Mesogen-jacketed liquid crystalline polymers
Xiao-Fang
Chen
,
Zhihao
Shen
,
Xin-Hua
Wan
,
Xing-He
Fan
,
Er-Qiang
Chen
,
Yuguo
Ma
and
Qi-Feng
Zhou
*
Beijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry and Physics at the Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: qfzhou@pku.edu.cn; Fax: +86 10 62751708; Tel: +86 10 62754518
First published on 18th June 2010
Abstract
This critical review covers the recent progress in the research of mesogen-jacketed liquid crystalline polymers (MJLCPs), special side-on side-chain liquid crystalline polymers with very short spacers or without spacers. MJLCPs can self-organize into supramolecular columnar phases with the polymer chains aligned parallel to one another or smectic phases with the backbones embedded in the smectic layers. The semi-rigid rod-like MJLCP with a tunable rod shape in both length and diameter provides an excellent building block in designing novel rod–coil liquid crystalline block copolymers which can self-assemble into hierarchical supramolecular nanostructures depending on the competition between liquid crystal formation and microphase separation (229 references).
 Yuguo Ma, Zhihao Shen, Er-Qiang Chen, Qi-Feng Zhou, Xin-Hua Wan, Xiao-Fang Chen and Xing-He Fan | Xiao-Fang Chen was born in 1975. She received a BS degree in Chemistry from Peking University in 1998. After working in Peking University for three years, she started her doctoral research under the supervision of Prof. Qi-Feng Zhou and received a PhD degree in Polymer Chemistry and Physics from Peking University in 2007. She is currently working in the College of Chemistry and Molecular Engineering at Peking University as a technician with expertise in X-ray diffraction. Her work is focused on the synthesis and self-assembly behavior of liquid crystalline polymers and block copolymers. |
Zhihao Shen was born in 1972. He received a BS degree in Polymer Science from Nanjing University under supervision of Gi Xue in 1994. After receiving a PhD degree in Polymer Science from The University of Akron under the supervision of S. Z. D. Cheng in May 2001, he worked as a postdoctoral researcher in both academia (with M. A. McHugh at Virginia Commonwealth University) and industry. He joined Peking University as an associate professor of polymer physics in October 2007. His main research interests are condensed matter physics of complex polymeric systems and applications of supercritical fluid technologies in polymer science. |
Xin-Hua Wan received his BCE (1985) degree from Hefei University of Technology. In 1985 he went to China Textile University to pursue his MA (1988) and PhD (1991) degrees. After working as a postdoctoral researcher at Peking University for two years, he became an associate professor (1993) and a full professor (1997) of polymer chemistry and physics there. He visited The University of Akron and Harvard University as a senior visiting scholar. His research interests include self-assembly of liquid crystalline polymers, block copolymers, and macromolecules of controlled architecture and well-defined shape, as well as chirality in polymers. |
Xing-He Fan was born in 1959. He received his BS degree in Chemical Engineering from Zhejiang University in 1982. He was a chemical engineer at the Chemistry Research Institute, Nanjing, China until 1996. He was awarded a PhD in 2000, majoring in polymer chemistry from Kagoshima University, Japan and has been an associate professor in the Department of Polymer Science and Engineering at Peking University since 2000. He is interested in research on novel multifunctional liquid crystalline polymers, and has published about 80 papers, 4 books, and 4 Chinese patents. |
Er-Qiang Chen received his BS (1988) and MS (1991) degrees from Fudan University in Shanghai, China. In 1994 he went to The University of Akron in USA and obtained his PhD degree in 1998 under the supervision of S. Z. D. Cheng. He spent the period of 1998–2000 as a postdoctoral researcher at The University of Akron and the Polymer Division of the National Institute of Standards and Technology. In 2000 he joined Peking University, where he is currently professor of polymer physics. His research focuses on phase transitions and structures of polymers including crystallization, liquid crystalline behavior and self-assembly. |
Yuguo Ma obtained his BSc in 1994 and MSc in 1997 from the College of Chemistry at Peking University under the supervision of Prof. Qi-Feng Zhou. He continued his graduate study at the University of Illinois at Urbana-Champaign with Prof. Steven C. Zimmerman, and obtained his PhD in 2002. From 2003 to 2005, he was a postdoctoral research associate with Prof. Geoffrey W. Coates at Cornell University. In September 2005, he returned to Peking University and has been an associate professor of chemistry. His current research interests include supramolecular chemistry, self-assembly and molecular recognition, and organic/polymer functional materials. |
Qi-Feng Zhou graduated from Peking University and stayed to become a teaching assistant in 1970. He worked there until 1980 when he was admitted by the Graduate School of the University of Massachusetts at Amherst, USA, where he was awarded a Master's degree in 1981 and a PhD in 1983, majoring in polymer science and engineering. Immediately afterwards he went back to China and returned to his post at Peking University. He has been involved in the study of liquid crystalline polymers. He is author of several books, including a textbook on liquid crystalline polymers. |
1. Introduction
Liquid crystals (LCs) are soft materials that can spontaneously self-organize into ordered phase structures while retaining the fluid properties at the same time. They are switchable in response to external stimuli,1 and have gained a lot of attention not only in materials science, but also in biosystems.2 Liquid crystals have a great potential in functional soft materials and, more importantly, are motivating us to get closer to the universal living systems, big liquid crystalline hierarchical structures.As a mesophase between three-dimensionally long-range ordered crystalline state and three-dimensionally short-range ordered liquid state, liquid crystals can exhibit phase structures with one- or two-dimensional order on the length scale of about 1∼10 nm. The phase structure of liquid crystals can be nematic (N), smectic (or layer-like), columnar, or cubic in the viewpoint of phase symmetry.3 The N phase has found wide, sophisticated applications in information display technology (thermotropic LC) and high-strength, high-modulus polymer fibers (lyotropic LC). Taking advantage of advanced synthetic technology and the fruitful collaborations between chemists and physicists ever since liquid crystals were first discovered,4 scientists have found more and more LC materials and new complex mesophases. The building blocks that self-organize into partially ordered mesophase structures are not restricted to rod-like molecules and amphiphilic compounds any more. Molecules, molecular parts, and the aggregates of self-assembled molecules with well-defined structures, shapes, and molecular architectures are able to self-organize into complex ordered nanostructures through weak intermolecular interactions, such as van der Waals and π–π interactions.
From the molecular weight (MW) point of view, liquid crystalline self-organization can take place not only in low molar mass systems or molecular oligomers with discrete structures, but also in polymeric systems. Numerous books and reviews on liquid crystals have been covering all the MW range.5 In this review we will focus on the liquid crystalline phase behavior in polymeric systems, especially discussing the phase structures of various mesogen-jacketed liquid crystalline polymers (MJLCPs).
When LC meets polymers, the resulting liquid crystalline polymers (LCPs) show both anisotropic properties which originate from mesogenic units and good mechanical properties which come from long-chain structures.6 LCPs have aroused considerable attention since 1960s because of their wide applications as engineering plastics, high-strength and high-modulus fibers, electro-optic or nonlinear optic materials, stationary phases, gas separation membranes, etc.7 The molecular engineering of LCPs is mostly carried out by introducing mesogenic units, such as rod-like or disc-like mesogens derived from low molecular weight compounds, into polymer chains, which has been proven to be an efficient way of obtaining LCPs with desired properties. In general, two typical strategies in incorporating mesogenic units into the polymer chain are frequently executed. One is embedding mesogens into the backbone of a polymer chain, and the resulting LCP can be named as main-chain liquid crystalline polymer (MCLCP). The other is attaching mesogens to the main chain as side groups, and the LCP obtained is named as side-chain liquid crystalline polymer (SCLCP).8 Depending on the position where the mesogens are linked to the backbone, the mesogen can be either longitudinally or laterally attached into the polymeric system (Table 1). Thus SCLCPs with a longitudinal connection (end-on) of the mesogen are end-on SCLCPs and those with lateral connection (side-on) are called side-on SCLCPs. Similarly, for MCLCPs, there are also end-on MCLCPs and side-on MCLCPs, as shown in Table 1. In particular, directly connecting rigid units along the backbone in an end-on fashion produces a fully rigid supramolecular rod. Actually, other liquid crystalline polymers with complex structures can be also designed on the basis of the above-mentioned consideration. Furthermore, in addition to covalent bonds, non-covalent interactions, such as hydrogen bonds,9,10 ionic bonds,11 and charge transfer interactions have recently been applied to synthesize supramolecular LCPs.12
| Side-chain LCP | Main-chain LCP | |
|---|---|---|
| End-on (longitudinally attached) |

|

|
| Side-on (laterally attached) |

|
|
1.1 The key role of the flexible spacer
It should be noted that thermodynamics determines the different behaviors of flexible polymer chains and rigid side-chain mesogens. The polymer backbone tends to keep the Gaussian chain conformation while the mesogenic side groups tend to align parallel to one another to form liquid crystalline phases. So the motion of the backbone disturbs the self-organization of the attached mesogenic side chains, resulting in more complex mesomorphic phase behavior than in low molar mass systems. In some cases, no LC phases appear in polymers synthesized, even though mesogens are incorporated into the systems.8 Finkelmann et al.13 first considered the influence of the connection between the side group and the polymer chain. If the mesogen is attached to the main chain directly, the tendency of the polymer to form random-coil chain conformation will disturb the parallel alignment of side chains. Thus, in end-on SCLCPs, flexible spacers are purposefully introduced to decouple the dynamics of the main chain and the mesogenic side groups in order to achieve liquid crystallinity. Therefore, the statistical movements and conformational changes of the polymer backbones will have limited effects on the mesomorphic organization of the mesogens. This decoupling rationale has been proven by numerous experiments and the decoupling concept has become a useful guide for the molecular design of SCLCPs.14 This concept was also a basis for the design of the first reported side-on SCLCPs, and the resulting polymers exhibited an N phase with biaxial optical properties.15,16 Percec further introduced the microphase separation concept into SCLCP systems by using immiscible polymer backbone and side groups, for example, polysiloxane as the backbone and dioxaborinane as the side group. In this way, highly or even completely decoupled side-chain liquid crystalline polymers could be realized.17,181.2 Jacketing effect
In the late 1980s, Zhou et al.19–21 reported that side-on SCLCPs with a short spacer or with a single covalent bond connecting the mesogen to the polymer backbone still showed stable liquid crystalline properties, and these polymers were termed MJLCPs. The monomers exhibited high polymerizability, even though the bulky side group was attached very close to the double bond which could polymerize to afford LCPs. Unlike the use of flexible spacers to decouple the interaction between the side chain and the backbone in SCLCPs, the underlying consideration in the design of these MJLCPs was that the torque through which the backbone exerted on the side chain should be minimal when the connection was at or near the gravity center of the side chain. The mesogen-jacketed model was proposed to vividly describe that the laterally attached mesogenic side groups wrapped closely around the flexible polymer backbone and forced the polymer chain to stretch along the main-chain direction.19 The whole polymer chain acts as a supramolecular rod, similar to fully rigid end-on MCLCPs, although chemically speaking, MJLCPs belong to side-on SCLCPs. So from this viewpoint of creating LCPs containing laterally attached side chains with the linkages located at the gravity centers of the mesogens, the use of a flexible spacer seems not necessary. From then on, many efforts have been devoted to revealing the structure–property relationship of this unique system. We will review the related research progress in this field.2. Side-on side-chain liquid crystalline polymers
Realizing the importance of flexible spacers, Finkelmann et al. first systematically investigated the properties of side-on SCLCPs by synthesizing a series of polymers with different mesogenic moieties, spacer lengths, and terminal groups.22 Different from end-on SCLCPs, the lateral substituent tended to depress the formation of smectic layers. All the polymers they synthesized showed a stable N phase with the nematic-to-isotropic transition temperature (TN−I) in the range of 20–93 °C. The biaxial optical properties of this N phase were first found by conoscopic observations of homeotropically aligned side-on SCLCPs. In this way, such kind of unique molecular architecture of side-on SCLCPs has been proven to be a successful model in designing biaxial nematic materials.16,22 Since then many researchers have investigated the structure–property relationship of side-on SCLCPs, mainly focusing on such aspects as the polymer backbone, grafting density of side chains, the side-chain moiety usually containing the rigid mesogenic core and the flexible aliphatic tail, and the length and chemical structure of the spacer. A schematic representation of a side-on SCLCP is shown in Fig. 1. | ||
| Fig. 1 Schematic illustration of a typical side-on side-chain liquid crystalline polymer. | ||
2.1 Structure–property relationship in side-on SCLCPs
The chemical structure which is associated with the flexibility of the polymer backbone is of crucial importance for the formation of the mesophase. Liquid crystalline polychloroacrylates with similar side-on fixed mesogenic groups to that of polymer 1 all exhibited a biaxial N phase.23 It is interesting to note that TN−I of polymer 1 with identical mesogenic moiety appeared independent of the tail length and the flexibility of the backbone. Hydrodynamic properties of polymer 1 (R = –CH3, m = 11, n = 6) in dilute solutions indicated as well that the steric hindrance of laterally attached side groups decreased the flexibility of the main chain.24,25
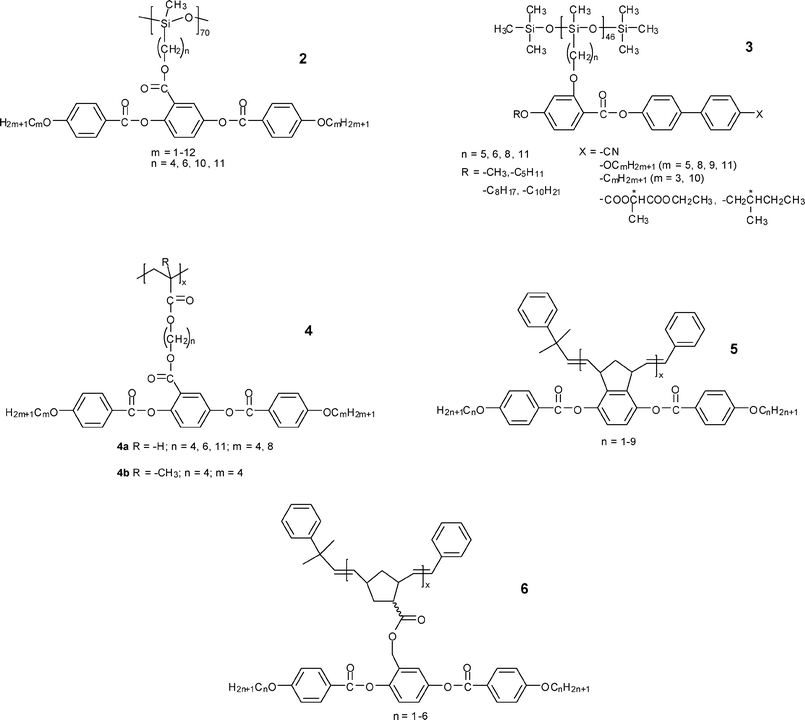
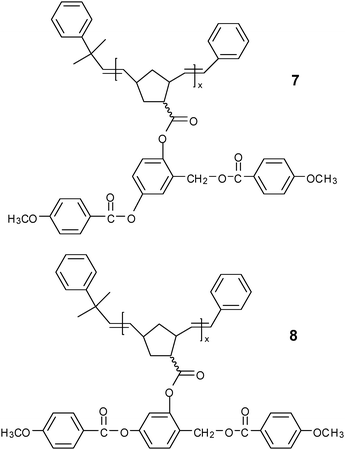
N phases have also been found in polysiloxanes 2 with similar calamitic mesogenic groups laterally attached to the backbone through flexible spacers.26,27 The two-dimensional wide-angle X-ray diffraction (2D WAXD) pattern of single domain in the N phase showed four diffuse spots located at low angles off the meridian, which had previously been interpreted as strong smectic C (SmC) fluctuations in polymers 2 (m = 4, n = 4) and 3.27,28 Similar diffuse patterns were also observed in both neutron diffraction and X-ray diffraction (XRD) of polyacrylates 4a (n = 4, m = 8).29 While detailed calculation revealed that the diffuse spots were different from SmC off-axis spots and their existence and position remained nearly unchanged when the spacer length was varied, it was finally proposed to originate from local correlations of the aliphatic tails of the mesogenic side chain.30 With increasing lengths of the spacer (n = 10) and aliphatic tails (m ≥ 6), additional diffuse spots corresponding to the characteristic diffraction pattern from the SmC structure appeared, indicating the formation of an SmC phase. Besides polyacrylates, polychloroacrylates, and polysiloxanes, other polymer backbones can also be used to construct side-on SCLCPs. Nematic polyoxetanes with laterally attached mesogenic groups were synthesized.31 First introduced by Pugh et al., polynorbornenes with controllable MW and fairly narrow polydispersity showed enantiotropic N mesophases in polymers 5 and 6.32 However, no mesophase was observed in polymer 7 with a bent-shaped mesogen or polymer 8 when the methylene group was inserted into the linear rigid mesogen.
In addition to the abovementioned flexible backbones, rigid polymer chains were also introduced into side-on SCLCPs. When the mesogens were laterally attached to the polypeptide backbone, the coexistence of the nematic ordering originating from side chains and the hexagonal packing from polymer backbones was first observed, which was quite different from the end-on attached liquid crystalline polypeptides.33 The flexible spacer influenced the melting temperatures (Tm's) and the degree of ordering of the rod-like polypeptides in the N state. The anisotropy of the backbone in polymer 9 favored the nematic arrangement of side-on mesogens along the backbone and prevented the polymer from melting directly to an isotropic phase until the helices were eventually disordered, resulting in high stability of the N phase. Another rigid polymer backbone, polyacetylene, was also laterally attached with calamitic mesogens. Polymer 10 with a relatively long tail (R = –C6H13) in the mesogens exhibited enantiotropic smecticity.34

Since the polymer backbone of many side-on SCLCPs was strongly stretched, the length of the polymer would influence the mesomorphic behavior, and the MW dependence of mesomorphic properties should be considered as well. Polymers 1 (R = –H, m = 11, n = 4) with different degrees of polymerization (DPs) were obtained. All of these polymers had similar TN−I and glass transition temperature (Tg).22 The study of MW dependence was further continued by using a set of fractionated polymer 2 samples.35 It was found that TN−I and Tg first increased with increasing DP, reached a plateau, then with DP further increasing, unexpectedly declined slightly. The unusual decrease in both TN−I and Tg for polymers with large DPs indicated that the jacketing effect appeared less pronounced for long polymer backbones. A similar phenomenon was also reported for side-on polysiloxane 3, which was attributed to the short persistence length or hairpins existing in high MW samples.36
The reported results have revealed that the attaching mode of the side chain to the polymer backbone, either side-on or end-on, does give rise to different characteristics of LCPs. In particular, the lateral attachment of the rod-like mesogenic groups favors the N state, exhibiting a prolate shape of the backbone conformation, while the end-on LCPs are predominantly smectogenic, exhibiting an oblate shape in the smectic A (SmA) phase, in which the backbones are confined between the adjacent mesogenic layers. If these two attaching modes appear in one polymer chain, the competition between the two chain conformations will certainly influence the final mesomorphic behavior of this kind of copolymers. Achard et al. investigated the properties of side-on and end-on side-chain LC copolysiloxanes, and found that the addition of side-on mesogens tended to lower the stability of the smectic phases.37 On the other hand, the addition of end-on mesogens restricted the chain extension in the N phase, which weakly reduced the N-to-I transition temperature and enthalpy change.
A similar phenomenon was also reported in Gray's work.38 In this system, 2–5% concentration of the laterally attached side chain decreased both Tm and the smectic-to-isotropic transition temperature. When the concentration was increased to 14%, the N phase appeared above the SmA phase. When the concentration was further increased to 16%, only an N phase existed. Therefore, the lateral side-chain component has a great effect on the terminally attached LCPs. The macroscopic alignment of copolymer 11, both homeotropically and planarly between two electrodes under AC field, was investigated using dielectric relaxation spectroscopy and optical microscopy.39,40
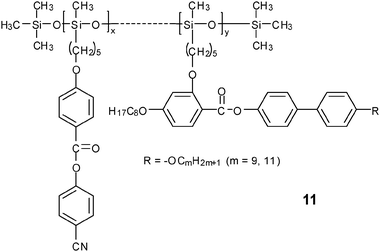
With a lower density of mesogenic side chains, the Tg of the copolysiloxane 12 decreased gradually, indicating a more flexible backbone than that in the homopolymers. The TN−I and ΔSN−I values systematically decreased from homopolymer to copolymer. For polymer 12 with n = 4 and m = 4, when x, which was a/(a + b), became 0.09 or lower, no mesophase could be formed any more. This implied that the nematic order was destabilized by lowering the grafting density of side-on mesogens, while the SmC phase was somewhat stabilized with decreasing composition of mesogenic side chains.41 The evolution is different in the case of smectic end-on SCLCP, where the smectic order tends to be depressed when the mesogenic density around the backbone is decreased.

Flexible spacers play an important role in the mesophase formation of side-on SCLCPs. The alkyl chain is frequently used to act as a flexible spacer linking the backbone and the side chain. The information from a small-angle neutron scattering (SANS) study indicated that the increase in the spacer length would dramatically reduce the prolate anisotropy of the backbone conformation in the N phase for side-on polymers.42 When the spacer was longer than 11 methylene units, the jacketing effect vanished.29
Other spacers such as oligooxyethylenic spacers were also used to connect a polysiloxane backbone and calamitic side chains.43 The resulting polymer 13 had an enantiotropic N phase. Meanwhile, LiCF3SO3/polymer complexes showed an N phase up to a value of 0.4 mole LiCF3SO3 per mole repeat unit of the polymer. On the other hand, if a disiloxane unit was added to the spacer, no mesophase was observed for homopolymers, and a mesophase could be observed only when such a monomer with the disiloxane unit was copolymerized with other end-on polymers.44 An N phase was observed along with a smectic phase when the two comonomers had the same siloxane spacer.45 When comonomers with different spacer structures were copolymerized, the resulting copolymers showed an enantiotropic smectic or N phase when the comonomer content was about 75 or 50 mol%, respectively.46

The phase behavior of side-on SCLCPs is also greatly influenced by the structure of the side chain, which comprises a rigid mesogenic core and flexible tails at both ends. Rod-shaped mesogens were frequently used, especially those constructed from three phenyl rings connected with short substituent groups through para connections. Aliphatic groups were commonly used as flexible tails attached to both ends.
As mentioned above, biaxial optical properties could be achieved with the lateral attachment of the mesogenic side chain. The central phenyl group was further replaced by a naphthalene ring in order to increase the anisotropy of polarizability.47–49 In addition to a biaxial N phase, biaxial SmA and chiral N (N*, or cholesteric) phases were first observed in polymer 14 and copolymer 15, respectively.48,50 Furthermore, if the central phenyl group was replaced by a carbazolyl group, the side-on polymers could present a metastable N phase.51 The azobenzene group which easily undergoes cis–trans isomerization with UV irradiation was introduced to the side-chain mesogen by Li et al., resulting in polymer 16 with an interesting optical response property.52 A nematic-to-isotropic transition was induced by UV irradiation, and the turnoff of the UV light led to spontaneous isotropic-to-nematic back transition.53

In most side-on SCLCPs, the connecting sites of mesogenic side groups were at the middle phenyl ring in three-ring systems. Gray et al.38,54 and Lewthwaite et al.55 moved the connecting site to the side part of the mesogen instead of the waist of the mesogen. An N phase was also found in polymer 3, even with –CN56 or long-chain alkyl or alkoxy groups.57,58 Under a magnetic field, both the mesogenic groups and the backbones were preferentially oriented parallel to the fiber axis which was also the direction of the magnetic field.28 An analysis of the diffuse peaks in the XRD pattern suggested that there was a tendency for the mesogenic units to wrap around the backbone as suggested by the mesogen-jacketed model.
The effect of the tail length on the LC behavior was also investigated in most systems. Generally, the Tg and the isotropilization temperature decreased with the increase in the flexible tail length. In polymer 1, no mesophase was found when R2 = –C3H7. Furthermore, the chiral moiety tended to depress the liquid crystalline formation of polymers, and only copolymers could show a chiral N phase. In polymer 17 with side chains containing tolane and naphthalene groups, the existence of the chiral group did not disturb the mesophase formation, and an N* phase was found in polymer 17b with a cholesteric-to-isotropic transition temperature of about 77.9 °C.59 In some cases, an SmC phase tended to appear with increasing aliphatic tail length.41 In addition, increasing the tail length more or less reduced the jacketing effect to a less extent, as revealed by SANS results.29
2.2 Chain conformation of side-on SCLCPs
When the jacketed model was first proposed by Zhou et al.19–21 to illustrate the unique cooperation between the side chains and the polymer backbone in side-on SCLCP systems, the chain conformation in the N phase of side-on polymers aroused much attention. SANS experiments were first carried out to study the orientation of polymer backbones and mesogens of polysiloxane 2 in the N phase using a pseudo-binary system which mixed 50 wt% of polymer 2 with the analogous polymer selectively deuterated on the aliphatic tails.60 A large prolate anisotropy of conformation in their N phases was observed, indicating that the mesogens were aligned parallel to the polymer backbone under the magnetic field (Fig. 2).36 This was also evidenced in side-on liquid crystalline polyacrylates 4 (n = 4, 6 and m = 4) selectively deuterated on the polymer backbone.42 The prolate shape of the backbone in an N phase was much stronger than that observed in the N phase lacking smectic A fluctuation in end-on SCLCPs61 and similar to the conformation of main-chain LCPs in the N phase. A form factor of cylinder was used to model the experimental scattering data as in the case of MCLCPs. The SANS data of polymer 4 (n = 4 and m = 4) revealed that the polymer backbone was totally extended, without hairpins, and the whole polymer chain was completely elongated like a rod.62 | ||
| Fig. 2 Schematic representation of a nematic jacketed side-on SCLCP and its orientation under a magnetic field. Both the mesogens and the polymer backbones were aligned parallel to the direction of the magnetic field.36 | ||
Furthermore, investigation of the same polymer system in isotropic, nematic, and glassy phases using neutron time off-light spectrometry revealed that the mesogenic motions were systematically reduced in the direction parallel to their orientation, but the rotational, translational, and vibrational modes of motions remained in the glassy state.63 SANS results showed that the anisotropic prolate shape of the polymer conformation was dramatically reduced with the decrease in grafting density.64 The anisotropy was less for the polysiloxane than for the polyacrylate when these two polymers with the same spacer length and similar DP were compared.65
Meanwhile, in some side-on SCLCPs having an SmC phase, the backbone conformation turns to an oblate shape. A reversible inversion of conformation of the polymer main chain from a weak prolate shape in the N phase (R///R⊥ ∼ 1.2) to a largely oblate shape in the SmC phase (R///R⊥ ∼ 0.5) was observed in a SANS study. In the SmC phase, the backbones could be confined either between the mesogenic layers or in the middle part of the smectic organization.66–68
Lecommandox et al. investigated the conformation evolution and thermodynamic behaviors of LC copolyacrylates through a SANS study.69 The SmA phase of end-on SCLCP was strongly depressed by incorporating a small amount of side-on mesogenic units. The N phase induced by the side-on mesogenic groups existed up to 80% of end-on mesogenic groups, and at the same time, the prolate shape was retained even for a copolymer with a majority of end-on mesogenic groups, although the anisotropy value, R///R⊥, decreased with increasing percentage of end-on mesogenic groups in copolymers. On the other hand, the SmA phase induced by the end-on mesogenic groups was strongly depressed with the addition of a small amount of side-on mesogens. The evolution was consistent with that of the side-on/end-on copolysiloxanse systems.
3. Molecular design and synthesis of MJLCPs
The decoupling of interaction between the side chain and the backbone in conventional side-on SCLCPs usually renders LCPs with an N phase. However, as mentioned before, the jacketing effect in some side-on SCLCPs, particularly in MJLCPs, actually induces the cooperation (coupling) of the backbone and the side chain, causing the polymer chain to take an extended-chain conformation.19 Thus, MJLCPs can behave like a rigid rod (Fig. 3) with certain shape stability. The diameter of such a supramolecular rod can be readily controlled by varying the size of the side chain, while the length of the rod is almost linearly proportional to the MW of the polymer. Different from fully rigid MCLCPs which are usually prepared by condensation polymerization with difficulty in controlling MW, MJLCPs, a special class of SCLCPs, can be synthesized by radical polymerizations which have the advantages of mild reaction conditions, less purity requirements of materials, and easy MW control if controlled or “living” radical polymerization techniques are employed. Rod-like MJLCPs have excellent tunability in molecular characteristics because the rod length can be controlled by the MW of the polymer, while the rod diameter can be changed by controlling the lengths of both the rigid core and the flexible tails in the mesogenic side chains. | ||
| Fig. 3 Schematic representation of a rod-like MJLCP. The steric hindrance of the bulky pendant groups forces the backbone to take an extended-chain conformation, resulting in a supramolecular rod. The rigid side chains are oriented at an angle with respect to the chain direction of the backbone. | ||
As revealed in section 2, the jacketing effect does exist in side-on SCLCPs and tends to become more obvious either by decreasing the spacer length or by increasing the grafting density. Other structural parameters, such as the length of the aliphatic tail and DP, also influence the jacketing effect to some extent. The N phase was a dominant phase in side-on SCLCPs. SANS and other measurements revealed that the chain in the N phase was strongly stretched to form a rigid cylinder. Thus a supranematic phase was proposed.42 MJLCP, as a specific example of side-on SCLCPs with short flexible spacers or even without spacers, will have the largest jacketing effect compared with other side-on SCLCP systems. From previous studies on MJLCPs, some characteristic properties of the MJLCPs imply that such a system will give us an opportunity to intensively understand the mesomorphic thermodynamics and structural properties of side-on SCLCPs, and new properties may be explored in this system. In terms of the molecular design of MJLCPs, several aspects can be considered, such as the shape and rigidity of the side chain, the mesogenic density, the polydispersity and MW of the polymer, and so on.
In general, the connection between the side chain and the polymer backbone has been kept with only one covalent bond in the recent study of MJLCPs in order to construct jacketed polymers, except for polymethacrylates 18–20 which contain a short linkage.19,20,70–72 In contrast to polyacrylates, the polymethacrylate backbone tended to depress the LC formation, indicating the conformation influence of the polymer backbone, which was quite similar to end-on SCLCPs without flexible spacers.73,74 With the same mesogen-jacketed model originally proposed,19–21 mesogenic side chains can be attached directly with only one covalent bond, like polystyrene derivatives. As shown in Table 2, several series of MJLCPs (21–26) have been synthesized. They were based on 2-vinylhydroquinone,21 2-vinyl-1,4-phenylene-diamine,75 2-vinylterephthalic acid,76,77 and vinyl terphenyl.78,79 The laterally attached calamitic mesogens were mostly constructed with three phenyl rings connected at the para positions with diverse linkages. Flexible aliphatic tails were mostly appended to the calamitic mesogens. All these polymers show LC phases, which is similar to other side-on SCLCPs, while the LC-to-isotropic transition temperatures are hardly observed below the decomposition temperatures, indicating very stable LC phases above Tg. A lyotropic LC phase was formed in polymer 21 with polar groups and in polymer 22.75,80

| Polymer | Molecular structure |
|---|---|
| 21 |

|
| 22 |

|
| 23 |

|
| 24 |

|
| 25 |

|
| 26 |

|
As mentioned before, for conventional side-on SCLCPs, most attention has been paid to the effect of the spacer, the grafting density, and the tail length of the side chain, while the research on the rigidity of the mesogenic units has not progressed much. In fact, since the steric hindrance of the side-chain mesogens plays an important role in inducing the jacketing effect on the flexible backbone, the rigidity of the side chain must be considered. Several approaches have been attempted to change the rigidity of the side chain. The first approach was to replace the alkoxyphenyl side group with the semi-rigid cyclohexyl group, and the resulting polymer still exhibited an LC phase when heated above Tg.81 Other cycloalkyl groups also had a similar effect on the formation of an LC phase.82 And the polymer with the cyclopentyl group not only showed a stable LC phase but was also found for the first time to exhibit a high isotropilization temperature in MJLCP systems.83 On the basis of these interesting results, a more audacious attempt was carried out to open the substituent alkyl ring in order to achieve more flexible side chains. The resulting polymer 27 showed a stable columnar LC phase.84 Other similar structures with Y-shaped alkoxy chains also showed columnar phases.85 Thus, it appeared necessary to further increase the flexibility of the laterally attached side chain, and linear aliphatic tails were directly attached to the central phenyl ring. Even with such a flexible side group, the polymer (28) with a suitable tail length still showed a stable LC phase.86,87
Another way to decrease the steric effect of the side chain on the backbone was to change the shape of the side-on mesogen from a rod-like to a bent shape. The steric effect of the side chain on the polymer backbone was dramatically lowered in this case. The liquid crystallinity of polymer 29 was weak and could only be enhanced by mechanical shear and long time annealing. This issue could be overcome by increasing the length of the arms of the bent-core mesogen, and polymer 30 showed a stable LC phase.88

Contrary to decreasing the rigidity of the side chain, the other consideration was to increase the steric effect of the side chain by extending the length of the mesogen. The resulting polymers 31 and 3289,90 still showed stable liquid crystalline properties, and the phase structure changed from columnar phases to smectic phases. When the 1,3,4-oxadiazole group replaced the ester group to construct a more rigid mesogen, polymer 33 turned from a columnar phase when the aliphatic tail was tert-butyl (t-Bu) group to smectic phases for longer linear aliphatic tails in the mesogenic group.91,92
Furthermore, recently a new series of special MJLCPs was synthesized,93 wherein biphenyl groups were introduced into the bulky pendant group by connecting them to the styrene unit through a spacer. The packing of the resulting MJLCPs could be expected to be even more complex.

Although most SCLCPs can be readily polymerized via conventional radical polymerization methods, the broad MW distribution resulting from these processes will lead to phase transitions much broader than those in low molar mass liquid crystals with phase transitions over a few degrees. Therefore, living polymerizations have been carried out to synthesize SCLCPs with well-defined polymers that are of great importance for the study of the structure–property relationship in SCLCP systems. Most living polymerizations, such as anionic, cationic, metalloporphyrin, and ring-opening metathesis polymerizations, have been carried out to synthesize SCLCPs with well-defined homopolymers, block and graft copolymers, and statistical copolymers.94 However, these living polymerizations require crucial conditions, and the tolerance of functional groups and impurities in the monomer is limited. The development of living/controlled free radical polymerization really offered an efficient technique to synthesize LCPs with controlled MW and low polydispersity.95–97 Side-on SCLCPs, polymers 4b and 16 were synthesized by atom-transfer free radical polymerization (ATRP) at ambient temperature.98
Polymerization methods other than free radical polymerization have been carried out to obtain side-on SCLCPs with different polymer backbones. For example, nematic polyoxetanes with laterally attached mesogenic groups were synthesized by cationic polymerization.31 Pugh et al. first synthesized polynorbornenes using living ring-opening metathesis polymerization to give polymers a with controllable MW and fairly narrow polydispersity.32
Monomers of MJLCPs are mostly vinyl monomers that usually undergo radical polymerization easily. MJLCPs can also be synthesized using nitroxide-mediated polymerization (NMP) (or stable free radical polymerization, SFRP) and ATRP methods, resulting in MJLCP polymers with a controlled MW and a relatively narrow MW distribution for a systematic study on the MW dependence of liquid crystalline properties. As a substituted styrenic monomer, 2,5-bis[(4-butylbenzoyl)oxy]styrene (BBOS), exhibited reactivity an order of magnitude higher than that of styrene under identical bulk polymerization conditions.99 The presence of a nematic phase, as well as steric factors and the electronic effect of the acetoxy groups, could cause BBOS to be more reactive than styrene.100 Another styrenic monomer, 2,5-bis[(4-methoxyphenyl)oxycarbonyl]styrene (MPCS), also showed high reactivity in ATRP and SFRP polymerizations to afford PMPCS (polymer 23 with R1 = R2 = –OCH3) homopolymer.101,102 First-order kinetics, a linear MW-conversion profile, and a narrow MW distribution were evidences used to verify the “living” ATRP of MPCS. Thus PMPCS with different MWs and narrow MW distributions were synthesized.102 A recent review summarized the synthetic aspects in the MJLCP systems.103
4. Mesophase behavior of MJLCPs
As mentioned in section 2, in laterally attached SCLCP systems with spacers, the N phase is the most frequently observed phase. The bulky rigid mesogens impose steric constraints on the polymer backbone and force the backbone into an extended chain with the mesogens jacketing it (Fig. 2). A SANS study of side-on SCLCPs showed that the N phase was imposed by the laterally attached mesogenic moieties to form highly anisotropic polymers; therefore, it was the supranematic ordering formed in this system or an NIII-type phase if the parallel alignment of the polymer backbone and the mesogenic side chain were considered as based on the Wang and Warner theory.104 The orientation was similar to the behavior observed for the thermotropic main-chain LC polymers.105The phase behavior of MJLCPs can be investigated using X-ray scattering (small-angle and wide-angle), polarized light microscopy (PLM), differential scanning calorimetry (DSC), Fourier transform infrared (FTIR) spectroscopy, and other characterization methods. Among these techniques, WAXD, especially 2D WAXD on oriented samples, is the main technique used to analyze the phase structures. The banded texture from sheared polymer samples is the evidence of low-ordered LC phases (N or SmA) or columnar phases. In addition, DSC can be employed to obtain transition temperatures and changes of enthalpy during phase transitions, although phase transitions involving LC phases are usually undetectable for many MJLCP systems. Previous studies have suggested that most MJLCPs form columnar phases. However, some MJLCPs can form smectic phases. It should also be mentioned that information obtained from MJLCP systems may also be applied to other systems, such as conventional side-on SCLCPs.
The first reported MJLCPs were assigned as nematic phases based on XRD results. However, the detailed phase structure and the phase behavior were not clear at that time. Banded texture was usually observed for MJLCPs under shear from PLM. And small-angle light scattering (SALS) measurements also showed typical grating diffraction patterns, indicating a rigid-rod feature of the polymer conformation. Infrared dichroism and 2D WAXD measurements indicated that the main-chain axes of the polymers tended to orient almost parallel to the shear direction, while the laterally attached mesogens around the backbone did not.99,106,107 This was more or less different from the orientation behavior of other side-on SCLCPs with both the backbones and the side chains maintaining parallel alignment under a magnetic field.24,26
Pragliola et al. reported that the orientational order parameters, 〈P〉's, of a shear oriented sample of PBBOS (polymer 21 with R = –C4H9) were about 0.5 for the polymer chain, and 0.38 for the mesogens, which indicated that the mesogens in the LC phase were relatively less oriented.99 The stiffness of the polymer 21 (R = –OCH3) in dilute solutions studied by static light scattering and viscometry in tetrahydrofuran (THF) indicated that the polymer in THF retained the worm-like chain and the persistence length was around 11.5 to 13.5 nm,108 suggesting a semi-rigid chain. In dilute solutions, polymer 26 also maintained a similar conformation.109 2D FTIR spectroscopy has been used to study PMPCS in order to investigate the mechanism of the LC phase development in MJLCPs. Before the phase transition, the individual side chains changed conformation sooner than the backbone due to their larger motional freedom. After the phase transition, the backbone gained enough energy and changed its conformation at a lower temperature than the side chain.110
4.1 Columnar phases of MJLCPs
Rod-like MJLCP chains tend to self-organize into columnar LC phases. The type of these columnar phases depends on the molecular properties including the shape, size, and flexibility of side chains. The columnar nematic (ΦN) phase has been found in many systems. More ordered columnar phases, such as 2D hexagonal columnar (ΦH) and rectangular columnar (ΦR) phases, have also been observed.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/31/2:1/41/2, indicating a hexagonal packing. And the diffractions could be assigned as (100), (110), and (200) of the ΦH LC phase, of which the calculated a parameter was comparable to the length of the side group. Such kind of packing information implies that the polymer chains are cylindrical, with the rod diameter determined by the side-chain size. In addition, banded texture in the PLM micrograph (Fig. 4b) of a uniaxially sheared sample, typical of rod-like molecules, also suggested the existence of a two-dimensional columnar phase, which confirmed the rod-like nature of this polymer. Furthermore, the results obtained from polarized FTIR experiments on a mechanically sheared sample indicated that the pendant group was more or less perpendicular to the shear direction, thus perpendicular to the polymer backbone. A proposed packing model is shown in Fig. 4c. Similar hexagonal or even rectangular columnar packing schemes were later proposed for other MJLCP systems.
:1/31/2:1/41/2, indicating a hexagonal packing. And the diffractions could be assigned as (100), (110), and (200) of the ΦH LC phase, of which the calculated a parameter was comparable to the length of the side group. Such kind of packing information implies that the polymer chains are cylindrical, with the rod diameter determined by the side-chain size. In addition, banded texture in the PLM micrograph (Fig. 4b) of a uniaxially sheared sample, typical of rod-like molecules, also suggested the existence of a two-dimensional columnar phase, which confirmed the rod-like nature of this polymer. Furthermore, the results obtained from polarized FTIR experiments on a mechanically sheared sample indicated that the pendant group was more or less perpendicular to the shear direction, thus perpendicular to the polymer backbone. A proposed packing model is shown in Fig. 4c. Similar hexagonal or even rectangular columnar packing schemes were later proposed for other MJLCP systems.
 | ||
| Fig. 4 The ΦH phase of polymer 27. (a) 2D WAXD pattern of an oriented fiber shows three low-angle diffractions on the equator with a d-spacing ratio of 1:1/31/2:1/41/2, characteristic of the hexagonal packing of rod-like molecules. (b) The banded texture of the sheared sample from PLM experiments indicates the possible existence of a 2D columnar phase. (c) Hexagonally packed supramolecular rods with the polymer backbone in the center of the rod. Helical conformation of the polymer chain has also been proposed. Reprinted with permission of the American Chemical Society.84 | ||
2D WAXD studies on PMPCS, which is a more typical and frequently used MJLCP, have revealed a hexatic columnar nematic (ΦHN) phase.111 Previous studies on this polymer only suggested the formation of a low-ordered columnar phase without more details. The 2D WAXD pattern on a PMPCS fiber with the X-ray incident beam aligned parallel to the fiber axis provided experimental evidence of the detailed structure. As shown in Fig. 5, six diffraction arcs were observed in the 2D WAXD fiber pattern. The azimuthal intensity profile showed six maxima which were 60° apart from the adjacent diffraction maxima, indicating a hexagonal lateral packing of cylinders. The lack of higher-order diffractions suggests that this hexagonal lateral packing does not have a long-range order perpendicular to the fiber axis, and the ordered structure can be assigned as a ΦHN phase, the order of which should be between those of the ΦN and ΦH phases.
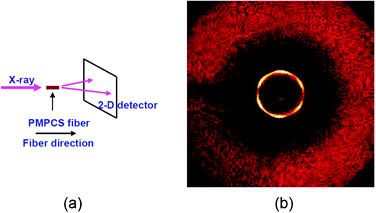 | ||
| Fig. 5 Identification of the phase structure of PMPCS. (a) Geometry of experimental setup in the 2D WAXD experiment. The X-ray incident beam is parallel to the fiber axis. (b) 2D WAXD fiber pattern of the fiber sample. The azimuthal scanning data of the low-angle diffraction of the sample shows a six-fold symmetry. The sample forms a ΦHN phase. Reprinted with permission of the American Chemical Society.111 | ||
Influence of side-chain structure. When the structure of the side chains in MJLCPs varies, such as chemical changes in the rigid core and the flexible tails, the resulting side chains will have different size, rigidity, and interaction type and strength. These side chains will affect the liquid crystallinity of the MJLCP polymers obtained, as mentioned in section 3.
The first series of MJLCPs contained calamitic mesogenic side groups. Since the MJLCP concept was proposed, many series of MJLCPs with different calamitic mesogens have been designed, synthesized, and investigated. Some of these MJLCPs are listed in Table 2. Depending on the structure of the side-chain mesogens, the rod-like polymers can form different columnar phases, including ΦN, ΦHN, ΦH, and ΦR phases. For example, PMPCS can exhibit a ΦHN phase,111 as described in section 4.1.1. The analogous MJLCP, PBPCS (polymer 23 with R1 = R2 = –OC4H9),112 also forms a ΦHN LC phase. The properties of these columnar phases, such as the size of the columns and transition temperatures, are also correlated with the side-chain structure. As expected, the diameter of the PBPCS cylinder is larger than that of the PMPCS one because of the larger side-chain length for PBPCS. Consequently, rod-like molecules with a controlled size can be constructed using MJLCPs.
As described in section 3, with the introduction of bent-core mesogens by changing the mesogenic substituting group from 2,5-positions to 3,5-positions of the phenyl group, the steric effect from side chains decreased dramatically. Thus, polymer 29 with bent-shaped side chains showed an N-like diffraction pattern and the packing of the rods could be enhanced by mechanical shear. The sample of polymer 29 (n = 4) exhibited a 2D ΦR phase with a = 2.50 nm and b = 2.76 nm. The weaker interaction between the backbone and the mesogenic groups increases the “free volume” of the polymer backbones for segmental motion and the jacketing effect is weakened.88 In addition to changing the position where the backbone and the mesogen were connected, the steric hindrance between the side chains and the polymer backbone in this MJLCP system can be altered by varying the size of the side groups. Larger side groups impose stronger steric hindrance, and a nearly extended chain can be achieved. Polymers 30 with n = 6, 8, 10, 12, 14, and 16 have been found to exhibit stable columnar phases. A ΦR was found in polymer 30 with relatively short aliphatic tails, whereas a ΦH was formed when the tail length was further increased. The phase transition from ΦR to ΦH was detected by annealing the sheared sample of polymer 30 (n = 12) at 180 °C for 12 h. The “softness” of the macromolecular rod surface might cause the difference in packing schemes. It should be noted that in this system the rod diameter of polymer 30 was about 3–4 nm, considerably larger than that of polymer 23 and other MJLCPs with three-ring mesogenic side chains or relatively flexible side chains.
In this way, well-defined shape-persistent macromolecules with the tunability both in shape and size could be realized in MJLCPs. Such shape controllability of MJLCPs somewhat resembled that of currently investigated dendronized polymers,113 in which the molecular shape could be varied from sphere to cylinder by increasing either the DP114 or the generation of side-attached monodendrons to create steric hindrance and stretch the coiled polymer backbone into an extended chain,115 forming the monodendron-jacketed polymers.116 Supramolecular columnar LC phases exist in both MJLCPs and dendronized polymers, but cubic phases which are frequently observed in dendronized polymers have not been found in MJLCPs yet, which is not surprising, given the rod-like shape of MJLCPs. Meanwhile, the driving force of self-assembly in dendronized polymers mostly originates not only from the steric hindrance of bulky dendritic side chains, but also from π–π stacking, hydrogen bonding, and other secondary interactions of side chains depending on their chemical structures. In addition, microphase separation of different moieties of columnar structures also induces self-assembly. Although the secondary interactions from mesogenic side chains in MJLCPs have not been thoroughly discussed in previously reported articles, it should be interesting to take them into account in designing novel jacketed polymers to reveal the nature of the jacketing effect not only in MJLCPs but also in other similar polymer systems.
In the research of MJLCPs, attention has been paid to the effect of the rigidity, thus also the flexibility, of the mesogenic unit on the chain conformation. It has been proven that when the flexibility of mesogens is increased step-wise from alkoxyphenyl to cycloalkyl81–83 substituent groups and finally to alkoxyl groups,84–87 the resulting polymers still maintained stable LC properties. The study on the origin of the mesomorphic phase behavior and the phase structure formed from MJLCPs with the laterally attached side chains not mesogenic any more thus provided hints on the mesomorphic behavior of flexible non-polar macromolecules without mesogenic moieties. As shown in Fig. 4, the 2D WAXD pattern of oriented fibers of polymer 27 is typical of a ΦH LC phase.84 These and other experimental evidences suggest that in MJLCP systems, the jacketing effect exists even when the side group is small or flexible, which is different from dendronized polymers.
Columnar phases appear in such flexible polymers with the mesomorphic phases originating from the parallel alignment of polymer chains. This kind of self-assembly behavior is quite similar to that of poly(di-n-alkylsiloxane)s,117,118 poly(di-n-alkoxyphosphazene)s, and other alkyl substituted polysilane, polypeptide, and cellulose polymers.119 In this case, although the polymer chains are stretched, both the backbone and the alkyl side chains are conformationally disordered. The length of the side chain has a profound effect on the stability of the mesophase. For example, poly(di-n-alkylsiloxane)s substituted with ethyl to hexyl side chains were able to form ΦH phases, while for poly(di-methylsiloxane)s and poly(di-n-alkylsiloxane)s substituted with heptyl to decyl side chains no mesophases were found between the crystalline phase and the isotropic phase.
As mentioned in section 3, columnar LC phases also existed in polymer 28 laterally attached with only one phenyl ring which was substituted with two flexible aliphatic tails through ester linkages.87 More interestingly, the phase structure of 28 could be manipulated by tailoring the length of aliphatic tails in the side group. Such a change in mesophase behavior with variation in the structure, such as length, of the flexible tails in the side groups has also been found in quite a few occasions for MJLCPs. As shown in Fig. 6, when the side chain contains propyl/isopropyl to hexyl tails, long-range 2D ΦH phases are obtained. Third-order diffractions with scattering vectors of 71/2 and 91/2 relative to that of the first-order diffraction can be clearly observed in this pattern, indicating a highly ordered hexagonal packing of cylinders. In addition, the peak value of the first-order diffraction moves to a lower angle when the tail length increases. If the n-alkyl tails are longer than hexyl, the polymer gradually loses its ability to form the LC phase. Substituting dodecyl groups at the ends leads to a completely disordered phase.
 | ||
| Fig. 6 Tail length dependence of the phase structure for polymer 28. (a) 1D WAXD pattern of polymer 28 (R = –CH(CH3)2) indicates a highly ordered 2D ΦH phase of the sample. (b) The phase structure of polymer 28 depends on the structure or length of the flexible tails in the side groups. The long-range 2D ΦH phase is only formed when the side chain contains propyl/isopropyl to hexyl tails. The polymer is less ordered for other tails. Reprinted and adapted with permission of the American Chemical Society.87 | ||
Molecular weight dependence. It has been proven that the MW and the MW distribution influence the liquid crystalline properties of SCLCPs. For example, liquid crystalline polymethacrylates, polynorbornenes, and poly(vinyl ether)s reach the limiting values of their thermal transition temperatures at less than 50 repeat units. The phase behavior of the columnar phase for poly(di-n-alkylsiloxane)s is also MW-dependent. A critical MW is required for the mesophase formation in poly(di-n-alkylsiloxane)s. So the similarity of these systems revealed that their self-assembly behaviors had the same possibility of an entropy driven mechanism.120,121
MW-dependent phase behavior has also often been observed in MJLCP systems. As shown in Fig. 7, the LC phase structures of this series of PMPCS samples have been found to be strongly MW-dependent.111 When the MW of the PMPCS sample is lower than a critical MW which is approximately 1.0 × 104 g mol−1 (measured by GPC calibrated with the polystyrene standards), PMPCS is amorphous. When the MW is in-between 1.0 × 104 and 1.6 × 104 g mol−1, a stable ΦN phase exists. Above 1.6 × 104 g mol−1, a ΦHN phase is observed. Furthermore, other MJLCPs with similar structures, such as PBPCS (polymer 23 with R1 = R2 = –OC4H9)112 and PMBPS (polymer 26 with R1 = R2 = –OCH2CH*(CH3)C2H5)122 with terphenyl as the rigid core, showed similar MW-dependent phase behavior. Within these two LC phases, the polyethylene backbone is strongly jacketed by the laterally attached rigid mesogenic side chains, and the cooperative assembly of these two parts makes the whole polymer chain cylindrically shaped. Such cylindrical polymer chains naturally self-organize into columnar LC phases by parallel alignment of the polymer chains. The dependence of the phase diagram on MW suggests a critical aspect ratio of the cylindrical building blocks required to stabilize these LC phases, as predicted by Flory.123 For PMPCS, the critical value of DP is 39∼42 based on Flory's criteria.
 | ||
| Fig. 7 MW dependence of LC formation for PMPCS. (a) WAXD powder patterns at 200 °C for PMPCS samples with different MWs. Patterns of samples with high MWs show a low-angle diffraction, indicating LC order, while patterns for samples with low MWs only have amorphous scattering. For samples with moderate MWs, the low-angle broad peak can be separated into two peaks. (b) The d-spacing from (a) as a function of MW. When the MW is lower than a critical MW of 1.0 × 104 g mol−1, PMPCS is amorphous. A ΦN phase is formed when the MW is in-between 1.0 × 104 and 1.6 × 104 g mol−1. Furthermore, above 1.6 × 104 g mol−1, a ΦHN phase is observed. Reprinted and adapted with permission of the American Chemical Society.111 | ||
Influence of the molecular architecture. On the basis of the research on MW-dependent LC behavior, it has been concluded that MJLCPs exhibit supramolecular columnar phases, in which MJLCP supramolecular rods are oriented parallel to one another. Therefore, the change in molecular architecture may influence the phase behavior resulting from the parallel packing of the rod-like polymer chains in these polymer systems. Branching may dramatically affect the phase behavior because the packing behavior can be favored or prohibited in different cases. On the contrary, most of the mesophases formed by conventional end-on SCLCPs are caused by parallel alignment of mesogenic units in the polymers, and the molecular architecture of the polymers may not have a great influence on the phase structure, which has been proven by Pugh's work.124
Star polymers with three (34a and 34b)125 and four arms (34c)126 of MJLCPs were synthesized successfully by the ATRP method, resulting in star-shaped PMPCS polymers of different MWs with low polydispersities. The LC behavior of these star polymers showed similar MW dependence to that of the corresponding linear homopolymers. All polymers showed one glass transition, and Tg increased with increasing MW. The critical Mn(GPC) of each arm was 0.9 × 104 g mol−1, above which the phase behavior became MW-independent. In this case, the chemical structures of the core did not affect the LC properties of MJLCPs.
To study how architecture affected the liquid crystalline behavior of MJLCPs, a novel series of eight-arm star PMPCS polymers 34d was synthesized via ATRP using initiator octakis(2-bromo-2-methylpropionoxypropyldimethylsiloxy)-octasilsesquioxane.127 The arms of the star polymers could be released by cleavage of the silsesquioxane core with hydrofluoric acid, and the precise octafunctionality of the star polymers was confirmed by GPC results obtained from GPC equipped with a multiangle laser light scattering (LLS) detector. It was found that when Mn was larger than 4.48 × 104 g mol−1, a ΦHN phase developed, and below this MW value, only an amorphous state existed. The calculated MW of each arm to stabilize the LC phase was lower than that of the linear hompolymers. Moreover, the ΦN was not found in eight-arm star PMPCS, only the more ordered ΦHN phase was obtained. This indicates that the LC phase is stabilized with such a molecular architecture which favors the parallel packing of semi-rigid polymer chains.

Hyperbranched PMPCS polymers were also synthesized by ATRP.128 The thermotropic properties of the copolymers strongly depended on MW and the feed ratio of MPCS to 4-chloromethylstyrene (CMS). For the copolymers to exhibit LC behavior, the feed ratio should be at least 30 and the polymers should have a high enough MW. For the copolymer obtained with a feed ratio of 32, the minimum Mn(GPC) was 1.21 × 104 g mol−1. Although linear PMPCS displays ΦN and ΦHN phases with a suitable MW, the hyperbranched polymer only shows a ΦN phase, indicating that the hyperbranched structure depresses the formation of the more ordered mesophase.
Random copolymerization. Radom copolymers with non-mesogenic monomers will dilute the grafting density of the mesogenic side group around the polymer backbone. The monomer of polymer 18 (R = –H and m = 1) was reported to copolymerize with methyl methacrylate (MMA), and the resulting copolymers showed LC phases when the non-mesogenic content did not exceed 60%. The Tg declined steadily as the concentration of MMA was increased.129 When another MJLCP monomer MPCS was copolymerized with styrene (St) or MMA, the tolerance was relatively low.130 The LC phase only existed when the molar fraction of MPCS exceeded 89% in PMPCS-co-PS and 84% in PMPCS-co-PMMA. The same result was obtained for copolymers containing polymer 21 (R = –OCH3) and PS, i.e., the LC phase was formed when the LC comonomer composition exceeded 89%.131 Allyl alcohol was copolymerized into the MJLCP system with monomer of polymer 21 (R = –OCH3). The LC phase could be maintained, and Tg increased slightly in this copolymer when the molar fraction of the MJLCP monomer ranged from 75 to 93%.132
Different from copolymers based on MPCS with St or MMA, which displayed no mesophase when the content of St or MMA exceeded a certain amount, copolymers 35 obtained with two different MJLCP monomers, MPCS and 2,5-di(n-butoxycarbonyl)styrene (BCS), using free radical polymerization formed stable LC phases regardless of the copolymer compositions.133 The LC phase structures of this series of copolymers were strongly composition-dependent. The molar fraction of MPCS in feed (xMPCS) influenced the type and size of the LC phase structure of the resulting copolymer. When xMPCS was above 0.5, the copolymer formed a ΦN phase with a constant d-spacing value of 1.58 nm, while with xMPCS below 0.1, a ΦH phase was observed with a corresponding d-spacing of approximately 1.44 nm. If xMPCS was in between 0.1 and 0.5, a ΦHN phase could be obtained with a tunable d-spacing varying with the copolymer composition.

In addition to copolymers from MPCS and BCS, other binary copolymers 36 from comonomers containing similar molecular structures but with different tail lengths were also synthesized using free radical polymerization.134 It was found that subtle difference in the alkoxy groups had a significant impact on the mesomorphic phase behavior of the copolymers. The LC structure could be retained over the whole composition range when only one CH2 length difference existed in the two comonomers, such as m = 3, n = 4 and m = 6, n = 5. When the length difference increased to two CH2 units, e.g., m = 6, n = 4 or m = 3, n = 5, the LC phase disappeared when the contents of the two comonomers were comparable, and only a ΦH phase could be observed in copolymers with dominant content of one comonomer. The LC phase would only be formed if the content of one comonomer of an amorphous homopolymer was below 0.25, e.g., in the case of m = 3 and n = 2 (Fig. 8). Similar investigations about the mesophase behavior of random copolymers with different chain lengths in copoly(di-n-alkylsiloxane/di-n-hexylsiloxane)s revealed that the structural irregularities tended to depress the stability of the mesophase.135

 | ||
| Fig. 8 The d-spacing value of copolymers in the low 2θ region as a function of the molar fraction of 2,5-di(n-propoxycarbonyl)styrene (x) at 180 °C. The LC structure formed over the whole composition range when there was only one CH2 length difference in the two comonomers. When the length difference was two CH2 units, the LC phase disappeared for copolymers with comparable comonomer contents, and only a ΦH phase was observed in copolymers with one dominant comonomer. The LC phase would only be formed when the content of one comonomer of an amorphous homopolymer was below 0.25. Reprinted with permission of Elsevier.134 | ||
 | ||
| Fig. 9 Unusual phase behavior of PBPCS (polymer 23 with R1 = R2 = –OC4H9). (a) DSC thermograms show enantiotropic transitions which have small but non-negligible latent heat. PLM results (inset micrographs) show that the sample is birefringent at high temperatures, indicating the existence of a mesophase, while the sample is isotropic at low temperatures. (b) The worm-like chain changes to a rod-like chain during heating, which is driven by entropy, and this transformation is reversible upon cooling. The rods self-organized into a ΦHN phase which has weak lateral hexagonal order. Reprinted and adapted with permission of the American Chemical Society.112 | ||
4.2 Smectic phases of MJLCPs
As described previously, the N phase was predominantly observed in side-on SCLCPs due to the hindrance to the rotation of mesogens, while other LC phases, such as SmA48 and SmC30 phases, were also found in some cases. Polysiloxane 2 and copolysiloxane with long spacers and long aliphatic tails tended to exhibit stable SmC phases. Pugh et al. proposed several other ways to realize smectic phases, especially SmC(*) phases, in side-on SCLCPs.141 The first one was incorporating mesogens 1,4-bis[(3′-fluoro-4′-n-alkoxyphenyl)ethynyl]benzenes, which possessed an SmC–N–I phase sequence, into polymers. Polymer 37 with a short spacer (p = 1) was designed in this way to show an SmC phase over a very narrow temperature range, while polymer 37 with a longer spacer (p = 11) only exhibited an N phase besides the glass transition.142,143 Further investigation revealed that the mesophase behavior of polymer 37 containing a one-carbon spacer was strongly dependent on the tail length of the side chain. When n = 1, where n was the number of carbons in the tail, only an N phase was observed.144 Polymer 37 with n = 2–7 exhibited an N phase with SmC fluctuations; when n was increased to 8–11, the resulting polymers showed an SmC structure at room temperature and could transform to an N phase.144,145 A second-order transition between SmC and SmA phases was detected by WAXD in polymer 37 with n = 12.145 The second way to achieve the smectic structure in side-on SCLCPs was incorporating n-alkoxy substituents terminated with fluorocarbon segments146,147 or hydrocarbon/oligodimethylsiloxane substituents.148 The incompatibility of the two segments would induce layered structures. On the basis of the experimental results, it could be concluded that inducing smectic layering in polymer 38 required a minimum of n = 8, m = 3 or n = 3, m = 4. Otherwise, only an N phase could be observed. And in the case of n = 3 and m = 2, no mesophase existed at all. MJLCPs 39 with mesogens having semi-fluorinated tails also showed an SmC phase, and a “herring-bone” model was proposed to illustrate the packing arrangement in this phase.149 An SmA phase was found in polymer 40 with the asymmetric substitution of semi-fluorinated groups.150 The third approach was copolymerizing one monomer containing an electron-rich mesogen with a second monomer containing an electron-poor mesogen to induce smectic layering by electronic donor–acceptor interactions.141

When the central phenyl ring in the side chain of an MJLCP was replaced by a biphenyl group, the phase structure of the resulting polymers 31 and 32 with whatever bonding positions dramatically changed from an N (ΦN) phase to an SmA phase.89 When the aliphatic tail length was further increased (n > 6), an SmC phase developed.90 The evolution from ΦHN to SmA was found in MJLCPs containing the 1,3,4-oxadiazole unit (Fig. 10).92 Polymer 33 with t-Bu group still forms a ΦHN phase, while an SmA phase appears with the sequence of SmA–N–I when the aliphatic tail is changed to a longer alkoxy group. Fig. 10a shows the 1D WAXD patterns of polymer 33 with R = –C10H21 during cooling. All the profiles exhibit two strong diffractions in the low-angle region, with a d-spacing ratio of 2:1, indicating a smectic ordering. In Fig. 10b, strong diffractions which originate from layer structures appear on the equator of a 2D WAXD fiber pattern for polymer 33 with R = –C8H17, with the X-ray incident beam perpendicular to the shear direction (along the meridian, solid line), also suggesting a lateral layer structure. On the other hand, the amorphous scattering is centered on the meridian, indicative of an SmA phase. Other polymers 33 with long linear alkoxy tails also exhibit SmA phases. As the mesogenic group is laterally attached to the polyethylene backbone through a single carbon–carbon bond, polymer 33 in the SmA phase should be more or less ribbon-like with the backbone, which is squeezed by the side chains, aligned parallel (Fig. 11). The flexibility of the side-chain tails is of crucial importance in determining the LC structures. More interestingly, when the rigid core in the side group of polymer 33 changes to a bent-type core with the two phenyl-1,3,4-oxadiazole groups connected to the center phenyl ring in meta positions instead of para positions, all polymers 41 form ΦH phases even when the linear alkoxy end group contains 16 carbons, and no smectic phases are found.151 The latter work suggests that both the molecular shape and the flexibility of the side groups may impose a big impact on the phase structures of MJLCPs.
 | ||
| Fig. 10 Smectic phases in polymer 33. (a) 1D WAXD patterns of polymer 33 with R = –C10H21 during cooling exhibit two strong diffractions in the low-angle region, with a d-spacing ratio of 2:1, which indicates a smectic ordering. The inset shows the enlarged patterns of the second-order diffractions. (b) For polymer 33 with R = –C8H17, when the X-ray incident beam is perpendicular to the shear direction (along meridian, solid line), strong diffractions that originate from the layer structure appear on the equator, while the amorphous scattering is centered on the meridian, which indicates an SmA phase. Reprinted and adapted with permission of the American Chemical Society.92 | ||
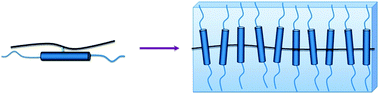 | ||
| Fig. 11 Schematic representation of a sheet-like MJLCP. The steric hindrance of the bulky rigid side chains forces the backbone to take an extended-chain conformation. The backbone lies in the middle of the sheet-like structure. And the rigid side chains are not oriented parallel to the chain direction of the backbone. | ||
The mechanism of the smectic phase formation in side-on SCLCPs appears more complicated than that in end-on SCLCPs. In polysiloxane 2, the conformation has been investigated using SANS. The chain conformation was detected to evolve from a prolate shape in the N phase to an oblate shape in the SmC phase. The flexible spacer connecting the polymer backbone and the mesogenic side chains makes the adjustment of the polysiloxane conformation relatively easy. Furthermore, the stabilization of the SmC phase by diluting the mesogen density in polymer 12 indicated that flexibility was needed in side-on SCLCPs for the side chains to self-assemble into the smectic phase. In MJLCPs and polynorbornenes, the spacer is rather short, and the conformation of the polymer backbone might not be altered as easily as in polymer 2. Therefore, it is interesting to find that the two contrast approaches to stabilize the smectic formation are both successful in one side-on SCLCP system. However, more investigation needs to be carried out to find the mechanism of the structural formation.
5. Chirality in MJLCPs
Chirality in liquid crystals can offer chiral LC structures, such as the cholesteric phase, the ferroelectric SmC phase, the blue phase, and so on.152 Meanwhile, if chirality is introduced into polymer systems, the helical conformation and other complex supramolecular structures may be obtained.153 A few polymers with helical chirality have been synthesized, such as polychlorals, polyisocyanates, polyisocyanides, poly(triarylmethyl methacrylate)s, and some substituted conjugated polymers. Among these helical polymers, the effect of the bulky side group which induces the chiral helical conformation is of great interest.In side-on SCLCPs, the addition of chiral groups appears to disturb the formation of the LC phase. Similar investigation was reported in MJLCPs previously when a chiral substituting group was attached to polymer 23.154 A cholesteric phase was observed in copolymers with chiral and non-chiral units.22,50,58 Lewthwaite et al. synthesized polymer 42 which showed chiral N phases and the chiral N range increased with increasing length of the rigid mesogenic side chain.155 An LC polysiloxane 43 with a cholesteric moiety laterally attached to the polymer backbone was previously reported, which showed an N or a cholesteric-like phase evidenced from DSC results, although no specific texture of a chiral N phase could be identified under PLM because of the high viscosity in the LC phase.156
Polymer 26 containing 4,4′-dialkoxyterphenyl mesogenic group laterally attached to the polyethylene main chain was able to maintain its LC phase when chiral groups were attached at the end of the mesogenic group. The resulting polymer PMBPS showed similar ΦN LC phase behavior as other non-chiral MJLCPs, and no chiral phase was detected so far. On the other hand, the chiroptical properties from the corresponding monomer, (+)-2,5-bis[4′-((S)-2-methylbutyloxy)phenyl]styrene (MBPS), to the polymer changed remarkably, indicating that a helical secondary structure was formed during the free radical polymerization. And this conformation could be maintained even after the initial chiral segment in the side group was chemically removed (Scheme 1).157

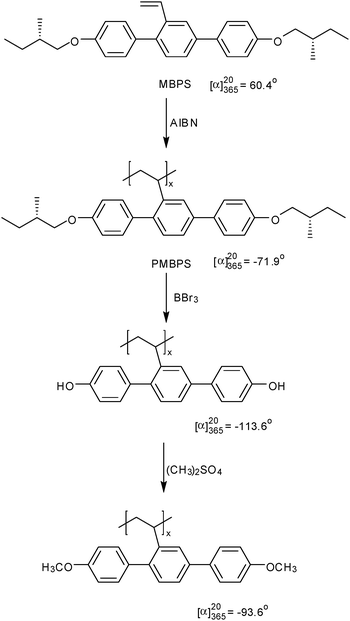 | ||
| Scheme 1 Synthesis of chiral MJLCPs and their chiroptical properties.157 | ||
The research on the MW dependence of chiroptical and thermotropic properties provided some hints on the development of the chain conformation. The monomer MBPS displayed a positive optical rotation, while the polymers gradually reversed the optical rotations to the negative value and reached a plateau when the DP of PMBPS was above 53. This critical DP was in coincidence with the formation of the ΦN phase, indicating that as soon as the stable helical conformation formed, the polymer chains were rigid enough to pack into a columnar phase. When MBPS was copolymerized with St, the compositions of copolymers were investigated to correlate with the liquid crystallinity and the helical chain structure of PMBPS.122 The optical rotations of copolymers remained positive in a large composition range and finally became negative when the composition of chiral units exceeded 90%, which was the similar critical composition value above which the copolymers were able to show a ΦN phase. The chirality transfer from monomers to polymers was further studied by designing a series of bulky vinyl monomers, 2,5-bis[(4′-alkoxycarbonyl)-phenyl]styrene, with different stereogenic centers. The absolute spatial configuration of the chiral atom and its position relative to the p-terphenyl group influenced the sign of the optical rotation of the resulting polymers.158
The solvent effect on the chiroptical properties was studied with glycopolymers, PTAGPS and PGPS (Scheme 2).159 PTAGPS had a thermodynamically controlled conformation which was not influenced by the polymerization conditions. However, the chiroptical properties of PGPS showed a solvent dependence during polymerization. An evolution of the conformation was observed by annealing the polymer in dimethyl sulfoxide at various temperatures. PGPS derived from PTAGPS showed the essential chiroptical features of both its precursor and PGPS with a kinetically controlled conformation obtained in N,N-dimethylformamide.

6. Rod–coil liquid crystalline block copolymers
LCPs can readily undergo self-organization on the length scale of 1–10 nm.160 Another most considered type of polymers with self-assembly behavior are block copolymers (BCPs) which can be constructed by covalently connecting two or more dissimilar polymer segments together. The immiscibility of different polymer blocks will induce microphase separation with ordered structures on the length scale of 5–100 nm.161,162 If LCPs are incorporated as one of the blocks in a BCP, a liquid crystalline block copolymer (LCBCP) is thus formed. The competition between microphase separation and liquid crystallinity induces LCBCPs to self-assemble into hierarchical nanostructures.163–166 This also provides a good opportunity to tune the functionality on different length scales in one polymer system. By definition, LC block copolymers contain at least one LC segment and may have structures including rod–rod, rod–coil, side-chain liquid crystalline (SCLC)–coil, and other combinations of blocks.Rod–coil BCP,167 with one block adopting a rigid rod-like conformation, is one unique type of LCBCP. Rigid polymer chains168 defined as the persistence length of about 10 nm in solution are both structurally unique and technologically important compared to their random-coil counterparts. Rigid polymers with a rigid-chain conformation of the backbone include helical-chain polymers, π-conjugated polymers, aromatic polymers, and so on. Self-assembly behavior of coil–coil BCPs is dictated by χ (Flory–Huggins parameter), N (degree of polymerization), and f (volume fraction of each block). For rod–coil BCPs, in addition to these parameters, the properties of rod blocks also critically affect the final phase formation. In general, the packing of the rod and coil blocks at the interface is controlled by the volume fraction of the coil (fcoil) and the area per junction of the coil (Acoil) and the rod (Arod). Because of the high asymmetry in the chain conformation, rod–coil BCPs can self-assemble into complex phase structures, which is quite different from coil–coil systems. Due to the spontaneous parallel aligning tendency of the rod block polymer chains, a flat inter-material dividing surface (IMDS) is preferred, which makes the lamellar morphology predominant over a large fcoil range. Only low molar mass rod–coil copolymers with asymmetric volume fractions have been found to form morphologies with highly curved interfaces, such as sphere (S) or gyroid (G). As the MW of the rod increases, the rigid rod becomes incompatible with the curved interface dictated by the S or G phase; thus, competition exists between liquid crystal formation of the rods and BCP microphase separation.167,169
From a synthetic point of view, only several types of rigid polymers have been used to construct rod–coil block copolymers with well-defined structures. And thorough research on their self-assembly and thermodynamics has been conducted. Polypeptides were first introduced into rod–coil systems.164,170 Chen et al. reported the syntheis of BCPs containing polyisocyanates (PHIC-b-PS) and their self-assembly behavior in films. Arrowhead, wavy lamellar, and zig-zag phases with layer-like structures were obtained in solution-cast films.171,172 BCPs with conjugated polymers, for example, poly(2,5-diethylhexyloxy-1,4-phenylenevinylene) (DEH-PPV), as the rod block and polyisoprene as the coil block were also synthesized.173–183
Block copolymers containing conventional side-on SCLCPs have seldom been reported. Li and Keller synthesized AB and ABA type block copolymers 44 and 45, respectively.184 In block copolymer 44, the TN−I of the LC block was the same as that of the homopolymer. While in 45, the TN−I was reduced by about 25 °C relative to that of the LC homopolymer, and the normalized clearing enthalpy change of the LC block was about 30–40% lower than the clearing enthalpy change of the LC homopolymer. The order–disorder transition temperature was the same as the TN−I in 45. The lamellar structure still remained in 44 when the LC block entered into the isotropic state, and thus a lamellar-to-lamellar transformation from an LC–coil structure to a coil–coil structure was observed at the TN−I. All the difference of the thermal behavior might originate from the different polymer backbone structure of the LC block, polynorbornene, which was rather different from the polyacrylate block, resulting in a strongly segregated system. The rheological study of 45 (copolymer Mn = 33, 200 g mol−1, polyacrylate block Mn = 17, 000 g mol−1; weight ratio 51/49) revealed that the lamellae could be aligned with the lamellar normal perpendicular to the shear direction as well as the velocity gradient direction and adopt perpendicular orientation under high frequency and strain amplitudes.185 Thin films of triblock copolymers 45 and 46 showed different surface alignment behavior. As for 45, the lamellar structure was aligned parallel to the silicon surface, while the lamellar structure of 46 was perpendicular to the silicon surface.186


It is evident that for most of the rod-forming systems discussed above, the rod length can be controlled by tuning the polymer MW. Other parameters such as the rod diameter and the surface chemistry of the rods can not be easily controlled. On the other hand, as a novel type of constructing unit to create rod-shaped macromolecules, MJLCPs are extremely attractive due to the structural tunability that they can offer: the length is determined by the MW of the MJLCP, and the diameter can be controlled by varying the lengths of both the rigid core and flexible tails in the mesogenic side group. Table 3 lists some d100 values of several MJLCPs. These values are indeed dependent on the size and the structure of the side group, which also further supports our packing models for MJLCPs. Furthermore, since the soft tails at the ends of the mesogenic side groups are flexible, while the mesogens are rigid, a novel core–shell cylinder can be achieved by using relatively long tails. Therefore, the surface chemistry can be readily adjusted with a variation in the tail structure. Thus, an MJLCP can be considered as a semi-rigid polymer chain which can be used to build up a novel kind of rod–coil block copolymers. In addition to the excellent shape tunability, well-defined MJLCPs can be easily prepared by controlled/living free radical polymerization with MW ranging from 10, 000 to 400, 000 g mol−1 and good solubility in most solvents. Various rod–coil block copolymers containing MJLCPs as the rod block have been successfully synthesized,187–192 with the coil block including amorphous polymers, such as PS, PMMA, poly(n-butyl acrylate) (PBA), polydimethylsiloxane, and polybutadiene, and crystalline polymers, such as poly(ethylene oxide) (PEO) and poly(ε-caprolactone) (PCL). The rod block candidates that are commonly used include PMPCS, PBBOS, PMBPS, and poly[{3,5-bis[(4′-((4′′-tetradecanoylbenzoyl)oxy)benzoyl)oxy]styrene}] (PTBOS). Rod–coil and rod–coil–rod as well as ABC-type193 and star rod–coil block copolymers194 have been synthesized. With such facile preparation of block copolymers, thorough study on the self-assembly of these rod–coil BCPs both in bulk and in solutions can be conducted. It should be noted that different from traditional rigid-chain polymers, MJLCPs exhibit unique characteristic LC properties, including MW-induced coil-to-rod conformational change, a very stable LC phase with most transition temperatures close to the decomposition temperatures, and re-entrant phase behavior, which further complicate the self-assembly behavior in these BCPs.

| Polymera | Molecular structure | Side-chain length (nm) | d 100 (nm) | LC Phaseb | Ref. |
|---|---|---|---|---|---|
| a Sample name in reference; b Φ HN = hexatic columnar nematic phase, ΦN = columnar nematic phase, ΦH = hexagonal columnar phase, SmA = smectic A, SmC = smectic C. | |||||
| PMPCS |

|
1.92 | 1.49 | Φ HN | 111 |
| 1.57 | Φ N | 111 | |||
| PBPCS |

|
2.75 | 1.86 | Φ HN | 112 |
| PCt |

|
2.10 | 1.85 | Φ H | 92 |
| 2C2Vp |
|
2.59 | 2.33 | SmA | 90 |
| 10C2Vp |

|
4.40 | 4.02 | SmC | 90 |
6.1 Self-assembly of MJLCP-based BCPs in bulk
In BCPs containing MJLCP blocks, the lamellar morphology in the bulk state is most frequently observed, and MJLCPs maintain the liquid crystallinity in the lamellae when the MW of the MJLCP block reaches the critical value similar to that of the homopolymer. In low MW samples of symmetric PS-b-PMPCS, the lamellar structure (Fig. 12) was confirmed with small-angle X-ray scattering (SAXS) and transmission electron microscopy (TEM) experiments.195 A uniform alignment of the lamellar structure could be obtained by a large-amplitude reciprocating shear, and the lamellar normal was perpendicular to the shear direction. Within the layers, PMPCS possessed a ΦN phase structure and the columns were parallel to the lamellar normal. Two layers of PMPCS columns were confined in each LC sublayer. Such a hierarchical structure resembles a “bilayer smectic A” structure of low molar mass LCs. In the asymmetric PS-b-PMPCS, the formation of a tetragonal perforated layer (TPL) structure, also shown in Fig. 12, was observed for the first time.196 In the TPL structure, coils (PS blocks) punctuated the PMPCS liquid crystalline layers, and the PS domains in the liquid crystalline layer possessed a tetragonal instead of hexagonal symmetry. The phase boundary between the lamellar and perforated structures critically depended on the MW of the PMPCS block.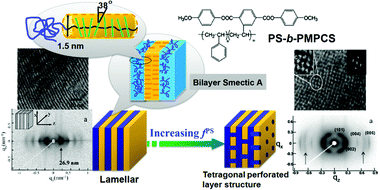 | ||
| Fig. 12 Hierarchical, self-assembled supramolecular structures of PS-b-PMPCS. For the symmetric BCP with a low MW, a lamellar microphase structure is formed, as shown in the TEM micrograph and the 2D SAXS pattern. The PMPCS columns are aligned parallel to the lamellar normal. The packing of the rod–coil BCP resembles a “bilayer smectic A” molecular structure. When the fPS is increased to obtain the asymmetric BCP, a tetragonal perforated layer structure is formed, in which coils (PS) punctuate the liquid crystalline layers, and the PS domains in the liquid crystalline layer are packed in a tetragonal instead of hexagonal lattice shown by the TEM and 2D SAXS results. Reprinted and adapted with permission of the American Chemical Society.195,196 | ||
Besides lamellar structures at moderate rod fractions, hexagonal coil cylinders in the rod matrix at high rod fractions were formed in rod–coil–rod triblock copolymers of PMPCS and polyisobutylene (PIB), PMPCS-b-PIB-b-PMPCS.197 This morphology with curved IMDS was seldom found in rod–coil block copolymer systems, especially in the case where the rod was on the convex side of the curvature. The morphology of coil spheres in the rod matrix was observed in PS-b-PTBOS, in which a soft shell was covered on the rod surface that reduced the interaction among rod blocks.198 When the curved IMDS formed, the liquid crystalline order of the rod block was reduced from ΦH to ΦN (Fig. 13). Such competition between liquid crystallinity of the rod block and microphase separation was also found in coil-rich sample PS567-b-PTBOS24, with PTBOS only showing a ΦN phase within a broken lamellar microphase structure. The lateral repulsion of the coil chains rendered a splaying stress field on the LC rods, which forced the rod to bend, thereby reducing the LC order. This repulsion could be released by blending a small amount of PS oligomers, and the LC block could return to the ΦH phase. Such a strategy of blending homopolymers with BCPs to adjust the microphase structure and size was also employed in a recent work, in which rod (PMPCS) or coil (PIB) homopolymer was added into PMPCS-b-PIB-b-PMPCS rod–coil–rod triblock copolymers.199
 | ||
| Fig. 13 TEM micrographs and corresponding schematic illustrations of three different hierarchical structures. (a) For PS567-b-PTBOS24 of low fPTBOS, the structure is ΦN-in-PL in which PTBOS forms a ΦN phase within the perforated layer structure of the self-assembled BCP. (b) For PS202-b-PTBOS35 with a relatively symmetric composition, it is ΦH-in-Lam with PTBOS exhibiting a ΦH phase in self-assembled BCP which had a lamellar structure. (c) For PS160-b-PTBOS126 of high fPTBOS, a ΦN-in-sphere structure, in which PTBOS forms a ΦN phase in spheres of the microphase separated BCP, is obtained. The self-assembly of BCP causes LC symmetry breaking in (b) and (c). Reproduced by permission of the Royal Society of Chemistry.198 | ||
The thermodynamic behavior of such rod–coil block copolymers was studied in several systems. In PS-b-PBBOS studied by Gopalan et al.,191 the order–disorder transition occurred between 120 and 150 °C, detected by temperature-dependent SAXS measurements, in which the intensity of the scattering peak gradually decreased on heating to 120 °C, and finally the peak disappeared at 150 °C, which was above the Tg's of both blocks. Slow cooling regenerated the morphology, indicating very slow ordering kinetics.
If the coil block is changed to a crystalline polymer, the self-assembly behavior becomes more complicated. Besides those structural parameters mentioned above, the additional crystallization of the coil part should be taken into account. For example, a lamellar-to-lamellar transition was triggered in PCL-b-PMPCS by the amorphous-to-LC transition of the PMPCS block, and the transition was irreversible due to the irreversible LC formation.200 The chain axes of the PCL blocks in the crystal and the PMPCS rods in the LC phase were all parallel to the lamellar normal of the microphase-separated structure. Since the melting temperature of the PCL crystal is well below the Tg of the PMPCS block, crystallization of the PCL block takes place in a one-dimensionally confined environment. In the confinement of LC PMPCS blocks, the PCL blocks are more stretched than those confined by the amorphous PMPCS blocks. And thus PCL crystallized faster with an increased fold length.
Crystallization and melting of PEO-b-PMPCS (PEO block Mn = 5, 300 g mol−1, PMPCS block Mn = 2, 100 g mol−1) in bulk201 and in ultrathin films202 have been studied as well. Since the MW of the PMPCS block in this sample was smaller than that of the critical MW required for LC formation, PMPCS was an amorphous, not rod-like but tablet-like polymer, which could significantly affect the crystallization and melting of the diblock copolymer. The sample studied could form monoclinic crystals with the structure identical to that of the PEO homopolymer. The PEO blocks were non-integral folded (NIF) in the crystals, and the PMPCS blocks which were rejected to the lamellar fold surfaces prevented the NIF PEO crystals from transforming into integral folded (IF) ones. In addition, the tablet-like PMPCS chains might adjust their neighboring positions up or down with respect to the lamellar surface normal, forming more than one PMPCS layer to accompany the increase in the PEO fold length with further crystallization.
6.2 Self-assembly of MJLCP-based BCPs in solution
The solution behavior of rod–coil PS-b-PMPCS BCPs has also been investigated. The rod-like PMPCS block can be dissolved in p-xylene, which is a good solvent for the PS coils, at temperatures higher than 100 °C. When a PS-b-PMPCS dilute solution was cooled, the copolymer chains could self-assemble into a core–shell nanostructure with the PMPCS segment as the core and PS as the shell. By combination of static and dynamic LLS techniques, the core radius (Rc) and the shell thickness (ΔR) can be calculated. With increasing solution concentration, the calculated Rc remains constant while ΔR becomes larger. Rc is predominantly associated with the length of the PMPCS block. When more chains are inserted into the core–shell structure, only the coil PS blocks in the shell have to be stretched to accommodate more chains (Fig. 14).203 Systematic study on the self-assembly behavior of PS-b-PMPCS with the same PS block length and different PMPCS lengths in p-xylene reveals that the critical association concentration (CAC) decreases as the MW of the rods increases. The molar mass of the self-assembled nanostructures increases very fast near CAC, and the dynamics slows at concentrations much higher than CAC.204,205 | ||
| Fig. 14 Schematic illustration of a core-shell nanostructure formed by the temperature-induced self-assembly of rod–coil diblock copolymer PS-b-PMPCS in dilute p-xylene solution. When more chains are assembled into the nanostructure, the core radius remains as a constant, close to the contour length of the PMPCS block, while the shell becomes thicker, indicating the PS blocks are forced to stretch in the shell. Reprinted and adapted with permission of the American Chemical Society.203 | ||
Another amphiphilic rod–coil block copolymer, PEO-b-PMBPS, has been proven to self-assemble into different supramolecular structures in aqueous solutions, including spherical micelles, vesicles, multilamellar vesicles, large compound vesicles, and tubules.206,207 The water-induced aggregation behavior of PEO104-b-PMBPS53 was further investigated in the common solvent THF and in the selective solvent dioxane.208 When water was added above the critical composition (6 wt%), narrowly distributed spheres were observed in dioxane, whereas in THF, a bimodal distribution was observed. When the hydrophilic block was changed to a brush-like polymer poly[poly(ethylene glycol)monomethyl ether methacrylate] (PPEGMA), the resulting amphiphlic block copolymer with the identical rod segment, PPEGMA37-b-PMBPS87 showed different self-assembled structures in THF–water. Spheres were easily formed regardless of the initial concentration and the rate of water addition. With increasing volume fraction of the rod block, the resulting PPEGMA37-b-PMBPS141 could self-assemble into primary spheres and large compound spherical or rod-like micelles at a low water addition rate. At a high water addition rate or even when the THF solution was poured into a large amount of water, only spherical aggregates were formed instead.209

The self-assembled monolayer behavior of PEO-b-PMBPS with predominantly hydrophobic contents at the air–water interface has been investigated. The morphological transition from individual spherical aggregates into long cylindrical aggregates with increasing surface pressure for PEO104-b-PMBPS17 was observed to accompany with the pancake-to-brush conformational transition of the block copolymer at the interface. The PEO chains desorbed from the air–water interface and went into the water subphase. Thus, the effective content of the rod block changed continuously from 61% at zero pressure to 93% at the start of the monolayer collapse by molecular reorganization. A similar morphological evolution was observed in all block copolymers PEO104-b-PMBPSm (m = 17, 30, 45, 53) with the rod content increased from 61 to 83% at zero pressure.210
7. Properties and applications
The applications of SCLCPs have been reviewed in several aspects and have been found to focus on the following several fields: optical data storage, nonlinear optics, chromatographic stationary phases, separation membranes,211 and other areas.7,8 In fact, similar to conventional end-on SCLCPs, many efforts have been devoted to developing the applications of side-on SCLCPs in almost the same fields mentioned above. For example, polymer 2 was first coated on silica as a stationary phase in HPLC, which showed excellent planarity and rod shape recognition capability for polynuclear aromatic hydrocarbon (PAH) solutes.212,213 The influence of molecular parameters, such as the spacer length, the aliphatic tail length, and the density of laterally attached mesogenic units along the polymer chain, were studied for the optimization of these stationary phases. The results revealed that the best chromatographic performances towards planarity and shape recognition for PAH solutes could be achieved when smectic side-on SCLCPs were used, exhibiting the local order effect on the shape recognition of stationary phases.214,215 The influence of fixation method of SCLCPs on the stability of stationary phases and column efficiencies,216 the retention behavior of SCLCP-based stationary phase,217 and the temperature effect in reverse phase HPLC218 were also investigated.SANS studies have indicated that side-on SCLCPs adopt a rod-like conformation in the N phase and turn to a coil-like conformation in the isotropic phase. The change in the average molecular shape induced by the conformational change due to the nematic-to-isotropic transition is quite similar to that of the main-chain LC polymers. When the sample was prepared as a liquid single crystal-like monodomain with all mesogens oriented uniformly, the shape change from the microscopic single chain to the macroscopic sample could be realized at the nematic-to-isotropic transition (Fig. 15a).219 Such reversible thermal-responsive elastic behavior makes the side-on SCLCPs possible candidates in applications as artificial muscles when nematic elastomers or glasses are used, on the basis of the original hypothesis proposed first for main-chain LC polymers by de Gennes.220 The first example of thermal-responsive materials with side-on SCLCPs was demonstrated with a typical contraction of around 35–45% and a generated stress of around 210 kPa.221 Subsequently, as mentioned in section 2.1, if the mesogenic side chain contained an azobenzene functional group, a photo-responsive material could be developed. Finkelmann et al. first demonstrated the opto-mechanical effect induced by photoisomerizing monodomain nematic elastomers, with reversible shape changes of between 10 and 400% in solids.222 Li et al. reported light-driven side-on nematic elastomer actuators with a fast contraction of around 20% induced by UV irradiation.53 Micrometer-sized artificial muscles were also prepared with a slightly crosslinked side-on SCLCP, which had the potential applications as micro-actuators.223
 | ||
| Fig. 15 (a) Schematic illustration of the shape change induced by the phase transition of side-on SCLCP between N and isotropic phases, making it thermally responsive.219 (b) Artificial muscle based on an RNR (R denotes rubber and N nematic phase) triblock copolymer exhibiting a lamellar microphase structure with a crosslinked R domain and the side-on SCLCP block forming the N domain.224 | ||
Another approach took advantage of the lamellar structure of a triblock copolymer RNR (R, classical elastomer; N, nematic polymer) (Fig. 15b). A new kind of muscle-like material based on a triblock copolymer containing a side-on nematic LC homopolymer as the N block instead of main-chain nematic LC polymers was obtained in this way.224
ABA-type triblock copolymer consisting of the PMPCS as hard A block and PBA as the soft B block had the potential as liquid crystalline thermoplastic elastomers (TPEs), which had been studied by dynamic mechanical analysis and tensile tests. For the curve of temperature dependence of the dynamic shear storage modulus (G′), two relaxation processes were detected, and a well-defined rubbery plateau was observed between them, which was consistent with the typical feature of TPEs. No other sharp drop associated with the LC-to-isotropic transition of G′was found within the temperature range studied, and the sample did not enter into the liquid flow region above the Tg of the PMPCS block. Both of these results indicated the presence of a very stable columnar nematic phase, and the physical network still persisted at least up to 200 °C.192
Fluorinated MJLCPs 39 and related block copolymers synthesized by Gopalan et al.149 could be used as surface-modifying agents. The polymer film showed high water contact angle value of around 120–122°, which was due to the high density of fluorinated groups on the surface. The hydrophobic surface is relatively stable, and hydrophobicity is anticipated to further increase with increasing order of the smectic phase.
The electro-optic properties and light emitting devices (LEDs) of polymer 33225 and its copolymers with polyvinylcarbazole (PVK)226,227 were investigated. In copolymers 47, photoluminescent (PL) peaks in the film showed a blue-shift compared with those in solutions, and the fluorescent quantum efficiency decreased with increasing content of N-vinylcarbazole (NVK), indicating an efficient energy transfer from NVK units to the oxadiazole units. Single-layer polymeric light emitting devices (PLEDs) were fabricated, which emitted a blue light around 450 and 490 nm with a maximum luminance of 703 cd m−2. The device performance varied with the content of NVK and the device configuration, and a best value of external quantum efficiency of 0.27% and luminous efficiency of 0.108 lm W−1 was achieved.226,227 Polymer 33 (R = -t-Bu) as a host material for an IrMDPP [Ir(III)bis(5-methyl-2,3-diphenylpyrazine)(acetyl acetonate)] doped polymer electrophosphorescent device has been investigated. The maximum luminance and external quantum efficiency were higher than those of devices with PVK. As the IrMDPP content was increased, the color in the electroluminescent (EL) spectra shifted from green-yellow to yellow-orange and the EL spectra were different from the PL spectra partly because direct charge-trapping and recombination in the EL process played a dominant role over the energy-transfer routes.228 The jacketed structure was also applied to polymers with rigid, conjugated backbones to explore their electro-optic properties. A thiophene-based monomer, 3-{2,5-bis[(4-hexadecyloxy-phenyl)-1,3,4-oxadiazole]phenyl}-2,5-dibromothiophene, was copolymerized with 9,9′-dioctylfluorene to obtain a series of new conjugated, jacketed copolymers 48, and the investigation on properties of PLED devices fabricated using these copolymers indicated that introducing a proper ratio of the thiophene-oxadiazole comonomer to polyfluorene could greatly improve the EL properties.229

8. Summary and outlook
From the above discussion, we can conclude that a new research area has been added to the field of polymer science, not only in liquid crystalline polymer science, but also in supramolecular chemistry. With only the change of the attachment method through the end-on to side-on type, the molecular dynamic behavior of the resulting LCPs shows a dramatic change. The jacketing effect of the rigid mesogenic side chain offers to stretch the flexible backbone to an extended-chain conformation. Such a jacketing effect has been proven to be greatly dependent on the length of the spacer and the grafting density of the side chain, and less on the length of aliphatic tails and the flexibility of the backbone. When the flexible spacer is removed and the connection between the msogenic side chain and the polymer backbone is further reduced to only one covalent bond, the resulting MJLCPs show tremendous variation in mesophase behavior. Supramolecular self-assembled columnar phases can be formed with the whole polymer chain packed parallel to one another. In the past several years, a rich variety of new MJLCPs as well as rod–coil diblock copolymers containing MJLCPs as rigid blocks have been designed and prepared. It can be foreseen that more ordered hierarchical structures will come out of these systems by rational design of building blocks and finely tuning self-assembling conditions. In addition, currently the concept of MJLCP first derived from side-on SCLCPs is not restricted in mesogenic side chains any more. Using flexible and apolar building blocks to construct LCPs has paved a versatile route toward novel organic materials. It is now clear that we have to take into account the dynamic variations in the molecular structure, such as conformational and geometrical changes. Thus, we can consider the formation and stabilization of the mesophase when designing liquid crystalline polymers. This strategy will not only be useful in the design of liquid crystalline polymers but also have implications in the exploration of other new materials. Apart from cylinder-like polymer chains of MJLCPs, which can self-assemble into columnar phases, the existence of new apparent smectic phases in MJLCPs may indicate the formation of sheet-like structures which require more systematic investigations. Since the highly controllable structural design and synthesis have been proven during the last two decades, the functional tunability can be achieved in MJLCP homopolymers and block copolymers on nano- and micro-sized scales.It should be noted that although various MJLCPs have been designed and synthesized, and their mesomorphic behaviors have been revealed and reported, the mechanism of the mesophase formation, including the chain conformation and the cooperative dynamics between the side chain and the polymer backbone, is not clear yet. SANS and other techniques, as well as modeling and simulations, are currently carried out in order to reveal the intrinsic molecular nature of MJLCP chains during the self-organization process. The research on the self-assembly behavior of MJLCPs will further help us understand more about the general interaction between the polymer backbone and pendant chains which also exists in numerous polymer systems, such as side-chain functional polymers, dendronized polymers, polymer brushes, and so on. Is the jacketing effect universal?
Acknowledgements
We thank all co-workers and collaborators for their contribution to this research. Financial support from the National Natural Science Foundation of China (Grant Nos.: 20504002, 20628405, 20634010, 20874002, and 20874003) and the ‘‘973 Program’’ (2003CB615605) is gratefully acknowledged.References
- I. W. Hamley, Introduction to Soft Matter: Polymers, Colloids, Amphiphiles and Liquid Crystals, John Wiley & Sons Ltd, Chichester, 2000 Search PubMed.
- D. G. Dervichian, Mol. Cryst. Liq. Cryst., 1977, 40, 19–31 CrossRef CAS.
- D. Demus, J. W. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Physical Properties of Liquid Crystals, Wiley-VCH, Weinheim, 1999 Search PubMed.
- T. J. Sluckin, D. A. Dunmur and H. Stegemeyer, Crystals That Flow, Classic Papers From The History of Liquid Crystals, Taylor & Francis, London and New York, 2004 Search PubMed.
- D. Demus, J. W. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Handbook of Liquid Crystals, Wiley-VCH, Weinheim, 1998 Search PubMed.
- X.-J. Wang and Q.-F. Zhou, Liquid Crystalline Polymers, World Scientific Publishing Co. Pte. Ltd., Singapore, 2004 Search PubMed.
- C. S. Hsu, Prog. Polym. Sci., 1997, 22, 829–871 CrossRef CAS.
- C. B. McArdle, Side Chain Liquid Crystal Polymers, Blackie and Son Ltd, Glasgow, 1989 Search PubMed.
- T. Kato, in Handbook of Liquid Crystals, ed. D. Demus, J. W. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Wiley-VCH, Weinheim, 1998 Search PubMed.
- T. Kato, in Supramolecular Polymers, ed. A. Ciferri, Taylor & Francis, London, 2005, pp. 131–152 Search PubMed.
- S. Ujiie and K. Iimura, Macromolecules, 1992, 25, 3174–3178 CrossRef CAS.
- M. R. Hammond and R. Mezzenga, Soft Matter, 2008, 4, 952–961 RSC.
- H. Finkelmann, H. Ringsdorf and J. H. Wendorff, Makromol. Chem. Macromol. Chem. Phys., 1978, 179, 273–276 Search PubMed.
- H. Finkelmann and G. Rehage, Adv. Polym. Sci., 1984, 60, 97–172.
- F. Hessel and H. Finkelmann, Polym. Bull., 1985, 14, 375–378 CAS.
- F. Hessel and H. Finkelmann, Polym. Bull., 1986, 15 Search PubMed.
- C. S. Hsu and V. Percec, Polym. Bull., 1987, 17, 49–54 CrossRef CAS.
- V. Percec and B. Hahn, Macromolecules, 1989, 22, 1588–1599 CrossRef CAS.
- Q. F. Zhou, H. M. Li and X. D. Feng, Macromolecules, 1987, 20, 233–234 CrossRef CAS.
- Q. F. Zhou, H. M. Li and X. D. Feng, Mol. Cryst. Liq. Cryst., 1988, 155, 73–82 CAS.
- Q. F. Zhou, X. L. Zhu and Z. Q. Wen, Macromolecules, 1989, 22, 491–493 CrossRef CAS.
- F. Hessel, R. P. Herr and H. Finkelmann, Makromol. Chem. Macromol. Chem. Phys., 1987, 188, 1597–1611 Search PubMed.
- F. Hessel and H. Finkelmann, Makromol. Chem. Macromol. Chem. Phys., 1988, 189, 2275–2283 Search PubMed.
- F. J. Bormuth and W. Haase, Liq. Cryst., 1988, 3, 881–891 CAS.
- J. Berghausen, J. Fuchs and W. Richtering, Macromolecules, 1997, 30, 7574–7581 CrossRef CAS.
- P. Keller, F. Hardouin, M. Mauzac and M. F. Achard, Mol. Cryst. Liq. Cryst., 1988, 155, 171–178 CAS.
- F. Hardouin, S. Mery, M. F. Achard, M. Mauzac, P. Davidson and P. Keller, Liq. Cryst., 1990, 8, 565–575 CrossRef CAS.
- A. S. Cherodian, N. J. Hughes, R. M. Richardson, M. S. K. Lee and G. W. Gray, Liq. Cryst., 1993, 14, 1667–1682 CrossRef CAS.
- S. Lecommandoux, L. Noirez, H. Richard, M. F. Achard, C. Strazielle and F. Hardouin, J. Phys. II, 1996, 6, 225–234 CrossRef CAS.
- M. F. Achard, S. Lecommandoux and F. Hardouin, Liq. Cryst., 1995, 19, 581–587 CrossRef CAS.
- Y. Kawakami, K. Takahashi, S. Nishiguchi and K. Toida, Polym. Int., 1993, 31, 35–40 CAS.
- C. Pugh and R. R. Schrock, Macromolecules, 1992, 25, 6593–6604 CrossRef CAS.
- K. E. Schaefer, P. Keller and T. J. Deming, Macromolecules, 2006, 39, 19–22 CrossRef CAS.
- L. Chen, Y. W. Chen, D. J. Zha and Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2499–2509 CrossRef CAS.
- N. Leroux, M. F. Achard, P. Keller and F. Hardouin, Liq. Cryst., 1994, 16, 1073–1079 CrossRef CAS.
- F. Hardouin, N. Leroux, S. Mery and L. Noirez, J. Phys. II, 1992, 2, 271–278 CrossRef CAS.
- M. F. Achard, N. Leroux and F. Hardouin, Liq. Cryst., 1991, 10, 507–517 CrossRef CAS.
- G. W. Gray, J. S. Hill and D. Lacey, Angew. Chem., Int. Ed. Engl., 1989, 28, 1120–1122 CrossRef.
- A. Nazemi, E. J. C. Kellar, G. Williams, F. E. Karasz, J. S. Hill, D. Lacey and G. W. Gray, Liq. Cryst., 1991, 9, 307–320 CrossRef CAS.
- G. Williams, A. Nazemi, F. E. Karasz, J. S. Hill, D. Lacey and G. W. Gray, Macromolecules, 1991, 24, 5134–5140 CrossRef CAS.
- S. Lecommandoux, M. F. Achard and F. Hardouin, Liq. Cryst., 1998, 25, 85–94 CrossRef CAS.
- N. Leroux, P. Keller, M. F. Achard, L. Noirez and F. Hardouin, J. Phys. II, 1993, 3, 1289–1296 CrossRef CAS.
- V. Percec and D. Tomazos, J. Mater. Chem., 1993, 3, 643–650 RSC.
- Y. Nagase, Y. Takamura, H. Abe, K. Ono, T. Saito and E. Akiyama, Makromol. Chem. Macromol. Chem. Phys., 1993, 194, 2517–2532 Search PubMed.
- Y. Nagase, T. Saito, H. Abe and Y. Takamura, Macromol. Chem. Phys., 1994, 195, 263–272 CrossRef CAS.
- E. Akiyama, Y. Takamura and Y. Nagase, Polym. J., 1994, 26, 1277–1285 CrossRef CAS.
- H. F. Leube and H. Finkelmann, Makromol. Chem. Macromol. Chem. Phys., 1990, 191, 2707–2715 Search PubMed.
- H. F. Leube and H. Finkelmann, Makromol. Chem. Macromol. Chem. Phys., 1991, 192, 1317–1328 Search PubMed.
- K. Severing and K. Saalwachter, Phys. Rev. Lett., 2004, 92, 125501 CrossRef.
- H. Ogawa, E. Stibal-Fischer and H. Finkelmann, Macromol. Chem. Phys., 2004, 205, 593–599 CrossRef CAS.
- A. Dobarro, D. Velasco, V. VonArnim and H. Finkelmann, Macromol. Chem. Phys., 1997, 198, 2563–2581 CrossRef CAS.
- M. H. Li, P. Auroy and P. Keller, Liq. Cryst., 2000, 27, 1497–1502 CrossRef CAS.
- M. H. Li, P. Keller, B. Li, X. G. Wang and M. Brunet, Adv. Mater., 2003, 15, 569–572 CrossRef CAS.
- G. W. Gray, W. D. Hawthorne, J. S. Hill, D. Lacey, M. S. K. Lee, G. Nestor and M. S. White, Polymer, 1989, 30, 964–971 CrossRef CAS.
- R. A. Lewthwaite, J. W. Goodby and K. J. Toyne, J. Mater. Chem., 1993, 3, 241–245 RSC.
- M. S. K. Lee, G. W. Gray, D. Lacey and K. J. Toyne, Makromol. Chem. Rapid Commun., 1989, 10, 325–331 CrossRef CAS.
- M. S. K. Lee, G. W. Gray, D. Lacey and K. J. Toyne, Makromol. Chem. Rapid Commun., 1990, 11, 109–115 CrossRef CAS.
- G. W. Gray, J. S. Hill and D. Lacey, Mol. Cryst. Liq. Cryst., 1991, 197, 43–55 CAS.
- N. Leroux and L. C. Chien, Liq. Cryst., 1996, 21, 189–195 CrossRef CAS.
- F. Hardouin, S. Mery, M. F. Achard, L. Noirez and P. Keller, J. Phys. II, 1991, 1, 511–520 CrossRef CAS.
- P. Davidson, L. Noirez, J. P. Cotton and P. Keller, Liq. Cryst., 1991, 10, 111–118 CrossRef CAS.
- S. Lecommandoux, M. F. Achard, F. Hardouin, A. Brulet and J. P. Cotton, Liq. Cryst., 1997, 22, 549–555 CrossRef CAS.
- S. Lecommandoux, F. Hardouin and A. J. Dianoux, Eur. Phys. J. B, 1998, 5, 79–85 CrossRef CAS.
- N. Leroux, M. Mauzac, L. Noirez and F. Hardouin, Liq. Cryst., 1994, 16, 421–428 CrossRef CAS.
- F. Hardouin, N. Leroux, L. Noirez, P. Keller, M. Mauzac and M. F. Achard, Mol. Cryst. Liq. Cryst., 1994, 254, 267–282 CrossRef CAS.
- S. Lecommandoux, L. Noirez, M. F. Achard and F. Hardouin, J. Phys. II, 1997, 7, 1417–1424 CrossRef CAS.
- S. Lecommandoux, L. Noirez, M. F. Achard, A. Brulet, J. P. Cotton and F. Hardouin, Phys. B, 1997, 234–236, 250–251 CrossRef CAS.
- S. Lecommandoux, L. Noirez, M. F. Achard and F. Hardouin, Macromolecules, 2000, 33, 67–72 CrossRef CAS.
- S. Lecommandoux, L. Noirez, M. F. Achard and F. Hardouin, J. Phys. II, 1996, 6, 1231–1242 CrossRef CAS.
- Q. D. Mi, H. L. Zhang and Q. F. Zhou, Chem. J. Chin. Univ.-Chin., 1999, 20, 1817–1819 Search PubMed.
- Q. D. Mi and Q. F. Zhou, Chin. J. Polym. Sci., 1999, 17, 499–502 CAS.
- Q. D. Mi and Q. F. Zhou, Chin. J. Polym. Sci., 2000, 18, 139–148 CAS.
- P. l. Magagnini, A. Marchetti, F. Matera, G. Pizzirani and G. Turchi, Eur. Polym. J., 1974, 10, 585–591 CrossRef CAS.
- B. Bresci, V. Frosini, D. Lupinacci and P. L. Magagnini, Makromol. Chem. Rapid Commun., 1980, 1, 183–186 CrossRef CAS.
- D. Zhang, Q. F. Zhou, Y. G. Ma, X. H. Wan and X. D. Feng, Polym. Adv. Technol., 1997, 8, 227–233 CrossRef CAS.
- Y. X. Liu, D. Zhang, X. H. Wan and Q. F. Zhou, Chin. J. Polym. Sci., 1998, 16, 283–288 CAS.
- D. Zhang, Y. X. Liu, X. H. Wan and Q. F. Zhou, Macromolecules, 1999, 32, 5183–5185 CrossRef CAS.
- Z. N. Yu, H. L. Tu, X. H. Wan, X. F. Chen and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1454–1464 CrossRef CAS.
- Z. G. Zhu, J. G. Zhi, A. H. Liu, J. X. Cui, H. Tang, W. Q. Qiao, X. H. Wan and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 830–847 CrossRef CAS.
- X. H. Wan, Q. F. Zhou, D. Zhang, Y. Zhang and X. D. Feng, Chem. J. Chin. Univ.-Chin., 1998, 19, 1507–1512 Search PubMed.
- D. Zhang, Y. X. Liu, X. H. Wan and Q. F. Zhou, Macromolecules, 1999, 32, 4494–4496 CrossRef CAS.
- X. H. Wan, H. L. Tu, Y. X. Liu, D. Zhang, X. F. Chen, H. L. Zhang and Q. F. Zhou, Chin. J. Polym. Sci., 2003, 21, 21–27 CAS.
- H. L. Tu, D. Zhang, X. H. Wan, X. F. Chen, Y. X. Liu, H. L. Zhang and Q. F. Zhou, Macromol. Rapid Commun., 1999, 20, 549–551 CrossRef CAS.
- H. L. Tu, X. H. Wan, Y. X. Liu, X. F. Chen, D. Zhang, Q. F. Zhou, Z. H. Shen, J. J. Ge, S. Jin and S. Z. D. Cheng, Macromolecules, 2000, 33, 6315–6320 CrossRef CAS.
- Y. Guan, X. F. Chen, Z. H. Shen, X. H. Wan and Q. F. Zhou, Polymer, 2009, 50, 936–944 CrossRef CAS.
- X. Y. Yin, E. Q. Chen, X. H. Wan and Q. F. Zhou, Chin. J. Polym. Sci., 2003, 21, 9–14 CAS.
- X. Y. Yin, C. Ye, X. Ma, E. Q. Chen, X. Y. Qi, X. F. Duan, X. H. Wan, S. Z. D. Cheng and Q. F. Zhou, J. Am. Chem. Soc., 2003, 125, 6854–6855 CrossRef CAS.
- X. F. Chen, K. K. Tenneti, C. Y. Li, Y. W. Bai, R. Zhou, X. H. Wan, X. H. Fan and Q. F. Zhou, Macromolecules, 2006, 39, 517–527 CrossRef CAS.
- S. Chen, L. C. Gao, X. D. Zhao, X. F. Chen, X. H. Fan, P. Y. Xie and Q. F. Zhou, Macromolecules, 2007, 40, 5718–5725 CrossRef CAS.
- S. Chen, L.-Y. Zhang, L.-C. Gao, X.-F. Chen, X.-H. Fan, Z. Shen and Q.-F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 505–514 CrossRef CAS.
- C. P. Chai, J. Wang, X. H. Fan, X. F. Chen and Q. F. Zhou, Acta Polymerica Sinica, 2006, 6, 532–535 Search PubMed.
- C. P. Chai, X. Q. Zhu, P. Wang, M. Q. Ren, X. F. Chen, Y. D. Xu, X. H. Fan, C. Ye, E. Q. Chen and Q. F. Zhou, Macromolecules, 2007, 40, 9361–9370 CrossRef CAS.
- H. L. Xie, T. H. Hu, X. F. Zhang, H. L. Zhang, E. Q. Chen and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7310–7320 CrossRef CAS.
- C. Pugh and A. L. Kiste, Prog. Polym. Sci., 1997, 22, 601–691 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- M. H. Li, P. Keller, E. Grelet and P. Auroy, Macromol. Chem. Phys., 2002, 203, 619–626 CrossRef CAS.
- S. Pragliola, C. K. Ober, P. T. Mather and H. G. Jeon, Macromol. Chem. Phys., 1999, 200, 2338–2344 CrossRef CAS.
- P. Gopalan and C. K. Ober, Macromolecules, 2001, 34, 5120–5124 CrossRef CAS.
- X. H. Wan, H. L. Tu, Y. F. Tu, D. Zhang, L. Sun, Q. F. Zhou, Y. P. Dong and M. Tang, Chin. J. Polym. Sci., 1999, 17, 189–192 CAS.
- H. Zhang, Z. Yu, X. Wan, Q. F. Zhou and E. M. Woo, Polymer, 2002, 43, 2357–2361 CrossRef CAS.
- L.-C. Gao, X.-H. Fan, Z.-H. Shen, X. Chen and Q.-F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 319–330 CrossRef CAS.
- X. J. Wang and M. Warner, J. Phys. A: Math. Gen., 1987, 20, 713–731 CrossRef CAS.
- J. P. Cotton and F. Hardouin, Prog. Polym. Sci., 1997, 22, 795–828 CrossRef CAS.
- G. Z. Xu, W. Wu, M. Xu and Q. F. Zhou, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 229–230 CrossRef CAS.
- G. Z. Xu, W. Wu, D. Y. Shen, J. N. Hou, S. F. Zhang, M. Xu and Q. F. Zhou, Polymer, 1993, 34, 1818–1822 CrossRef CAS.
- X. H. Wan, F. Zhang, P. Q. Wu, D. Zhang, X. D. Feng and Q. F. Zhou, Macromol. Symp., 1995, 96, 207–218 CAS.
- J. G. Zhi, A. H. Liu, Z. G. Zhu, X. C. Lu, X. H. Fan, X. F. Chen, X. H. Wan and Q. F. Zhou, Acta Polym. Sin., 2005, 774–778 Search PubMed.
- Y. Shen, E. Q. Chen, C. Ye, H. L. Zhang, P. Y. Wu, I. Noda and Q. F. Zhou, J. Phys. Chem. B, 2005, 109, 6089–6095 CrossRef CAS.
- C. Ye, H. L. Zhang, Y. Huang, E. Q. Chen, Y. L. Lu, D. Y. Shen, X. H. Wan, Z. H. Shen, S. Z. D. Cheng and Q. F. Zhou, Macromolecules, 2004, 37, 7188–7196 CrossRef CAS.
- Y. F. Zhao, X. H. Fan, X. H. Wan, X. F. Chen, Y. Yi, L. S. Wang, X. Dong and Q. F. Zhou, Macromolecules, 2006, 39, 948–956 CrossRef CAS.
- H. Frauenrath, Prog. Polym. Sci., 2005, 30, 325–384 CrossRef CAS.
- V. Percec, C. H. Ahn, G. Ungar, D. J. P. Yeardley, M. Moller and S. S. Sheiko, Nature, 1998, 391, 161–164 CrossRef CAS.
- S. Forster, I. Neubert, A. D. Schluter and P. Lindner, Macromolecules, 1999, 32, 4043–4049 CrossRef.
- A. F. Zhang, L. J. Shu, Z. S. Bo and A. D. Schluter, Macromol. Chem. Phys., 2003, 204, 328–339 CrossRef CAS.
- A. Molenberg, M. Moller and E. Sautter, Prog. Polym. Sci., 1997, 22, 1133–1144 CrossRef CAS.
- T. Ganicz and W. A. Stanczyk, Prog. Polym. Sci., 2003, 28, 303–329 CrossRef CAS.
- G. Ungar, Polymer, 1993, 34, 2050–2059 CAS.
- G. Ungar, Mol. Cryst. Liq. Cryst., 2003, 396, 155–168 CrossRef CAS.
- G. Allegra and S. V. Meille, Macromolecules, 2004, 37, 3487–3496 CrossRef CAS.
- A. H. Liu, J. Zhi, J. X. Cui, X. H. Wan and Q. F. Zhou, Macromolecules, 2007, 40, 8233–8243 CrossRef CAS.
- P. J. Flory, Proc. R. Soc. London, Ser. A, 1956, 234, 73–89 CrossRef CAS.
- A. M. Kasko, A. M. Heintz and C. Pugh, Macromolecules, 1998, 31, 256–271 CrossRef CAS.
- X. Z. Wang, H. L. Zhang, D. C. Shi, J. F. Chen, X. Y. Wang and Q. F. Zhou, Eur. Polym. J., 2005, 41, 933–940 CrossRef CAS.
- X. Z. Wang, H. L. Zhang, G. Q. Zhong and X. Y. Wang, Polymer, 2004, 45, 3637–3642 CrossRef CAS.
- Q. W. Pan, L. C. Gao, X. F. Chen, X. H. Fan and Q. F. Zhou, Macromolecules, 2007, 40, 4887–4894 CrossRef CAS.
- X. Mei, A. H. Liu, J. X. Cui, X. H. Wan and Q. F. Zhou, Macromolecules, 2008, 41, 1264–1272 CrossRef CAS.
- E. A. Whale, F. J. Davis and G. R. Mitchell, J. Mater. Chem., 1996, 6, 1479–1485 RSC.
- Y. F. Zhao, Y. Yi, X. H. Fan, X. F. Chen, X. H. Wan and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2666–2674 CrossRef CAS.
- Z. F. Li, C. X. Zhang, S. Y. Zhang and Q. F. Zhou, Polym. Mater. Sci. Eng., 1995, 11, 36–40 CAS.
- Z. F. Li, C. X. Zhang, S. Y. Zhang, X. K. Li and Q. F. Zhou, Acta Polym. Sin., 1996, 350–356 Search PubMed.
- H. Tang, Z. G. Zhu, X. H. Wan, X. F. Chen and Q. F. Zhou, Macromolecules, 2006, 39, 6887–6897 CrossRef CAS.
- H. Tang, H. Q. Cao, Z. G. Zhu, X. H. Wan, X. F. Chen and Q. F. Zhou, Polymer, 2007, 48, 4252–4263 CrossRef CAS.
- G. J. J. Out, A. A. Turetskii, M. Moller and D. Oelfin, Macromolecules, 1995, 28, 596–603 CrossRef CAS.
- Z. N. Yu, X. H. Wan, H. L. Tu, X. F. Chen and Q. F. Zhou, Acta Polym. Sin., 2003, 430–433 Search PubMed.
- J. P. Lin, Polymer, 1997, 38, 4837–4841 CrossRef CAS.
- S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hagele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS.
- U. Pietrasik, J. Szydlowska and A. Krowczynski, Chem. Mater., 2004, 16, 1485–1492 CrossRef CAS.
- V. S. Papkov, M. N. Ilina, V. P. Zhukov, D. J. Tsvankin and D. R. Tur, Macromolecules, 1992, 25, 2033–2040 CrossRef CAS.
- C. Pugh, S. Arehart, H. Liu and R. Narayanan, J. Macromol. Sci. Pure Appl. Chem., 1994, A31, 1591–1607 CrossRef CAS.
- C. Pugh, J. Dharia and S. V. Arehart, Macromolecules, 1997, 30, 4520–4532 CrossRef CAS.
- C. Pugh, J. Shao, J. J. Ge and S. Z. D. Cheng, Macromolecules, 1998, 31, 1779–1790 CrossRef CAS.
- G. H. Kim, C. Pugh and S. Z. D. Cheng, Macromolecules, 2000, 33, 8983–8991 CrossRef CAS.
- G. H. Kim, S. Jin, C. Pugh and S. Z. D. Cheng, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 3029–3037 CrossRef CAS.
- S. V. Arehart and C. Pugh, J. Am. Chem. Soc., 1997, 119, 3027–3037 CrossRef CAS.
- A. C. Small and C. Pugh, Macromolecules, 2002, 35, 2105–2115 CrossRef CAS.
- C. Pugh, J. Y. Bae, J. Dharia, J. J. Ge and S. Z. D. Cheng, Macromolecules, 1998, 31, 5188–5200 CrossRef CAS.
- P. Gopalan, L. Andruzzi, X. F. Li and C. K. Ober, Macromol. Chem. Phys., 2002, 203, 1573–1583 CrossRef CAS.
- J. Mu, H. Okamoto, T. Yanai, S. Takenaka and X. S. Feng, Colloids Surf., A, 2001, 181, 303–313 CrossRef CAS.
- Y. D. Xu, Q. Yang, Z. H. Shen, X. F. Chen, X. H. Fan and Q. F. Zhou, Macromolecules, 2009, 42, 2542–2550 CrossRef CAS.
- H.-S. Kitzerow and C. Bahr, Chirality in Liquid Crystals, Spinger-Verlag New York, Inc., New York, 2001 Search PubMed.
- M. M. Green, R. J. M. Nolte and E. W. Meijer, Materials-Chirality, Wiley-Interscience, Hoboken, 2003 Search PubMed.
- Q. D. Mi, X. H. Wan and Q. F. Zhou, Acta Polym. Sin., 2000, 253–256 Search PubMed.
- R. A. Lewthwaite, G. W. Gray and K. J. Toyne, J. Mater. Chem., 1992, 2, 119–124 RSC.
- H. Leube and H. Finkelmann, Polym. Bull., 1988, 20 Search PubMed.
- Z. N. Yu, X. H. Wan, H. L. Zhang, X. F. Chen and Q. F. Zhou, Chem. Commun., 2003, 974–975 RSC.
- J. G. Zhi, Z. G. Zhu, A. H. Liu, J. X. Cui, X. H. Wan and Q. F. Zhou, Macromolecules, 2008, 41, 1594–1597 CrossRef CAS.
- J. X. Cui, A. H. Liu, J. Zhi, Z. G. Zhu, Y. Guan, X. H. Wan and Q. F. Zhou, Macromolecules, 2008, 41, 5245–5254 CrossRef CAS.
- P. G. de Gennes and J. Prost, The Physics of Liquid Crystals, Oxford University Press, New York, 1993 Search PubMed.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS.
- I. W. Hamley, The Physics of Block Copolymers, Oxford University Press, Oxford, 1998 Search PubMed.
- M. Walther and H. Finkelmann, Prog. Polym. Sci., 1996, 21, 951–979 CrossRef CAS.
- B. Gallot, Prog. Polym. Sci., 1996, 21, 1035–1088 CrossRef CAS.
- G. Mao and C. K. Ober, Acta Polym., 1997, 48, 405–422 CrossRef CAS.
- S. Poser, H. Fischer and M. Arnold, Prog. Polym. Sci., 1998, 23, 1337–1379 CrossRef CAS.
- B. D. Olsen and R. A. Segalman, Mater. Sci. Eng., R, 2008, 62, 37–66 CrossRef.
- M. Ballauff, Angew. Chem., Int. Ed. Engl., 1989, 28, 253–267 CrossRef.
- M. Lee, B. K. Cho and W. C. Zin, Chem. Rev., 2001, 101, 3869–3892 CrossRef CAS.
- H. A. Klok, J. F. Langenwalter and S. Lecommandoux, Macromolecules, 2000, 33, 7819–7826 CrossRef CAS.
- J. T. Chen, E. L. Thomas, C. K. Ober and S. S. Hwang, Macromolecules, 1995, 28, 1688–1697 CrossRef CAS.
- J. T. Chen, E. L. Thomas, C. K. Ober and G. P. Mao, Science, 1996, 273, 343–346 CrossRef CAS.
- B. D. Olsen and R. A. Segalman, Macromolecules, 2005, 38, 10127–10137 CrossRef CAS.
- B. D. Olsen, S. Y. Jang, J. M. Luning and R. A. Segalman, Macromolecules, 2006, 39, 4469–4479 CrossRef CAS.
- B. D. Olsen and R. A. Segalman, Macromolecules, 2006, 39, 7078–7083 CrossRef CAS.
- B. D. Olsen and R. A. Segalman, Macromolecules, 2007, 40, 6922–6929 CrossRef CAS.
- B. D. Olsen, X. F. Li, J. Wang and R. A. Segalman, Macromolecules, 2007, 40, 3287–3295 CrossRef CAS.
- Y. F. Tao, B. D. Olsen, V. Ganesan and R. A. Segalman, Macromolecules, 2007, 40, 3320–3327 CrossRef CAS.
- Y. F. Tao, H. Zohar, B. D. Olsen and R. A. Segalman, Nano Lett., 2007, 7, 2742–2746 CrossRef CAS.
- B. D. Olsen, M. F. Toney and R. A. Segalman, Langmuir, 2008, 24, 1604–1607 CrossRef CAS.
- B. D. Olsen, D. Alcazar, V. Krikorian, M. F. Toney, E. L. Thomas and R. A. Segalman, Macromolecules, 2008, 41, 58–66 CrossRef CAS.
- B. D. Olsen, M. Shah, V. Ganesan and R. A. Segalman, Macromolecules, 2008, 41, 6809–6817 CrossRef CAS.
- Y. F. Tao, B. W. Ma and R. A. Segalman, Macromolecules, 2008, 41, 7152–7159 CrossRef CAS.
- M. H. Li, P. Keller and P. A. Albouy, Macromolecules, 2003, 36, 2284–2292 CrossRef CAS.
- V. Castelletto, P. Parras, I. W. Hamley, P. Davidson, J. Yang, P. Keller and M. H. Li, Macromolecules, 2005, 38, 10736–10742 CrossRef CAS.
- W. Deng, P. A. Albouy, E. Lacaze, P. Keller, X. G. Wang and M. H. Li, Macromolecules, 2008, 41, 2459–2466 CrossRef CAS.
- X. H. Wan, Y. F. Tu, D. Zhang and Q. F. Zhou, Chin. J. Polym. Sci., 1998, 16, 377–380 CAS.
- X. H. Wan, Y. F. Tu, D. Zhang and Q. F. Zhou, Polym. Int., 2000, 49, 243–247 CrossRef CAS.
- H. L. Zhang, Y. F. Tu, X. H. Wan, Q. F. Zhou and E. M. Woo, J. Polym. Res., 2002, 9, 11–15 Search PubMed.
- Y. Yi, X. H. Wan, X. H. Fan, R. Dong and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1799–1806 CrossRef CAS.
- P. Gopalan, Y. M. Zhang, X. F. Li, U. Wiesner and C. K. Ober, Macromolecules, 2003, 36, 3357–3364 CrossRef CAS.
- Y. Yi, X. H. Fan, X. H. Wan, L. Li, N. Zhao, X. F. Chen, J. Xu and Q. F. Zhou, Macromolecules, 2004, 37, 7610–7618 CrossRef CAS.
- H. L. Zhang, X. Y. Sun, X. Y. Wang and Q. F. Zhou, Macromol. Rapid Commun., 2005, 26, 407–411 CrossRef.
- X. Z. Wang, H. L. Zhang, S. Mao, X. Y. Wang and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 733–741 CrossRef CAS.
- C. Y. Li, K. K. Tenneti, D. Zhang, H. L. Zhang, X. H. Wan, E. Q. Chen, Q. F. Zhou, A. O. Carlos, S. Igos and B. S. Hsiao, Macromolecules, 2004, 37, 2854–2860 CrossRef CAS.
- K. K. Tenneti, X. F. Chen, C. Y. Li, Y. F. Tu, X. H. Wan, Q. F. Zhou, I. Sics and B. S. Hsiao, J. Am. Chem. Soc., 2005, 127, 15481–15490 CrossRef CAS.
- L. C. Gao, C. L. Zhang, X. Liu, X. H. Fan, Y. X. Wu, X. F. Chen, Z. H. Shen and Q. F. Zhou, Soft Matter, 2008, 4, 1230–1236 RSC.
- K. K. Tenneti, X. F. Chen, C. Y. Li, X. H. Wan, X. H. Fan, Q. F. Zhou, L. X. Rong and B. S. Hsiao, Soft Matter, 2008, 4, 458–461 RSC.
- L. C. Gao, J. H. Yao, Z. Shen, Y. X. Wu, X. F. Chen, X. H. Fan and Q. F. Zhou, Macromolecules, 2009, 42, 1047–1050 CrossRef CAS.
- X. B. Liu, Y. F. Zhao, E. Q. Chen, C. Ye, Z. H. Shen, X. H. Fan, S. Z. D. Cheng and Q. F. Zhou, Macromolecules, 2008, 41, 5223–5229 CrossRef CAS.
- Y. Huang, J. Wang, X. B. Liu, H. L. Zhang, X. F. Chen, W. C. Zhuang, X. Chen, C. Ye, X. H. Wan, E. Q. Chen and Q. F. Zhou, Polymer, 2005, 46, 10148–10157 CrossRef CAS.
- Y. Huang, X. B. Liu, H. L. Zhang, D. S. Zhu, Y. J. Sun, S. K. Yan, J. Wang, X. F. Chen, X. H. Wan, E. Q. Chen and Q. F. Zhou, Polymer, 2006, 47, 1217–1225 CrossRef CAS.
- Y. F. Tu, X. H. Wan, D. Zhang, Q. F. Zhou and C. Wu, J. Am. Chem. Soc., 2000, 122, 10201–10205 CrossRef CAS.
- Y. F. Tu, X. H. Wan, H. L. Zhang, X. H. Fan, D. N. Lu, X. F. Chen and Q. F. Zhou, Chin. J. Polym. Sci., 2003, 21, 569–573 CAS.
- Y. F. Tu, X. H. Wan, H. L. Zhang, X. H. Fan, X. F. Chen, Q. F. Zhou and K. C. Chau, Macromolecules, 2003, 36, 6565–6569 CrossRef CAS.
- J. Zhang, Y. Yu, X. H. Wan, X. F. Chen and Q. F. Zhou, Acta Polym. Sin., 2005, 305–308 Search PubMed.
- J. Zhang, Z. N. Yu, X. H. Wan, X. F. Chen and Q. F. Zhou, Macromol. Rapid Commun., 2005, 26, 1241–1245 CrossRef CAS.
- J. Zhang, W. R. Lin, A. H. Liu, Z. N. Yu, X. H. Wan, D. H. Liang and Q. F. Zhou, Langmuir, 2008, 24, 3780–3786 CrossRef CAS.
- H. Cao, W. Lin, A. Liu, J. Zhang, X. Wan and Q. Zhou, Macromol. Rapid Commun., 2007, 28, 1883–1888 CrossRef CAS.
- J. Zhang, H. Q. Cao, X. H. Wan and Q. F. Zhou, Langmuir, 2006, 22, 6587–6592 CrossRef CAS.
- H. Kawakami, Y. Mori, H. Abe and S. Nagaoka, J. Membr. Sci., 1997, 133, 245–253 CrossRef CAS.
- I. Terrien, M. F. Achard, G. Felix and F. Hardouin, J. Chromatogr., A, 1998, 810, 19–31 CrossRef CAS.
- I. Terrien, G. Felix, M. Laguerre, M. F. Achard and F. Hardouin, Mol. Cryst. Liq. Cryst., 1999, 331, 431–2298 CrossRef.
- F. Gritti, G. Felix, M. F. Achard and F. Hardouin, J. Chromatogr., A, 2000, 897, 131–143 CrossRef CAS.
- F. Gritti, G. Felix, M. F. Achard and F. Hardouin, J. Chromatogr., A, 2001, 913, 147–157 CrossRef CAS.
- F. Gritti, I. Terrien, S. Menu, E. J. Dufourc, G. Felix, M. F. Achard and F. Hardouin, J. Chromatogr., A, 2001, 922, 37–50 CrossRef CAS.
- F. Gritti, G. Felix, M. F. Achard and F. Hardouin, J. Chromatogr., A, 2001, 922, 51–61 CrossRef CAS.
- F. Gritti, G. Felix, M. F. Achard and F. Hardouin, Chromatographia, 2002, 56, 9–17 CAS.
- M. H. Li and P. Keller, Philos. Trans. R. Soc. London, Ser. A, 2006, 364, 2763–2777 CrossRef CAS.
- P. G. de Gennes, C. R. Acad. Sci. Ser. IIb: Mec. Phys. Chim. Astron., 1997, 324, 343–348 Search PubMed.
- D. L. Thomsen, P. Keller, J. Naciri, R. Pink, H. Jeon, D. Shenoy and B. R. Ratna, Macromolecules, 2001, 34, 5868–5875 CrossRef.
- H. Finkelmann, E. Nishikawa, G. G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 87, 015501 CrossRef CAS.
- A. Buguin, M. H. Li, P. Silberzan, B. Ladoux and P. Keller, J. Am. Chem. Soc., 2006, 128, 1088–1089 CrossRef CAS.
- M. H. Li, P. Keller, J. Y. Yang and P. A. Albouy, Adv. Mater., 2004, 16, 1922–1925 CrossRef CAS.
- P. Wang, C. P. Chai, Y. T. Chuai, F. Z. Wang, X. F. Chen, X. G. Fan, Y. D. Xu, D. C. Zou and Q. F. Zhou, Polymer, 2007, 48, 5889–5895 CrossRef CAS.
- P. Wang, C. P. Chai, F. Z. Wang, Y. T. Chuai, X. F. Chen, X. H. Fan, D. C. Zou and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1843–1851 CrossRef CAS.
- P. Wang, C. P. Chai, Q. Yang, F. Z. Wang, Z. H. Shen, H. Q. Guo, X. F. Chen, X. H. Fan, D. C. Zou and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5452–5460 CrossRef CAS.
- P. Wang, Y. T. Chuai, C. P. Chai, F. Z. Wang, G. L. Zhang, G. P. Ge, X. H. Fan, H. Q. Guo, D. C. Zou and Q. F. Zhou, Polymer, 2008, 49, 455–460 CrossRef CAS.
- Q. Yang, H. Jin, Y. D. Xu, P. Wang, X. C. Liang, Z. H. Shen, X. F. Chen, D. C. Zou, X. H. Fan and Q. F. Zhou, Macromolecules, 2009, 42, 1037–1046 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |