. Yang and M. J. Madou, Anal. Chem., 2004, 76, 1832–1837 CrossRef CAS.
- C. Lu, Y. Xie, Y. Yang, M. M. Cheng, C. G. Koh, Y. Bai, L. J. Lee and Y. J. Juang, Anal. Chem., 2007, 79, 994–1001 CrossRef CAS.
- L. Riegger, J. Steigert, M. Grumann, S. Lutz, G. Olofsson, M. Khayyami, W. Bessler, K. Mittenbühler, R. Zengerle and J. Ducrée, Proceeding of the 19th IEEE International Conference on Micro Electro Mechanical Systems, Istanbul, 2006 Search PubMed.
- S. Lutz, P. Lang, I. Malki, D. Mark, J. Ducrée, R. Zengerle and F. von Stetten, Proceedings of the 12th International Conference on Miniaturized Systems for Chemistry and Life Sciences, 2008 Search PubMed.
- B. S. Lee, J. N. Lee, J. M. Park, J. G. Lee, S. Kim, Y. K. Cho and C. Ko, Lab Chip, 2009, 9, 1548–1555 RSC.
- Y. K. Cho, J. G. Lee, J. M. Park, B. S. Lee, Y. Lee and C. Ko, Lab Chip, 2007, 7, 565–573 RSC.
- SpinX Technologies, Switzerland, http://www.spinx-technologies.com, accessed 2006.
- C. A. Burtis, N. G. Anderson, J. C. Mailen, C. D. Scott, T. O. Tiffany and W. F. Johnson, Clin. Chem., 1972, 18, 753–761 CAS.
- J. Ducrée, S. Haeberle, T. Brenner, T. Glatzel and R. Zengerle, Microfluid. Nanofluid., 2006, 2, 97–105 CrossRef.
- J. Ducrée, T. Brenner, S. Haeberle, T. Glatzel and R. Zengerle, Microfluid. Nanofluid., 2006, 2, 78–84 CrossRef CAS.
- M. Grumann, J. Steigert, L. Riegger, I. Moser, B. Enderle, K. Riebeseel, G. Urban, R. Zengerle and J. Ducrée, Biomed. Microdevices, 2006, 8, 209–214 CrossRef CAS.
- S. Haeberle, S. Pausch, R. Burger, S. Lutz, F. von Stetten, R. Zengerle and J. Ducrée, Proceedings of the 11th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Paris, 2007 Search PubMed.
- J. Hoffmann, D. Mark, R. Zengerle and F. von Stetten, Proceedings of the 15th IEEE International Conference on Solid-State Sensors, Actuators and Microsystems, Denver, 2009 Search PubMed.
- A. P. Wong, M. Gupta, S. S. Shevkoplyas and G. M. Whitesides, Lab Chip, 2008, 8, 2032–2037 RSC.
- H. Kido, M. Micic, D. Smith, J. Zoval, J. Norton and M. Madou, Colloids Surf., B, 2007, 58, 44–51 CrossRef CAS.
- S. A. Lange, G. Roth, S. Wittemann, T. Lacoste, A. Vetter, J. Grassle, S. Kopta, M. Kolleck, B. Breitinger, M. Wick, J. K. H. Horber, S. Dubel and A. Bernard, Angew. Chem., Int. Ed., 2006, 45, 270–273 CrossRef CAS.
- M. Focke, B. Faltin, T. Hoesel, C. Mueller, J. Ducrée, R. Zengerle and F. von Stetten, Proceedings of the 12th International Conference on Miniaturized Systems for Chemistry and Life Sciences, San Diego, 2008 Search PubMed.
- R. A. Potyrailo, W. G. Morris, A. M. Leach, T. M. Sivavec, M. B. Wisnudel and S. Boyette, Anal. Chem., 2006, 78, 5893–5899 CrossRef CAS.
- A. Penrose, P. Myers, K. Bartle and S. McCrossen, Analyst, 2004, 129, 704–709 RSC.
- N. Sasaki, T. Kitamori and H. B. Kim, Lab Chip, 2006, 6, 550–554 RSC.
- P. W. Atkins, Physikalische Chemie, 1987, pp. 781 Search PubMed.
- C. Y. Lee, G. B. Lee, J. L. Lin, F. C. Huang and C. S. Liao, J. Micromech. Microeng., 2005, 15, 1215–1223 CrossRef CAS.
- D. E. Raymond, A. Manz and H. M. Widmer, Anal. Chem., 1994, 66, 2858–2865 CrossRef CAS.
- H. Morgan, M. P. Hughes and N. G. Green, Biophys. J., 1999, 77, 516–525 CrossRef CAS.
- U. Zimmermann and J. Vienken, J. Membr. Biol., 1982, 67, 165–182 CrossRef CAS.
- A. Valero, J. N. Post, J. W. van Nieuwkasteele, P. M. ter Braak, W. Kruijer and A. van den Berg, Lab Chip, 2008, 8, 62–67 RSC.
- R. Krupke, F. Hennrich, H. von Lohneysen and M. M. Kappes, Science, 2003, 301, 344–347 CrossRef CAS.
- D. J. Harrison, K. Fluri, K. Seiler, Z. H. Fan, C. S. Effenhauser and A. Manz, Science, 1993, 261, 895–897 CrossRef CAS.
- C. S. Effenhauser, A. Manz and H. M. Widmer, Anal. Chem., 1993, 65, 2637–2642 CrossRef CAS.
- L. Wang, J. Lu, S. A. Marukenko, E. S. Monuki, L. A. Flanagan and A. P. Lee, Electrophoresis, 2009, 30, 782–791 CrossRef CAS.
- P. Patel and G. H. Markx, Enzyme Microb. Technol., 2008, 43, 463–470 CrossRef CAS.
- C. J. Huang, A. L. Chen, L. Wang, M. Guo and J. Yu, Biomed. Microdevices, 2007, 9, 335–343 CrossRef.
- J. M. Ramsey, S. C. Jacobson and M. R. Knapp, Nat. Med., 1995, 1, 1093–1096 CAS.
- Caliper Life Sciences, USA, http://www.caliperls.com, accessed 2007.
- Agilent Technologies Inc., USA, http://www.agilent.com, accessed 2007.
- J. Cheng, E. L. Sheldon, L. Wu, A. Uribe, L. O. Gerrue, J. Carrino, M. J. Heller and J. P. O'Connell, Nat. Biotechnol., 1998, 16, 541–546 CAS.
- W. Kohlrausch, in Leitfaden der Praktischen Physik, BiblioBazaar, Charleston, SC, USA, 31st edn, 1875 Search PubMed.
- G. Lippmann, Ann. Chim. Phys., 1875, 5, 494–549 Search PubMed.
- F. Mugele and J. C. Baret, J. Phys.: Condens. Matter, 2005, 17, R705–R774 CrossRef CAS.
- J. Lee, H. Moon, J. Fowler, T. Schoellhammer and C. J. Kim, Sens. Actuators, A, 2002, 95, 259–268 CrossRef.
- M. G. Pollack, R. B. Fair and A. D. Shenderov, Appl. Phys. Lett., 2000, 77, 1725–1726 CrossRef CAS.
- J. Lee and C. J. Kim, J. Microelectromech. Syst., 2000, 9, 171–180 CrossRef CAS.
- D. Chatterjee, B. Hetayothin, A. R. Wheeler, D. J. King and R. L. Garrell, Lab Chip, 2006, 6, 199–206 RSC.
- V. Srinivasan, V. K. Pamula and R. B. Fair, Lab Chip, 2004, 4, 310–315 RSC.
- H. Ren, R. B. Fair and M. G. Pollack, Sens. Actuators, B, 2004, 98, 319–327 CrossRef.
- J. Berthier, P. Clementz, O. Raccurt, D. Jary, P. Claustre, C. Peponnet and Y. Fouillet, Sens. Actuators, A, 2006, 127, 283–294 CrossRef.
- M. G. Pollack, A. D. Shenderov and R. B. Fair, Lab Chip, 2002, 2, 96–101 RSC.
- P. Paik, V. K. Pamula, M. G. Pollack and R. B. Fair, Lab Chip, 2003, 3, 28–33 RSC.
- P. Paik, V. K. Pamula and R. B. Fair, Lab Chip, 2003, 3, 253–259 RSC.
- A. R. Wheeler, H. Moon, C. J. Kim, J. A. Loo and R. L. Garrell, Anal. Chem., 2004, 76, 4833–4838 CrossRef CAS.
- Y. H. Chang, G. B. Lee, F. C. Huang, Y. Y. Chen and J. L. Lin, Biomed. Microdevices, 2006, 8, 215–225 CrossRef CAS.
- R. B. Fair, Microfluid. Nanofluid., 2007, 3, 245–281 CrossRef CAS.
- A. Wixforth, Superlattices Microstruct., 2003, 33, 389–396 CrossRef CAS.
- A. Wixforth, C. Strobl, C. Gauer, A. Toegl, J. Scriba and Z. von Guttenberg, Anal. Bioanal. Chem., 2004, 379, 982–991 CrossRef CAS.
- D. Beyssen, L. Le Brizoual, O. Elmazria and P. Alnot, Sens. Actuators, B, 2006, 118, 380–385 CrossRef.
- M. K. Tan, J. R. Friend and L. Y. Yeo, Lab Chip, 2007, 7, 618–625 RSC.
- Z. Guttenberg, H. Muller, H. Habermuller, A. Geisbauer, J. Pipper, J. Felbel, M. Kielpinski, J. Scriba and A. Wixforth, Lab Chip, 2005, 5, 308–317 RSC.
- S. Fox, S. Farr-Jones, L. Sopchak, A. Boggs and J. Comley, J. Biomol. Screening, 2004, 9, 354–358 CrossRef CAS.
- R. P. Hertzberg and A. J. Pope, Curr. Opin. Chem. Biol., 2000, 4, 445–451 CrossRef CAS.
- O. Ramstrom, T. Bunyapaiboonsri, S. Lohmann and J. M. Lehn, Biochim. Biophys. Acta, Gen. Subj., 2002, 1572, 178–186 Search PubMed.
- D. M. Brown, M. Pellecchia and E. Ruoslahti, ChemBioChem, 2004, 5, 871–875 CrossRef CAS.
- U. F. Vogel and B. D. Bueltmann, Am. J. Clin. Pathol., 2006, 126, 342–348 CrossRef.
- J. F. Desnottes, Trends Biotechnol., 1996, 14, 134–140 CrossRef CAS.
- S. B. Rawool and K. V. Venkatesh, BioSystems, 2007, 90, 636–655 CrossRef CAS.
- G. H. W. Sanders and A. Manz, TrAC, Trends Anal. Chem., 2000, 19, 364–378 CrossRef CAS.
- A. Brazma, TheScientificWorldJOURNAL, 2009, 9, 420–423 Search PubMed.
- P. Pantano and D. R. Walt, Chem. Mater., 1996, 8, 2832–2835 CrossRef CAS.
- M. Margulies, M. Egholm, W. E. Altman, S. Attiya, J. S. Bader, L. A. Bemben, J. Berka, M. S. Braverman, Y. J. Chen, Z. T. Chen, S. B. Dewell, L. Du, J. M. Fierro, X. V. Gomes, B. C. Godwin, W. He, S. Helgesen, C. H. Ho, G. P. Irzyk, S. C. Jando, M. L. I. Alenquer, T. P. Jarvie, K. B. Jirage, J. B. Kim, J. R. Knight, J. R. Lanza, J. H. Leamon, S. M. Lefkowitz, M. Lei, J. Li, K. L. Lohman, H. Lu, V. B. Makhijani, K. E. Mcdade, M. P. McKenna, E. W. Myers, E. Nickerson, J. R. Nobile, R. Plant, B. P. Puc, M. T. Ronan, G. T. Roth, G. J. Sarkis, J. F. Simons, J. W. Simpson, M. Srinivasan, K. R. Tartaro, A. Tomasz, K. A. Vogt, G. A. Volkmer, S. H. Wang, Y. Wang, M. P. Weiner, P. G. Yu, R. F. Begley and J. M. Rothberg, Nature, 2005, 437, 376–380.
- D. R. Walt, Science, 2000, 287, 451–452 CrossRef CAS.
- A. C. Pease, D. Solas, E. J. Sullivan, M. T. Cronin, C. P. Holmes and S. P. A. Fodor, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5022–5026.
- S. A. Dunbar, C. A. Vander Zee, K. G. Oliver, K. L. Karem and J. W. Jacobson, J. Microbiol. Methods, 2003, 53, 245–252 CrossRef CAS.
- Handbook Luminex Beadarray, http://www.luminexcorp.com/, accessed 2009.
- K. L. Gunderson, S. Kruglyak, M. S. Graige, F. Garcia, B. G. Kermani, C. F. Zhao, D. P. Che, T. Dickinson, E. Wickham, J. Bierle, D. Doucet, M. Milewski, R. Yang, C. Siegmund, J. Haas, L. X. Zhou, A. Oliphant, J. B. Fan, S. Barnard and M. S. Chee, Genome Res., 2004, 14, 870–877 CrossRef CAS.
- J. B. Fan, K. L. Gunderson, M. Bibikova, J. M. Yeakley, J. Chen, E. W. Garcia, L. L. Lebruska, M. Laurent, R. Shen and D. Barker, Methods Enzymol., 2006, 410, 57–73 CAS.
- M. Ronaghi, S. Karamohamed, B. Pettersson, M. Uhlen and P. Nyren, Anal. Biochem., 1996, 242, 84–89 CrossRef CAS.
- VeraCode Research Guide, http://www.illumina.com, accessed 2007.
- D. Ryan, M. Rahimi, J. Lund, R. Mehta and B. A. Parviz, Trends Biotechnol., 2007, 25, 385–389 CrossRef CAS.
Footnotes |
| † Part of the themed issue: From microfluidic application to nanofluidic phenomena. |
| ‡ All authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2010 |