DOI:
10.1039/B809912J
(Critical Review)
Chem. Soc. Rev., 2010,
39, 712-733
Organopalladium(IV) chemistry
Received 3rd December 2008
First published on 2nd September 2009
Abstract
Although the chemistry of Pd(0), Pd(I) and Pd(II) is well established, high oxidation state Pd(IV) complexes are less well-known. This situation has highly changed in recent years. Many well-defined Pd(IV) complexes has been isolated and characterized, providing evidence for a series of proposed Pd(II)/Pd(IV) catalytic reactions. A deep understanding of the behavior of Pd(IV) complexes could lead to the design and development of novel reactions that could not be accessed by traditional Pd(0)/Pd(II) chemistry. This critical review describes the stoichiometric reactions of Pd(IV) complexes and discusses their potential mechanism in catalytic reactions (137 references).
 Ling-Min XuThese authors equally contributed to this work. Ling-Min XuThese authors equally contributed to this work. | Ling-Min Xu was born in Guangdong, China, in 1983. She received her BS degree from Peking University in 2007. She is currently a second-year graduate student in the research group of Professor Yang at Peking University. |
 Bi-Jie Li Bi-Jie Li | Bi-Jie Li was born in Hubei, China, in 1985. He received his BS degree from Peking University in 2007. He is currently a second-year graduate student in the research group of Professor Shi at Peking University. |
 Zhen Yang Zhen Yang | Zhen Yang was born in China in 1959. He received his BS degree and MS degree in medicinal chemistry from Shenyang College of Pharmacy, P. R. China in 1982 and in 1986, respectively. He went on to pursue his PhD in chemistry in The Chinese University of Hong Kong in 1989. He then joined K. C. Nicolaou’s group in The Scripps Research Institute as a postdoctoral fellow. He continued his research career as an assistant professor in The Scripps Research Institute and then he moved to Harvard Institute of Chemistry and Cell Biology in 1998. He has become a Changjiang Professor in College of Chemistry, Peking University since 2001. |
 Zhang-Jie Shi Zhang-Jie Shi | Zhang-Jie Shi was born in Anhui, China, in 1974. He obtained his BS degree at East China Normal University in 1996, an PhD degree at Shanghai Institute of Organic Chemistry (SIOC), CAS in 2001. After serving as Postdoctoral studies in Department of Chemistry and Chemical Biology, Harvard University and at the Department of Chemistry, The University of Chicago, he joined the chemistry faculty of Peking University in 2004, where he was promoted to a full Professor in 2008. |
1. Introduction
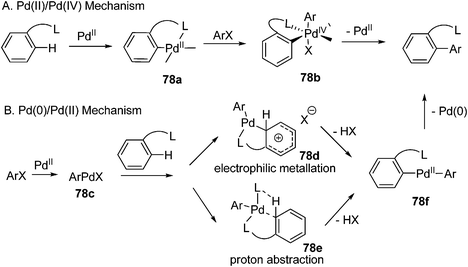
In the past several decades, transition-metal catalyzed cross-coupling reactions have revolutionalized our ability to construct C–C bonds as well as C–heteroatom bonds.1 In particular, palladium-catalyzed cross-coupling reactions are the most intensively investigated, providing numerous methods that greatly enriched the toolbox for synthetic chemists.2 As a result, a great number of natural products and synthetically useful molecules have been efficiently synthesized through judicious choice of these methodologies.3 Generally, the intrinsic feature of the palladium catalyzed cross-coupling reactions is the Pd(0)/Pd(II) catalytic cycle, which relies on the facile interconversion between Pd(0) and Pd(II) of palladium complexes.In contrast to the high oxidation state platinum (Pt(IV)) complexes known as early as 1907,4 the corresponding Pd(IV) complexes remained scarce. It was in the 1970s that several isolatable Pd(IV) complexes were isolated and characterized, providing stoichiometric reactivity patterns of these novel complexes. However, these early investigations were mainly limited to stoichiometric transformations. The lack of catalytic versions was largely attributed to the poor stability of Pd(IV) complexes and the difficulty to engage in catalytic turnover. These challenges notwithstanding, recent years have witnessed dramatic changes in this situation, largely driven by the use of various novel oxidants.5 Furthermore, several well-defined Pd(IV) complexes have been isolated, allowing detailed studies of the Pd(IV) structures as well as the insight into their catalytic reactivity. These accumulated knowledge, in its turn, can help us to better understand and design new Pd(II)/Pd(IV) catalyzed reactions.
Exploiting the novel Pd(II)/Pd(IV) catalysis presents us new sceneries in terms of both mechanistic information and synthetic applications. By overviewing recent advances of Pd(IV) chemistry, significant advantages have emerged over those of Pd(0)/Pd(II) catalysis: (1) Pd(IV) species are often resistant to β-H elimination process, allowing diverse transformations of Pd(IV) intermediates (β-H elimination is often a competing process with reductive elimination or multiple bond insertion). (2) Usually, Pd(IV) species undergo facile reductive elimination. Various bond forming reductive eliminations have been observed and most of them occur under mild conditions. Thus, bond constructions through Pd(IV) chemistry exhibit a multitude of diversity compared to the typical Pd(0)/Pd(II) catalyzed processes. (3) Pd(II)/Pd(IV) catalyzed reactions are operationally simple and do not require the careful exclusion of air (especially O2) and moisture; (4) Pd(II)/Pd(IV) catalyzed reactions exhibit complementary functional group tolerance to Pd(0)/Pd(II) catalyzed transformations. Functional groups, such as C–Br, assumed to be highly reactive in Pd(0)/Pd(II) catalysis might be stable under Pd(II)/Pd(IV) catalytic conditions. (5) In some cases, Pd(II)/Pd(IV) catalysis can realize transformations that are hardly accessible by Pd(0)/Pd(II) catalyzed reactions.
This review is not aimed to be exhaustive, but mainly deals with recent advances. First, we will introduce the stoichiometric preparation of Pd(IV) complexes as well as their reactivities. Then we will discuss catalytic reactions involving Pd(IV) intermediates, with a special emphasis on direct C–H functionalizations.
2. Organopalladium(IV) species—general mechanistic aspects
Unlike platinum whose oxidation state of +4 was known since the report of [PtIMe3]4 in 1907,4 palladium has been generally confined to the formal oxidation states of 0, +1 and +2. Although several examples of palladium(IV) complexes such as PdCl3(C6F5)(bpy)6 existed for quite a long time, little attention has been paid to them. Organopalladium(IV) complexes had hardly been studied until 1986 that the first unequivocal trialkylpalladium(IV) complex, [PdIMe3(bpy)], was isolated and characterized by X-ray analysis.7 Since then, efforts were made to speed up the studies on preparation and characterization of Pd(IV) complexes.8 By employing suitable oxidants, Pd(IV) complexes, traditionally thought to be transient intermediates, were isolated and characterized by single-crystal X-ray crystallography. Since reductive elimination readily occurs at or below room temperature, the presence of the strong donor ligands is essential to stabilize Pd(IV) complexes.2.1 Preparation of Pd(IV) complexes
Various oxidants have been applied to prepare Pd(IV) complexes from Pd(II) compounds, including inorganic oxidants such as dihalogens, oxone and organic oxidants, such as alkyl halides, PhI(OAc)2, peroxide. Below, various Pd(IV) complexes were summarized according to the use of different oxidants.2.1.1 Oxidation of Pd(II) complexes by dihalogens, peroxides, (PhE)2 (E = S, Se). The first known organopalladium(IV) complexes 2 which were stable at room temperature were prepared by oxidation of (C6F5)2Pd(II) by chlorine in 1975.6 Chelating dinitrogen ligands were crucial for the stabilization of the high valent intermediates and thereby facilitated the isolation. However, the configuration of the Pd(IV) complexes were not well determined (eqn (1)).| |  | (1) |
Pd(II) complexes supported by other chelating ligands, such as 3, also underwent the oxidative addition by dihalogens to form Pd(IV) complexes, which were only characterized by 1H NMR spectroscopy due to their relatively low stability (eqn (2)).9
| |  | (2) |
Pd(IV) complexes were also obtained by Elsevier and co-workers. In their studies, palladacyclopentadienes 7 containing bidentate nitrogen ligands could be oxidized by dihalogens at low temperature to give Pd(IV) species 8, which were also detected by 1H NMR spectroscopy (eqn (3)).10,11
| |  | (3) |
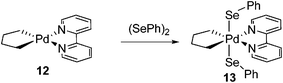
Peroxides, for example (PhCOO)2, were also applied as efficient oxidants to produce Pd(IV) complexes at very low temperature (eqn (4)). Other than peroxides, PhSSPh and PhSeSePh could play similar roles to form Pd(IV) complexes, however, at higher temperature due to their relatively lower redox potential compared to peroxides (eqn (5) and (6)). In this system, strong σ-donor alkyl ligands together with chelating nitrogen ligands were used to stabilize the Pd(IV) complexes. Notably, the incorporation of heteroatoms in the Pd(IV) complexes is important since it provides an opportunity for the study of C–heteroatom formation by reductive elimination.12
| |  | (4) |
| |  | (5) |
| |  | (6) |
Carbopallada(II)cycle 14 can be oxidized by H2O2 to form dihydrocarbylhydroxylpalladium(IV) complexes 15 with support of hydrotris(pyrazolyl)borate (TP).13 Surprisingly, even with the use of water instead of H2O2 as oxidant, the same adduct 15 was obtained upon release of hydrogen. This can be attributed to the low reductive potential of Pd(II) complexes 14, which were supported by the high electron donating alkyl and TP group (eqn (7)). Similar oxidation of 14 by dihalogens such as Cl2, Br2, I2 was also studied, providing various dihydrocarbylpalladium(IV) complexes 16 which were stable at room temperature (eqn (8)).
| |  | (7) |
| |  | (8) |
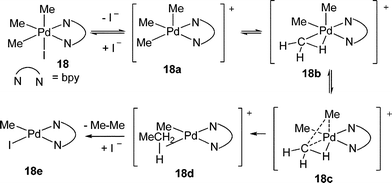
2.1.2 Oxidative addition of alkyl halides to Pd(II) complexes. The pioneering studies in this area have been done by Canty and co-workers.8 During the study of the chemistry of mono- and di-methylpalladium(II) complexes with nitrogen donor ligands, they found that PdMe2(bpy) reacted rapidly with MeI to give fac-PdIMe3(bpy) 18, which was sufficiently stable at room temperature (eqn (9)).7In situ1H NMR studies of this oxidative addition of PdMe2(bpy) by iodomethane in (CD3)2CO indicated that a cationic species was formed prior to the formation of neutral PdIMe3(bpy), suggesting that the oxidative addition went through a general SN2 pathway.14 Most importantly, the first single-crystal X-ray structure of a palladium(IV) complex was obtained, which provided the most straightforward evidence to support the previously suggested transient alkylpalladium(IV) intermediate.15| |  | (9) |
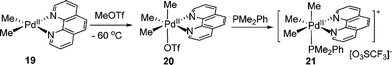
With the use of methyl triflate instead of MeI, the oxidative addition could also processed at low temperature. Since the triflate ligand in 20 was readily displaced, the first monodentate phosphine supported cationic organopalladium(IV) complex 21 was isolated and structurally determined (eqn (10)).16,17 By this method, various analogous Pd(IV) phosphine complexes were prepared.
| |  | (10) |
The method for the preparation of fac-PdIMe3(bpy) was extended to the preparation of allyl-, benzyl- and ethyl Pd(IV) complexes 23 by using allyl, benzyl bromide and ethyl iodide as oxidant, respectively.18 However, Pd(IV) complexes containing bidentate ligands (such as tmeda) less rigid than bipy and phenanthroline, are typically unstable. Considering that cationic Pd(IV) complexes are more stable towards reductive elimination, Canty and co-workers designed new tripod nitrogen ligands. With the support of this ligand, both PdIVEt and PdIVPr complexes were isolated, which were even stable in (CD3)2CO at ∼60 °C (eqn (11)).
| |  | (11) |
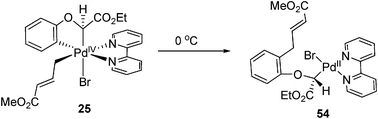
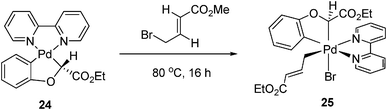
Similarly, with bipyridine and a C-based σ-donor ligand, pallada(II)cycle 24 reacted with allyl bromide to form allyl-Pd(IV) complexes 25, whose structure was characterized by X-ray crystallography (eqn (12)).19 A rigid bipyridine ligand was crucial for the reaction. By contrast, a palladacycle with diimine as ligands did not react at all. This Pd(IV) complex was proved as to adopt cis-coordination by X-ray crystallography.
| |  | (12) |
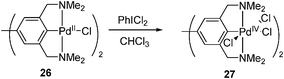
2.1.3 Oxidation of the Pd(II) complexes by hypervalent iodine(III) reagents. Hypervalent iodine reagents were proved efficient to oxidize suitable Pd(II) complexes to Pd(IV) complexes.20 These reactions are classified according to different types of hypervalent iodine reagents.2.1.3.1 Oxidation with PhIX2. Van Koten and co-workers first reported that pincer complex 26 bearing bridging ligands could be oxidized by PhICl2 to afford the dipalladium(IV) complex 27 (eqn (13)).21 However, the resulting complex 27 was relatively unstable and decomposed in a few minutes after its formation.
| |  | (13) |
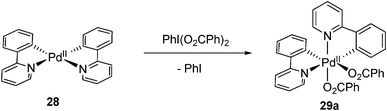
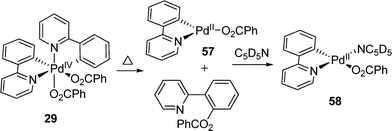
Very recently, a significant contribution in this field was make by Sanford and co-workers. A series of Pd(IV) complexes 29 containing two rigid cyclometalated bidentate pyridine ligands were synthesized by oxidation of the corresponding Pd(II) complex by a hypervalent iodine(III) reagent PhI(O2CPh)2 (eqn (14)).22 The electronic donating ability of this carbon–nitrogen ligand stabilized the high oxidation state of Pd(IV) species 29. Meanwhile, the ligand exchange could be prevented by the chelating aryl group, thus resulting in the stable Pd(IV) complexes at ambient temperature. The 1H NMR spectrum showed that two benzoate ligands were in cis-geometry, which was further confirmed by X-ray crystallography.
| |  | (14) |
Similarly, PhICl2 can also oxidize Pd(II) to form Pd(IV) complex 30, which was stable at room temperature (eqn (15)).23 NMR spectra showed that the formed octahedral Pd(IV) complex 30 had a cis geometry with two inequivalent phenylpyridinyl ligands.
| | 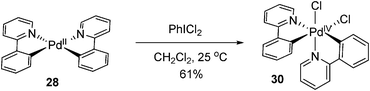 | (15) |
2.1.3.2 Oxidation with [ArI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) X]n. The reactions of Pd(II) complexes with another type of hypervalent iodine reagent (ArI
X]n. The reactions of Pd(II) complexes with another type of hypervalent iodine reagent (ArI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X)n (X = O, NR) resulted in the insertion of X group into the Pd–C bond with concomitant release of ArI. Although no high oxidation state Pd intermediates were observed in any of these cases, a mechanism involving oxidation to generate the Pd(IV)-oxo intermediate followed by intramolecular insertion of the heteroatom into the Pd(IV)-aryl bond was highly preferred (eqn (16)).24,25 Similarly, amination of C–H bonds was also reported by Sanford using PhINTs as the oxidant (eqn (17)).26
X)n (X = O, NR) resulted in the insertion of X group into the Pd–C bond with concomitant release of ArI. Although no high oxidation state Pd intermediates were observed in any of these cases, a mechanism involving oxidation to generate the Pd(IV)-oxo intermediate followed by intramolecular insertion of the heteroatom into the Pd(IV)-aryl bond was highly preferred (eqn (16)).24,25 Similarly, amination of C–H bonds was also reported by Sanford using PhINTs as the oxidant (eqn (17)).26| |  | (16) |
| | 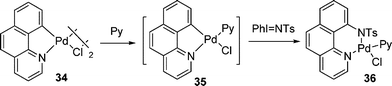 | (17) |
2.1.3.3 Oxidation with [ArIAr]X. Canty and co-workers reported the first example to oxidize Pd(II) complex 12 to Pd(IV) species by diphenyliodine(III) triflate.27 The resulting oxidation product 37 was obtained as a 1 : 1 ratio of trans (37a) : cis (37b) isomers (eqn (18)). A putative mechanism of this Ph+ transfer involved a nucleophilic attack of Pd(II) to the iodine(III) center. The absence of reactivity of phenyl triflate toward Pd(C4H8)(bpy) highlighted the unique Ar+ group transfer ability of the electrophilic iodine(III) reagent.| |  | (18) |
2.1.3.4 Oxidation with [PhI(alkenyl)]X and [PhI(alkynyl)]X. [PhI(alkenyl)]X and [PhI(alkynyl)]X reacted in a similar fashion with selective transfer of the alkenyl/alkynyl group. Starting from phosphine-coordinated complex PdMe2(dmpe) 38, PdMe2(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CSiMe3)(dmpe)(OTf) 39 was cleanly obtained in good yield through the alkynyl group transfer from PhI(CCSiMe3)OTf at −50 °C, whose configuration was interpreted by NMR spectroscopy (eqn (19)). Pincer palladium complex reacted slightly slower but gave similar results.28
CSiMe3)(dmpe)(OTf) 39 was cleanly obtained in good yield through the alkynyl group transfer from PhI(CCSiMe3)OTf at −50 °C, whose configuration was interpreted by NMR spectroscopy (eqn (19)). Pincer palladium complex reacted slightly slower but gave similar results.28| |  | (19) |
Pallada(II)cycle 24 reacted with vinyl- and alkynyl-(phenyl)iodinium reagents to give benzofurans. During this transformation, a similar Pd(IV) intermediate 40, detected by 1H NMR, was hypothesized as a key intermediate (eqn (20)).29 These results further supported that the alkenyl/alkynyl group were preferentially transferred to the Pd center over the phenyl group during the oxidation process.
| | 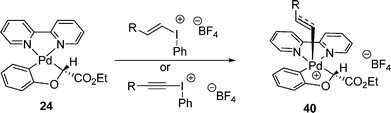 | (20) |
2.1.4 Oxidation of Pd(II) complexes with other oxidants. Interestingly, by heating Pd(dppe)(Me)238 in the presence of 1,2-disilylbenzene, a new silylpalladium(IV) complex 42 was formed. This is the first isolation of silyl group supported Pd(IV) species. Presumably, the silylpalladium(IV) hydride intermediate 41 was an intermediate (eqn (21)).30 The configuration of the C2 symmetry was clearly established by X-ray structure. These results also provided proof for the involvement of a tetrakis(silyl)palladium(IV) intermediate in Pd-catalyzed metathesis reactions of disilanes.31,32| |  | (21) |
With a strong electron donating Cp or Cp* as a ligand, Cp(η3-allyl)palladium(II) and Cp*(η3-allyl)palladium(II) were oxidized to afford tris(organosilyl)Pd(IV) complexes 44 by spirotrisilane at room temperature in benzene. These Pd(IV) complexes were air stable and could be isolated (eqn (22)). The configuration of Pd bonding to three silyl and one Cp groups was tetrahedral with only slight deviation.33
| |  | (22) |
Yamamoto and co-workers reported that the reaction of Pd2(dba)3 (dba = trans,trans-dibenzylideneacetone) with tetrachloro-1,2-benzoquinone (o-chloranil) and norbornene directly produced a novel spirocyclic palladium(IV) dialkyl complex 45 (eqn (23)).34 This is the first example to produce a Pd(IV) complex directly from a Pd(0) species. More importantly, the stability of the trigonal bipyramidal complexes in the solid state was remarkable since there were no bidentate or tripodal donor ligands present. Both o-chloranil and norbornene were essential for this transformation, suggesting that the strong oxidizing ability of o-chloranil and the ring strain of norbornene played critical roles in the oxidative cyclization.
| |  | (23) |
Oxidation of Pd(II) by diazald (N-methyl-N-nitroso-p-toluensulfonamide) or NO2 was investigated in detail by Cámpora and co-workers for the preparation of Pd(IV)-(NO) complex 47 from the same anionic pallada(II)cycle. The strong electron donating Tp ligand (hydrotris(pyrazolyl)borate) is important for this process (eqn (24)–(26)).35 The octahedral geometry of the resulting complex 47 was demonstrated by the X-ray structure. Moreover, treatment of this complex with dioxygen generated the Pd(IV)-nitrate complex 48. Due to the weak coordinating ability of the nitrate ligand, the displacement of the nitrate by PMe3 easily took place to afford the cationic Pd(IV) complex 49.
| |  | (24) |
| | 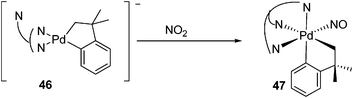 | (25) |
| |  | (26) |
Anionic Pd(II) complex 46 could also be oxidized by [FeCp2][PF6]. The resulting product was in situ trapped by DMAP to afford a cationic Pd(IV) complex 50 which showed characteristic spectroscopic features of Pd(IV) complexes (eqn (27)).36
| |  | (27) |
Interestingly, the oxidizing capability of N-chlorosuccinimide (NCS) was also demonstrated in the formation of Pd(IV) complexes. Sanford and co-workers reported that chloropalladium(IV) complex 51 was easily obtained through the oxidation of Pd(II) complex 28 by NCS. The structure of 51 was confirmed by X-ray crystallography, which showed that the chloride is trans to a pyridine nitrogen (eqn (28)). This was the first observation of direct oxidative insertion of a Pd(II) complex into the N–X bond of an N-halosuccinimide.23
| |  | (28) |
In their effort to clarify the possibility of carbon–fluorine bond formation from high-valent palladium complexes, Ritter and co-workers successfully prepared two Pd(IV) fluoride complexes 53a and 53b (eqn (29) and (30)).37,38 Oxidation of the Pd(II) complex 52 containing rigid, chelating benzoquinolinyl and phenylamido ligands by Selectfluor™ and XeF239 gave Pd(IV) mono- and di-fluoride complexes, respectively.40 The configuration of the latter complex was established by X-ray crystallography, which indicated that the two fluoride substituents were located in mutually cis position.
| |  | (29) |
| |  | (30) |
Based on the above review of the preparation of Pd(IV) complexes, it is obvious that the ligands play very important roles. Generally, bidentate or tripodal ligands with C-σ-donor and/or N-σ-donor atoms are required to support the high valent Pd(IV) structure. The existence of negative charge on these ligands strongly increases the electron density of the Pd center, which facilitates the oxidation process. The chelating effects of multiple coordination sites were beneficial for the stabilization of five-coordinated trigonal bipyramidal or six-coordinated octahedral Pd(IV) complexes. Undoubtedly, the high redox potential of the employed oxidants is also important for the oxidation of Pd(II) to Pd(IV) species.
2.2 Stoichiometric reactivities at the Pd(IV) center
Generally, Pd(IV) complexes exhibit high reactivity. In most cases, reductive elimination at the Pd(IV) center occurs predominantly, allowing various C–C and C–heteroatom bond formation, which is similar to Pd(II) chemistry. After the reductive elimination, highly reactive Pd(IV) species transformed to the thermodynamically more stable Pd(II) complexes.2.2.2 C–O Bond forming reductive elimination. In contrast to the formation of various C–C bonds, studies on C–heteroatom bond formation from Pd(IV) complexes were hampered by the relatively lower stability of the heteroatom supported Pd(IV) complexes as well as the competing C–C bond forming reductive elimination and other side reactions.Dimethylpalladium(IV) complex 10 containing oxygen coordinated benzoate ligands readily underwent reductive elimination soon after its formation. The C(sp3)–O coupling product was obtained with nearly equal amount as the C–C coupling product (eqn (34)). Similarly, the decomposition of dimethyl Pd(IV) selenolate complexes 56 gave C(sp3)–Se product by reductive elimination (eqn (35)).12
| | 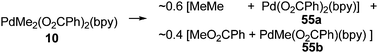 | (34) |
| |  | (35) |
The detailed mechanistic investigation of sp2 C–O reductive elimination at the Pd(IV) center was carried out with bicyclometallic Pd(IV) complex 29, a compound that has sufficient stability at room temperature.22 Upon heating at 60 °C for 1 h, 29 readily underwent C–O bond-forming reductive elimination without observation of C–C bond-forming reductive elimination product (eqn (36)). This study gave a direct support to the C–O reductive elimination of Pd(IV) complexes.
| |  | (36) |
Three distinct mechanistic pathways for this reductive elimination process were envisaged: mechanism A involves the dissociation of a benzoate ligand prior to the reductive elimination to form five-coordinated Pd(IV) complexes; mechanism B is the direct reductive elimination from the six-coordinate Pd(IV) complex; the final pathway (C) requires the dissociation of a pyridyl arm to form five-coordinated Pd(IV) species before C–O formation (Scheme 2).
 |
| | Scheme 2 Possible mechanisms of C–O reductive elimination. | |
Pathway A was ruled out by the solvent-independence of the rate of reductive elimination and a negative Hammett ρ value. This exclusion was further verified by cross-over experiments, in which no cross-over products were obtained. On the other hand, the comparison of the rates of reductive elimination of complex 29b-1 and 29b-2 indicated that the nitrogen dissociation was hampered by the more rigid benzoquinoline ligand, thus leading to an overall rate decrease (Scheme 3). These results seemed to support mechanism C. However, the high calculated free energy barriers for this process (+44.3 kcal mol−1) seemed to disfavor this conclusion. Pathways A and B had much lower energy barriers (+31.4 and +26.4 kcal mol−1 for pathways A and B, respectively). Therefore, mechanism B was preferred.41
 |
| | Scheme 3 Rate differentiation of C–O bond-forming reductive eliminations. | |
2.2.3 C–X (X = halogen) Bond forming reductive elimination. Elsevier and co-workers observed the (dihalo)(diorgano)Pd(IV) complexes 8 containing bidentate nitrogen ligands by NMR when the corresponding Pd(II) complexes were oxidized by molecular halogens, such as Cl2, Br2 and even I2. Reductive elimination of these Pd(IV) complexes formed the different C–X bonds (eqn (37)). With an excess amount of dihalogen, (E,E)-1,4-dihalobutadienes were readily afforded.10,11| |  | (37) |
As discussed above, heating complexes 30 and 51 induced the reductive elimination to produce C–C bonded homocoupling product. However, the products distribution via the reductive elimination of those complexes strongly depended on the solvents.23 The major product in pyridine was the C–C coupled product, while C–Cl bond-forming product became predominant in AcOH (Scheme 4). This result represented the first example of carbon–halogen coupling occurring in preference to C–C coupling at the Pd(IV) center. Notably, a small amount of C–N forming product by reductive elimination was also observed, suggesting the potential feasibility of such a process to approach C–N construction.
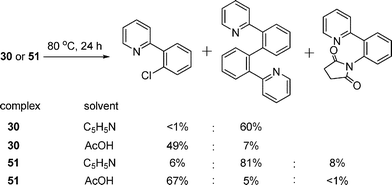 |
| | Scheme 4 Products distribution in different solvents. | |
The behavior of the novel Pd(IV) complexes with fluoride as a ligand was investigated by Ritter and co-workers. Interestingly, reductive elimination of 53a with a labile ligand occurred at 50 °C while 53b with difluoride at 150 °C (Scheme 5). The increased thermal stability of 53b compared to 53a is consistent with the formation of a pentacoordinated palladium(IV) fluoride prior to the reductive elimination. These studies provided valuable mechanistic insight into C–F reductive elimination reactions and the possibility to produce the monofluorinated products.38,40
 |
| | Scheme 5 Reductive elimination of PdIV-fluoride complexes. | |
2.2.4 Alkyl/aryl transfer to platinum(II) and palladium(II). Pt(IV) complexes are relatively stable compared to Pd(IV) complexes. Thus, Canty and co-workers studied the reaction between the labile Pd(IV) with Pt(II) complex.15 Interestingly, redox reactions involving alkyl and halide groups transfer from Pd(IV) to Pt(II) were observed (eqn (38)). When Pd(IV) complex contained methyl, benzyl, and aryl groups, selective transfer of the benzyl group was observed. Further studies indicated that selective group transfer was also feasible between Pd(II) and Pd(IV) complexes (eqn (39)).| |  | (38) |
| |  | (39) |
3. Catalytic reactions involving Pd(IV) intermediates
Once the feasibility of the oxidative addition and reductive elimination has been established in stoichiometric reactions of Pd(IV) complexes, the catalytic turnover quickly emerged. Recently, many novel reactions to construct new C–C and C–heteroatom bonds were developed through a Pd(II)/Pd(IV) catalytic cycle either with or without convincing evidence. Particularly, the unique reactivity of Pd(IV) species in the context of C–H activation and multiple bond difunctionalization has been unveiled as the sharpening stone for the Pd(IV) chemistry. The following paragraphs review recent advances in direct C–H functionalization and difunctionalization of alkenes and alkynes. We will not deal with the cross-coupling reactions in which iodonium salts were used as electrophiles42–44 as well as palladium migration reactions45,46 due to their mechanistic ambiguity.In fact, although the Pd(IV) intermediate was proposed in many cases, convincing evidence was lacking. According to analysis of stoichiometric transformations of Pd(IV) chemistry, it is obvious that suitable strong oxidants and structurally featured ligands are crucial for the generation of Pd(IV) species. This information was applied to roughly judge whether a Pd(IV) mediated mechanism was involved. It should definitely be noted that this judgement can not always hold true since there is substantial difference between the stoichiometric reaction and catalytic one. Clearly, there is no such conclusion as to which oxidants are sufficient to generate Pd(IV) species while which are not. Thus, a clear cut for the involvement of Pd(IV) intermediates in the catalytic reactions is difficult to achieve. The readers should always keep in mind that the mechanism may vary from case to case.
C–H activation has the vast potential to streamline organic synthesis and remains a great challenge for organic chemists. Over the past decades, numerous efficient methodologies were developed to functionalize aromatic as well as aliphatic C–H bonds.47,48 Among them, palladium catalyzed reactions have attracted much attention. Palladium catalyzed C–H activation could proceed through either a Pd(0)/Pd(II) catalytic cycle or Pd(II)/Pd(IV) catalytic cycle. Presumably, the Pd(II)/Pd(IV) catalytic cycle starts with C–H activation at the Pd(II) center by electrophilic attack, followed by oxidation of the resulting Pd(II) intermediate and ligand transfer to afford the Pd(IV) complex. Then reductive elimination delivers the coupling product and regenerates the Pd(II) catalyst to facilitate the catalytic cycle (Scheme 6). The diversity of bond-forming reductive elimination from the Pd(IV) center would allow the installation of different functionalities from C–H bonds.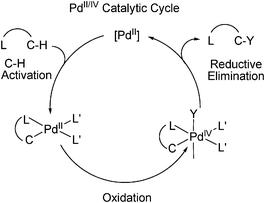 |
| | Scheme 6 C–H Activation through Pd(II)/Pd(IV) mechanism. | |
Many proposed Pd(II)/Pd(IV) catalyzed direct C–H functionalizations have emerged. Introduction of chelating ligands into the substrates can not only bring about a high level of regio-control and enhance the reactivity of the target C–H bonds, but also increase the electron density of the Pd(II) center to facilitate its oxidation to Pd(IV). This strategy has been employed for a number of direct C–H transformations which are usually highly chemo-, regio-, and even diastereoselective. These transformations were classified according to the different types of C–H bond that activated and the different types of bond that formed.
3.1.1 Functionalization of aromatic C–H bond. 3.1.1.1 Alkylation of aromatic C–H bonds. In pioneering studies, Catellani reported a complicated catalytic cycle for the synthesis of ortho-disubstituted vinylarenes.49 The reaction proceeded with an incredibly high degree of chemoselectivity in which palladium interconverted between three distinct oxidation states (Scheme 7). First, the arylpalladium(II) 65a generated by oxidative addition of PhI reacted selectively with strain norbonene to give the alkylpalladium species 65b. The lack of cis-β-hydride blocked the β-hydride elimination. Alternatively, it underwent direct metallation with ortho aryl C–H bond to form intermediate 65c. Then, this intermediate underwent oxidative addition by alkyl iodide to give Pd(V) intermediate 65d. The following selective reductive elimination formed the C–C bond. Another similar process introduced the second C–C bond at the ortho-position. Induced by the steric repulsion, the dialkylated palladium intermediate 65h underwent the β-carbon elimination to form aryl palladium 65i upon extrusion of norbornene. Finally, the aryl palladium species 65i participate in various cross-coupling reactions including Heck, Suzuki and Sonogashira coupling (eqn (40)–(43)).50,51| |  | (40) |
| |  | (41) |
| |  | (42) |
| |  | (43) |
Recently, the protocol was modified by Lautens and co-workers to synthesize fused ring systems. The incorporation of two types of functionalities in the same molecule allowed the facile formation of various carbocycles and heterocycles starting from different substrates. Starting from ortho-substituted iodobenzene, bicyclic compounds 67 with six- or seven-membered carbocycles were produced under modified condition with difunctional acceptor 66. It was shown that the nature of the ortho substituent played a critical role to tune the efficiency (eqn (44)).52 The reaction was also successfully applied to synthesize oxacycles.53,54
| |  | (44) |
Similarly, a new three-component domino reaction by using 68, alkyl halide and a Heck acceptor was developed by Lautens and co-workers. The reaction produced polysubstituted bicyclic molecules 69 in good yields, in which two ortho positions of the iodobenzene were functionalized by different alkyl halides (eqn (45)).55
| | 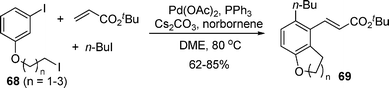 | (45) |
A recent key extension of this strategy was also made by Lautens and co-workers, who first employed enantioenriched secondary halides in this alkylation (eqn (46)).56 It turned out that the reaction proceeded with an overall inversion of the configuration. Assuming the reductive elimination from Pd(IV) species proceeded with retention of the configuration, the authors proposed that the inversion of the stereochemistry most likely occurred during the oxidative addition step by SN2-type substitution. This study provided valuable insight into the stereochemistry of oxidative addition to form Pd(IV) species.
| | 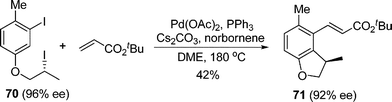 | (46) |
Lautens and co-workers further extended this transformation to bromoalkyl indole. Thus, the ortho alkylation was followed by intramolecular direct arylation. Two C–C bonds were formed starting from two C–H bonds, highlighting the efficiency of this transformation (eqn (47)).57 A wide range of polycyclic indoles 74 can be easily synthesized from simple starting materials.
| |  | (47) |
Similarly, the catalytic procedure can be applied to a one-pot alkylation/cyanation sequence. Steric hindered bi- and tricyclic benzonitriles 76 were readily obtained in a highly efficient manner (eqn (48)).58,59
| |  | (48) |
3.1.1.2 Arylation of aromatic C–H bonds. Biaryl scaffold is a privileged structural units, existing in numerous natural products, synthetic molecules and material chemistry. Among various methods, palladium-catalyzed reactions are particularly attractive. This part outlines the arylation of aromatic substrates through putative Pd(II)/Pd(IV) catalytic cycle with different aryl reagents and oxidants.3.1.1.2.1 Oxidative arylation of aromatic C–H bonds. High valent iodinium reagents are efficient oxidants to produce Pd(IV) species (section 2.1.3). Having established that PhI(OAc)2 was an efficient oxidative C–H functionalization reagent which can transfer an acetate group (section 3.1.1.3), Sanford and co-workers further reasoned that it might be possible to modify this reagent to function as a carbon transfer reagent. They successfully validated this strategy by developing the Pd-catalyzed C–H arylation of 2-phenylpyridine substrates using [Ph2I]BF4 as the phenyl transfer reagent (eqn (49)).60 When unsymmetrical hypervalent iodine(III) reagents [Mes–I–Ar]BF4 were used, the reaction proceeded with completely selective transfer of the sterically less hindered aryl group (eqn (50)). It should be noted that an alternative Pd(0)/Pd(II) catalytic cycle might also be possible.| | 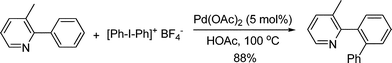 | (49) |
| | 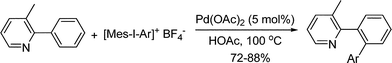 | (50) |
This novel palladium-catalyzed C–H arylation protocol was successfully applied to the direct arylation of heterocycles, such as indole scaffold (eqn (51)).61 NHC ligand was used in order to prevent catalyst deactivation at the expense of the reaction rate due to the diminished electrophilicity of the coordinated palladium center.
| |  | (51) |
Concurrently, a similar strategy was adopted by Daugulis and co-workers in which anilide derivatives was regioselectively arylated by diphenyliodonium salt (eqn (52)).62 In order to overcome the limited commercial availability of diaryliodonium salts, an alternative arylation protocol was further developed. A combination of aryl iodide and silver acetate was also effective as arylation reagents in the presence of palladium catalyst.
| |  | (52) |
Besides direct arylation with aryliodonium derivatives, the homocoupling of aromatic substrates using strong oxidant was also proposed to proceed through Pd(IV) mechanism. Thus, Sanford and co-workers developed a regioselective homocoupling of chelating aromatic substrates using oxone as the oxidant under mild conditions (eqn (53)).63
| |  | (53) |
The mechanism of this transformation was deeply investigated. Based on elegant mechanistic investigations, Sanford and co-workers exclude three out of four possible mechanisms. Finally, it was strongly suggested that the mechanism involved two distinct C–H activation steps: one at the initiated Pd(II) center and the other at the oxidized Pd(IV) center (eqn (54)). The C–H activation at the Pd(IV) center was particularly intriguing since it provided valuable insight into the fundamental reactivity of high valent palladium complexes.
| |  | (54) |
3.1.1.2.2 Arylation with aryl halides. Arylation of C–H bonds with aryl halides, known as direct arylation,64 has experienced significant development in recent years. However, the mechanistic ambiguity remains and mechanisms65–67 either with or without Pd(IV) intermediates have been invoked. This area has been reviewed extensively; thus, we mainly focus on selected examples in which Pd(IV) intermediates might be involved.For the mechanism favoring Pd(IV) intermediates, it involves the oxidative addition of aryl halide to the pallada(II)cycle generated by electrophilic aromatic substitution (path A, Scheme 8). Unfortunately, there is no solid evidence yet on Pd(IV) species formation through oxidative addition of aryl halide to Pd(II) species. Alternatively, other mechanisms such as electrophilic metallation and especially proton abstraction should be seriously considered since they have been supported by both experimental and computational results (path B, Scheme 8).65,66 Thus, the controversial issue of Pd(IV) intermediacy in C–H arylation reactions is still under debate.
 |
| | Scheme 8 Arylation of C–H bond with aryl halide. | |
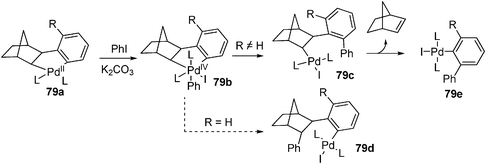
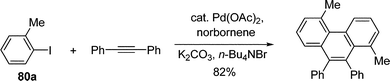
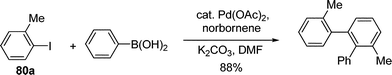
By using aryl halide instead of alkyl halide, Pd-catalyzed aromatic C–H alkylation with alkyl halide mediated by norbornene was extended to the C–H arylation reaction. Through stoichiometric studies, Catellani and co-workers determined that the presence of an ortho substituent on the iodobenzene played an vital role in controlling the selectivity of phenyl transfer in the postulated Pd(IV) intermediate (Scheme 9).68–70
By fine tuning of the reaction condition, a catalytic pathway to construct phenanthrene scaffold from ortho-substituted aryl iodides and diphenylacetylenes was developed.71 It was suggested that the reaction proceeded through a intermediate similar to 79c. Similarly, a tandem arylation/Suzuki coupling and arylation/Heck reaction were further developed (eqn (55)–(57)).72
| |  | (55) |
| |  | (56) |
| |  | (57) |
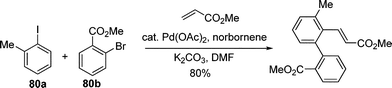
Interestingly, two different aryl halides, an aryl iodide 80a bearing an ortho electron-donating group and an aryl bromide 80b containing electron-withdrawing substituent engaged in a Pd-catalyzed sequential unsymmetrical aryl coupling and Heck alkenylation (eqn (58)).73 It was suggested that the o-substituted iodobenzene reacted much faster than bromobenzene with Pd(0) while the substituted bromobenzene preferentially reacted with Pd(II) species to form a proposed Pd(IV) intermediate. The reactivity of different aryl halides was responsible for the selectivity of such unsymmetrical aryl couplings.
| |  | (58) |
Based on similar design, intramolecular amination could also ensue with the ortho arylation (eqn (59)).74 In addition, Lautens and co-workers developed an unsymmetrical aryl coupling in which the final aryl palladium intermediate was trapped with cyanide (eqn (60)).59
| |  | (59) |
| |  | (60) |
Dyker and co-workers have described earlier the self-condensation of o-iodoanisole to form aryl–aryl coupling product in the absence of norbonene.75 Intramolecular cyclometallation of the arylPd(II) via alkyl C–H activation produced five-membered palladacycle 81a. After oxidative addition of ArI, a key Pd(IV) intermediate 81b was formed. Further reductive elimination formed the aryl–aryl bond. Then cyclometallation of the ortho C–H bond followed by repeating the oxidative addition and reductive elimination, biarylated intermediate 81d was obtained, which was further transformed to final product (Scheme 10). This strategy was successfully extended to aryl–alkenyl coupling as well.76,77
Carretero also reported an interesting case in which a similar pathway may be involved. During the investigation of the Heck reaction of aryl halides with vinyl sulfones, the normal Heck olefination product was not observed. Unexpectedly, 1-phenyl-9,10-dihydrophenanthrenes was isolated.78 A pathway involving the arylation of the palladacycle 82bvia a similar Pd(II)–Pd(IV)–Pd(II) sequence was proposed (Scheme 11).
Daugulis and co-workers also made significant contribution to approach the directed arylation of various aromatic substrates via C–H bond functionalization with aryl halides.79–81 Although they proposed Pd(IV) intermediates in the catalytic cycle, the exact catalytic species and mechanistic pathway remained to be clarified.
| |  | (61) |
3.1.1.3 Oxygenation of aromatic C–H bonds. Besides C–C bond formation, C–X (X = O, Cl, etc.) formation from C–H bonds has also drawn much attention in Pd(IV) chemistry. By using PhI(OAc)2 as the oxidant, an early work done by Crabtree and co-workers indicated that benzene could be converted to phenyl acetate (eqn (62)).82 However, regioisomers were obtained when substituted benzene derivatives were employed. In this transformation, Pd(IV) species was proposed as the key intermediate.| |  | (62) |
This field was studied in depth by Sanford and co-workers recently. They developed the highly selective aromatic C–H acetoxylation by using directing group assisted strategy. 2-Arylpyridine derivatives were used as substrates and PhI(OAc)2 was used as both oxidant and OAc source (Scheme 12).83,84 These transformations exhibit high functional group tolerance and exclusive ortho regioselectivity. Furthermore, several directing groups were systematically studied.85
 |
| | Scheme 12 Highly regioselective ligand-directed C–H bond acetoxylation. | |
Simple modification of such a catalytic system were performed in the presence of various alcohol solvents, allowing regioselective installation of alkoxy groups (eqn (63)).83 It was proposed that the in-situ generated PhI(OR)2 was responsible for the C–H alkoxylation reaction.
| | 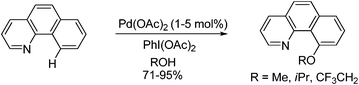 | (63) |
Furthermore, Pd-catalyzed direct acetoxylation with oxime ether as directing group was explored with various oxidants by utilizing AcOH as external acetate source in the same group (eqn (64)).86 In all cases, ortho-acetoxylated products were generated regioselectively. Notably, when inexpensive, safe, and environmentally benign oxone was employed, comparable yields with respect to Ph(IOAc)2 were obtained. The C–O formation was proposed through reductive elimination of Pd(IV) intermediates, which has found support from the stoichiometric studies.
| |  | (64) |
It should be mentioned that Fujiwara has reported the hydroxylation of benzene by palladium acetate under oxygen atmosphere.87 Thus, a Pd(0)/Pd(II) catalytic cycle should also be considered in the C–H oxidation reactions since oxygen could not oxidize Pd(II) to Pd(IV).
3.1.1.4 Halogenation of aromatic C–H bonds. Several groups reported the halogenation of aromatic C–H bond nearly at the same time. Shi and co-workers reported Pd-catalyzed halogenation of acetanilide derivatives by using CuX2 (X = Cl, Br) as both the halogen source and the oxidant (eqn (65)).88 The pallada(II)cycle also showed comparable catalytic ability. However, such Pd(II) species could not furnish the desired products in the presence of various oxidants unless Cu(OAc)2 or PhI(OAc)2 was used. Thus, a Pd(II)/Pd(IV) catalytic cycle was preferred although this conclusion is still premature. In fact, no clear evidence strongly supports this hypothesis.| | 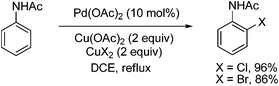 | (65) |
Sanford and co-workers explored Pd-catalyzed halogenation of sp2 C–H bonds by using electrophilic N-halosuccinimides (NXS) as the halogen source and oxidant (eqn (66)).83,89,90 A wide range of aromatic compounds can be regioselectively halogenated in good yields. In this studies, Pd(IV) intermediate was highly preferred, which was strongly supported by the isolation of Cl- and N-binding Pd(IV) complex and its following reductive elimination (section 2.2.3).
| |  | (66) |
Pd-catalyzed C–F bond formation was impeded by the difficulty of C–F reductive elimination at a Pd(II) center. Recently, Sanford and co-workers reasoned that it might be possible to obtain C–F reductive elimination at a Pd(IV) center. The feasibility of such a process was demonstrated through the effective fluorination of aromatic C–H directed by pyridinyl group (eqn (67)).91 Direct evidence for the mechanism was later provided by the isolation of a palladium(IV) fluoride complex, which readily underwent C–F reductive elimination.38,40
| |  | (67) |
Very recently, Yu and co-workers demonstrated the first Pd-catalyzed ortho-iodination/bromination of carboxylic acids, a complementary method to the traditional ortho lithiation/halogenation sequence.92 The IOAc was considered as an efficient oxidant to oxidize Pd(II) 85a to Pd(IV) 85b, at which center C–I bond was formed upon the reductive elimination (Scheme 13). Interestingly, tetraalkyl ammonium cations were found to effect the efficiency of C–H activation and enhance the monoselectivity.
 |
| | Scheme 13 lodination of carboxylic acids. | |
The similar iodination protocol was further extended to ortho iodination of phenethylamine triflate.93 Combining the ortho iodination and the CuI catalyzed C–N formation, a direct C–H activation/C–N bond formation reaction can be realized in one pot to approach valuable heterocycles (eqn (68)). However, during these transformations, strong evidence for the Pd(IV) intermediates was lacking.
| |  | (68) |
3.1.1.5 C–N formation of aromatic C–H bonds. Although many examples of Pd-catalyzed direct C–N bond formation of C–H bond have appeared, whether a Pd(0)/Pd(II) or Pd(II)/Pd(IV) catalytic cycles was involved still remains elusive.94–98Yu recently reported the direct C–N bond formation via C–H activation to synthesize β, γ and δ lactams, in which a Pd(IV) intermediate was preferred (eqn (69)).99 However, there is no evident to support the sufficient oxidation of Pd(II) to Pd(IV) by either Ag+ or Cu2+.
| |  | (69) |
More recently, Gaunt reported an intramolecular C–H amination reaction for the synthesis of carbazoles. PhI(OAc)2 was used as the oxidant and the reaction proceeded under extremely mild condition (eqn (70)). A stable carbopalladation complex was isolated, which gave the C–N coupling product when treated with PhI(OAc)2, thus suggesting the involvement of a Pd(IV) intermediate.100
| |  | (70) |
3.1.2 Functionalization of aliphatic C–H bonds. 3.1.2.1 Oxygenation of aliphatic C–H bonds. It is well known that O-methyl oxime is an efficient directing group for the synthesis of five-membered palladacycles. Sanford and co-workers utilized such directing group for the regioselective acetoxylation of unactivated sp3 C–H bonds by using PhI(OAc)2 as a stoichiometric oxidant (eqn (71)).101 The transformation could be applied to a series of substrates and afforded good yields with excellent chemo-, regio- and even diastereoselectivity. A Pd(II)/Pd(IV) catalytic cycle was highly favored since the reaction involved a donating ligand and a strong oxidant, both of which are important for efficient oxidation of Pd(II) to Pd(IV) complex.| |  | (71) |
Yu and co-workers also disclosed an oxazolyl group-assisted Pd-catalyzed acetoxylation of unactivated methyl groups using inexpensive peroxyester MeCOOOtBu as the stoichiometric oxidant (eqn (72)).102 Ac2O was crucial for this transformation as it was required for both steps of the oxidation of Pd(II) to Pd(IV) and regeneration of the Pd(OAc)2 catalyst. The isolated intermediate 87 may be oxidized to Pd(IV) to facilitate the further reductive elimination.
| | 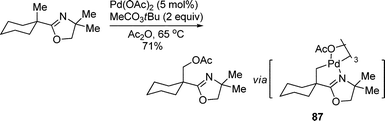 | (72) |
Later on, using the in-situ generated IOAc as the oxidant, Yu similarly developed an efficient Pd-catalyzed acetoxylation of C–H bond adjacent to N atom (eqn (73)).103 A synthetically useful Boc-protecting group was used to bring Pd to proximity of the target alkyl group.
| |  | (73) |
Selective acetoxylation of the naturally existing amino acids is of special note. Thus, Corey and co-workers recently achieved the β-acetoxylation of protected amino acids derivatives diastereoselectively (eqn (74)).104 Oxone was an efficient oxidant and Mn(OAc)2 exerted a noteworthy rate acceleration. Both these processes were proposed to proceed through Pd(IV) intermediate.
| |  | (74) |
3.1.2.2 Arylation of aliphatic C–H bonds. During the study of Pd-catalyzed arylation of aromatic C–H bonds with [Ph2I]BF4 directed by pyridinyl group, Sanford and co-workers found that such arylation could also be applied to benzylic sp3 C–H arylation.60 It is proposed that such sp3 C–H arylation proceed through similar Pd(II)/Pd(IV) catalytic cycle (eqn (75)).| |  | (75) |
Analogously, mechanism on the arylation of aliphatic C–H bonds with aryl halides is not firmly established. For example, highly regioselective arylation of sp3 C–H bond was achieved by Daugulis by using pyridine as directing group and ArI as arylation reagent (eqn (76)).105,106 The same strategy was also applied to the arylation of unprotected aliphatic acid and protected amino acid derivatives by Yu107,108 (eqn (77)) and Corey,104 respectively. Although in all these reactions a Pd(II)/Pd(IV) catalytic cycle was suggested, further investigation is required to clarify whether Pd(IV) intermediates are involved or not since formation of Pd(IV) species by oxidative addition of aryl halide to Pd(II) complexes has never been addressed.
| | 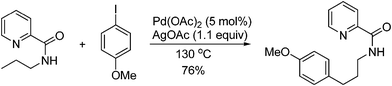 | (76) |
| | 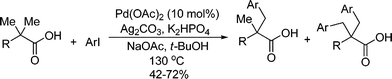 | (77) |
3.1.2.3 Halogenation of aliphatic C–H bonds. Similarly, the system for fluorination of aromatic C–H bond developed by Sanford could also be extended to benzylic sp3 C–H fluorination (eqn (78)).91 The benzylic C–H bond proximal to the directing group was selectively fluorinated by N-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate 84b catalyzed by Pd(OAc)2. A Pd(II)/Pd(IV) catalytic cycle can be envisaged since a similar fluorination compound was known to effect oxidation of Pd(II) to Pd(IV).38,40| |  | (78) |
Yu developed an efficient method to selectively introduce iodine group to the terminal methyl group by an oxazolinyl-directed iodination of aliphatic acids, in which preferential activation of primary alkyl group over secondary one was observed.109,110 Subsequently, they succeeded in the diastereoselective iodination of prochiral alkyl substrates. After deprotection of the oxazolinyl group, functionalized aliphatic acids can be obtained in excellent ee (Scheme 14). This elegant design provided an attractive method that met the formidable challenge of asymmetric functionalization of unactivated sp3 C–H bonds. Notably, the carbopalladacycle intermediate and the presence of the PhI(OAc)2 as oxidants may imply that Pd(IV) intermediates were involved in the catalytic cycle. Using such a catalytic protocol, Yu further developed the diiodination of the two methyl groups of 2-(1,1-dimethylalkyl)dimethyloxazolines via Pd-catalyzed double C–H activations.111 A sequential radical cyclization successfully converted the diiodination products to cyclopropane derivatives.
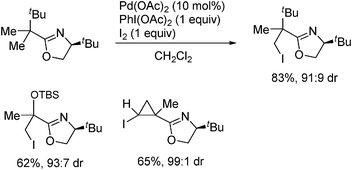 |
| | Scheme 14 Asymmetric iodination of aliphatic C–H bonds. | |
3.2 Difunctionalization of alkenes and alkynes
Vicinal oxidative difunctionalizations of alkenes, such as Sharpless dihydroxylation and aminohydroxylation, are efficient and powerful transformations in organic synthesis.112,113 The comprehensive and vast scope of the palladium catalyzed reaction would provide great opportunity for difunctionalization of such unsaturated bonds. Generally, a Pd(II)/Pd(IV) catalyzed difunctionalization of unsaturated bonds comprise two key steps: first, multiple bonds coordinate to Pd(II) and accept the nucleophilic attack to form a alkyl/alkenyl palladium(II) intermediate 88 with incorporation of the first functionality. After oxidation, the second functional group was installed by the elaboration of the resulting alkyl/alkenyl palladium(IV) species (Scheme 15).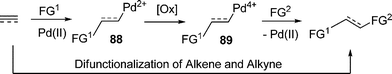 |
| | Scheme 15 Difunctionalization of alkene/alkynes. | |
Specifically, alkyl/alkenyl palladium(II) intermediate is generated by means of nucleopalladation or carbopalladation at the Pd(II) center. If Pd(II) intermediate 88a or 88b is trapped by a suitable oxidant, palladium(IV) species 89 is formed. Further transformation at the Pd(IV) center would afford the functionalization products with incorporation of two identical or different groups, such as diamination, aminohydroxylation, carboamination, and so on, on the unsaturated bonds (Scheme 16). Oxidative interception of the C–Pd(II) bond at the first stage by Cu(II) salt could also lead to difunctionalization of multiple bonds. However, this type of reaction is not discussed here since its mechanism may not involve the Pd(IV) species.114–118
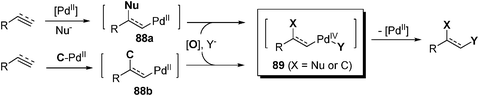 |
| | Scheme 16 Difunctionalization of alkene/alkynes. | |
3.2.1 Heteroatom–heteroatom difunctionalization of alkenes. Sorensen and co-workers first developed the Pd(II) catalyzed ring-forming intramolecular aminoacetoxylation of γ-aminoolefins. PhI(OAc)2, a common oxidant for the production of Pd(IV) species, was employed, thereby suggesting a potential Pd(II)/Pd(IV) catalytic cycle.119 In order to explain the overall trans difunctionalization stereochemical outcome, a mechanism involving reversible trans-aminopalladation of the alkene and C–O bond forming reductive elimination from the Pd(IV) center with retention of the configuration was proposed (eqn (79)–(80)). However, another pathway involving cis-aminopalladation followed by reductive elimination to form a C–O bond with inversion of the configuration could not be excluded.| |  | (79) |
| | 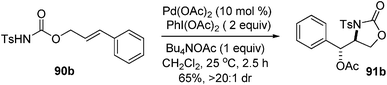 | (80) |
Soon after, Muñiz and co-workers reported the first example of Pd(II)-catalyzed intramolecular diamination of unactivated alkenes using ω-alkenyl-substituted ureas as substrates (eqn (81)).120 Again, PhI(OAc)2 was found as an efficient oxidant. This methodology offered an efficient way for the construction of 5,5-, 6,5- and 7,5-fused cyclic urea derivatives, which could be further transformed to diamine derivatives.121–123
| |  | (81) |
| |  | (82) |
| |  | (83) |
According to the observed reaction stereochemistry (eqn (82) and (83)), the formation of second sp3 C–N would require the dissociation of tosylamide group from Pd(IV) center and a single bond rotation to reach the exact triangular bipyramidal geometry for the SN2-like nucleophilic attack (eqn (84)). This C–N bond forming process was facilitated through the enhancement of electrophilicity of the carbon center by Pd(IV) center. During this process, such dissociation of the tosylamide group from the Pd(IV) center increased the rate of the SN2 type substitution by the generation of Pd(IV) zwitterion and simultaneously promoted the nucleophilicity of the nitrogen atom.123
| |  | (84) |
Furthermore, the diastereoselective intermolecular aminoacetoxylation of terminal alkenes with phthalimide as nitrogen nucleophile source was achieved by Stahl and co-workers.124 These transformations proceeded with exquisite regioselectivity. Allyl ethers proved to be effective substrates which gave high diastereoselectivity (typically >20 : 1) (eqn (85) and (86)).
| |  | (85) |
| |  | (86) |
During this transformation, the use of 1,2-disubstituted (Z)-olefins under identical condition predominantly afforded syn-difunctionalized product. Only two proposed mechanistic pathways could explain the stereochemical outcome of the products: (1) trans-aminopalladation and direct reductive elimination with retention of stereochemistry or (2) cis-aminopalladation and SN2-like C–O bond formation with inversion of stereochemistry (Scheme 17). To distinguish between these two pathways, the substrates were subjected to the condition which favors β-hydride elimination for the formed alkylpalladium intermediates. The resulting (Z)-alkene provided evidence for cis-aminopalladation, thus favoring the latter mechanism.
 |
| | Scheme 17 Mechanism of the aminoacetoxylation of alkenes. | |
The same strategy was also employed by Sanford and co-workers for the diastereoselective construction of substituted tetrahydrofurans through aminooxygenation of alkenes (eqn (87)).125
| |  | (87) |
Primary mechanistic investigation also implied a cis-aminopalladation process, which was again supported by performing the reaction under conditions that favor β-hydride elimination. These experiments were similar to those done by Stahl, and delivered identical results. Therefore, the C–O bond formation step would operate through a direct reductive elimination pathway, which stood in contrast to an SN2-like attack in Muñiz’s studies (Scheme 18).123 This difference may be attributed to either the intramolecularity of the ether-forming reductive elimination or the higher nucleophilicity of the alkoxide relative to acetate ligand. The latter was more likely because it has been proved that C–O bond-forming reductive elimination from Pd(IV) aryl benzoate complexes proceeded significantly faster with electron-donating substituents on the benzoate ligand.
 |
| | Scheme 18 Mechanism study of the aminooxygenation of alkene. | |
Very recently, Dong and co-workers reported the in situ generated cationic palladium diphosphine complex [Pd(dppp)(H2O)2](OTf)2 catalyzed dioxygenation of olefins (eqn (88)).126 On the basis of the isotopic labeling studies and the observation of syn diastereoselectivity, the proposed mechanism was depicted in Scheme 19. trans-Acetoxypalladation followed by the oxidation with hypervalent iodine reagent afforded the key Pd(IV) intermediate 97e, which was trapped by the pendant acetyl via a SN2-like attack. Subsequent hydrolysis delivered the final syn-acetoxylation product. This catalytic system was not limited to terminal olefins, and the presence of a directing group for the enhancement of reactivity is not necessary. The successful introduction of phosphine ligand in the proposed Pd(II)/Pd(IV) catalysis is remarkable since there is great potential for the application of chiral ligands to access the enantioselective synthesis.
| |  | (88) |
Branco and co-workers reported the Pd(II)-catalyzed aziridination of olefins with bromamine T as the nitrogen transfer reagent.127 The electron deficient olefins in general reacted to give better yields than the simple alkenes. A proposed mechanism involved trans-aminopalladation, followed by intramolecular oxidation to give alkyl palladium(IV) intermediate 98a (Scheme 20). In this transformation, low diastereoselection was obtained using chiral auxiliaries.128
 |
| | Scheme 19 Proposed mechanism for dioxygenation of alkene. | |
 |
| | Scheme 20 Proposed mechanism for aziridination of alkene. | |
3.2.2 Carbon–carbon and carbon-heteroatom difunctionalization of alkenes. Recently, Sanford and co-workers developed Pd-catalyzed cascade cyclization of enynes under oxidative conditions to afford cyclopropyl fused butenolide products 100 (eqn (89)).129 Concurrently, Tse and co-workers reported an almost identical result starting from the same substrate.130 These methodologies could be employed for the construction of lactones, lactams, tetrahydrofurans and pyrrolidines.| |  | (89) |
A proposed mechanism depicted in Scheme 21 rationalized the stereochemical outcome with a key Pd(IV) intermediate. The Pd(II) intermediate 102b was formed by insertion of the pendant alkene to the vinylic C–Pd intermediate 80a, which was generated by trans-acetoxylpalladation of the alkyne. σ-Bond rotation followed by oxidation with PhI(OAc)2 would afford the Pd(IV) intermediate 102d. A SN2-type attack by the electron-rich tethered alkene moiety to the Pd(IV)-bound carbon with inversion of the configuration delivered the cyclopropane product 100 upon further hydrolysis. Formation of several side products were properly explained by this mechanism. Further evidence came from the isolation of C–OAc bond-forming product 101c with the use of phenyl substituted substrate 99c in which the cyclopropane formation was inhibited by the increased steric hindrance of the phenyl group in intermediate 102e (eqn (90)).
| |  | (90) |
Subsequently, Liu and co-workers reported an efficient palladium-catalyzed oxidative cyclization of enynes in which H2O2 was used as the stoichiometric oxidant at room temperature.131 This reaction appeared to proceed through a mechanism involving the oxidation of sp3 C–Pd(II) species by H2O2, and the formation of C–Cl bonds by a direct reductive elimination from the Pd(IV) center that led to the retention of the configuration at the carbon center (eqn (91)).
| |  | (91) |
The alkyl–palladium bond can also be generated by means of transmetallation–alkene insertion sequence. Such common Heck type alkyl-palladium intermediates can be intercepted by oxidative functionalization by reacting with PhICl2.132,133 Again, β-H elimination, a featured process associated with palladium(II) species, was suppressed, suggesting the involvement of Pd(IV) intermediate. The small amout of isomer observed in this reaction can be explained by a β-hydride elimination and reinsertion sequence of the alkyl-Pd intermediate and found experimental support (eqn (92)).
| |  | (92) |
3.2.3 Difunctionalization of alkynes. In contrast to the well developed difunctionalization of alkenes, there are only limited reports on the difunctionalization of alkynes that presumably proceed through Pd(IV) intermediates.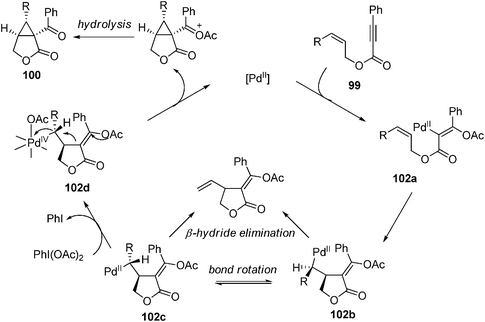 |
| | Scheme 21 Proposed mechanism for cycloisomerization of 1,6-enynes. | |
Elsevier and co-workers pioneered this work and developed a catalytic three-component synthesis of conjugated dienes from alkynes, organohalides and tetramethyltin reagent. Based on stoichiometric studies, a catalytic cycle was further envisaged. Supposedly, oxidative cyclometallation gave the palladacyclopentadienes 7, which further reacted with organohalide to generate the Pd(IV) intermediate 105a. Then C–C forming reductive elimination on Pd(IV) center resulted in a vinyl Pd(II) intermediate 105b, which underwent Stille coupling with tetramethyltin to give the final product (Scheme 22). Using molecular halogens instead of organohalides could lead to stereoselective synthesis of 1,4-dihalo-1,3-dienes.11
 |
| | Scheme 22 Three-component coupling for the synthesis of conjugated dienes. | |
During the investigation of diamination of internal alkenes, Muñiz and co-workers found that the protocol was also amendable to one example of diamination of internal alkyne, a process lead to annulated indole derivatives.121 It was suggested that the second alkenyl C–N bond formation proceeded through direct reductive elimination from Pd(IV) intermediate instead of a SN2-type mechanism (eqn (93)).
| |  | (93) |
Another successful example reported by Li and co-workers showed a sequential intermolecular aminopalladation/ortho-arene C–H activation reactions of N-phenylpropiolamides with phthalimide (eqn (94)).134 This reaction could be further modified to the acetoxypalladation/ortho-arene C–H activation by using PhI(OAc)2 alone. under the same condition.135 During this process, although a Pd(II)/Pd(IV) catalytic cycle was favored, the pathway through the traditional Pd(0)/Pd(II) catalytic cycle could not be ruled out (Scheme 23).
| |  | (94) |
4. Conclusion and perspective
The research field of intriguing Pd(IV) chemistry is relatively new and investigations are speeding up. These studies not only provide novel elementary organometallic reactions, but also facilitate many transformations that are not easily available by Pd(0)/Pd(II) catalysis. A series of novel reactions have been developed based on the understanding of the inherent reactivity of Pd(IV) complexes. |
| | Scheme 23 Possible mechanisms for the difunctionalization of alkynes. | |
Although organopalladium(IV) chemistry has attracted much attention from the chemical community in recent years, much limitation still remains. For example, stoichiometric reaction at the Pd(IV) center is mainly limited to reductive elimination. Other fundamental elementary reactions of Pd(II) chemistry, such as transmetallation with organometallic reagents and β-elimination, are essentially unknown for Pd(IV) complexes. In addition, unlike the formidable knowledge accumulated for ligands of Pd(0) and Pd(II) complexes, little is known about the ligand effect of Pd(IV) complexes especially in catalytic reactions. In most cases, nitrogen and carbon atoms in the substrates function as the ligand. Despite limited application of NHC (N-heterocyclic carbene)61 and phosphine126 ligands in Pd(II)/Pd(IV) catalyzed reactions that appeared recently, enantioselective synthesis using chiral ligands through Pd(IV) intermediates has not been forthcoming.136 Furthermore, the reaction patterns of the Pd(II)/Pd(IV) catalyzed methodologies are somewhat limited and application to natural product synthesis are lacking. Finally, although many structurally diverse Pd(IV) complexes have been isolated and studied, detailed mechanistic investigation in catalytic reactions which were proposed to proceed through Pd(IV) intermediates are also lacking.137
In conclusion, the fast development of organopalladium(IV) chemistry has led to many synthetically useful reactions and valuable mechanistic insights, both of which have greatly enriched palladium chemistry. Undoubtedly, the numerous advantages and vast potential associated with Pd(IV) chemistry will promote more and more research interest in this area.
Acknowledgements
Support of this work by a starter grant from Peking University and the grant from National Sciences of Foundation of China (No. 20672006, 20821062, GZ419) and the “973” Project from the MOST of China (2009CB825300) is gratefully acknowledged.References
- Metal-catalyzed Cross-coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, New York, 1998 Search PubMed
 ; J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 Search PubMed ; J. F. Hartwig, Nature, 2008, 455, 314 CrossRef CAS .
; J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 Search PubMed ; J. F. Hartwig, Nature, 2008, 455, 314 CrossRef CAS . - A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS ; D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS ; A. R. Muci and S. L. Buchwald, Top. Curr. Chem., 2002, 219, 131 Search PubMed ; D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234 CrossRef CAS ; Palladium Reagents and Catalysts: New Perspectives for the 21st Century, ed. J. Tsuji, Wiley and Sons, New York, 2003 Search PubMed .
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS .
- W. J. Pope and S. J. Peachey, Proc. Chem. Soc., London, 1907, 23, 86 Search PubMed .
- E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS .
- R. Uson, J. Fornies and R. Navarro, J. Organomet. Chem., 1975, 96, 307 CrossRef CAS .
- P. K. Byers, A. J. Canty, B. W. Skelton and A. H. White, J. Chem. Soc., Chem. Commun., 1986, 1722 RSC .
- A. J. Canty, Acc. Chem. Res., 1992, 25, 83 CrossRef CAS .
- P. L. Alsters, P. F. Engel, M. P. Hogerheide, M. Copijn, A. L. Spek and G. van Koten, Organometallics, 1993, 12, 1831 CrossRef CAS .
- R. van Belzen, H. Hoffmann and C. J. Elsevier, Angew. Chem., Int. Ed. Engl., 1997, 36, 1743 CrossRef CAS .
- R. van Belzen, C. J. Elsevier, A. Dedieu, N. Veldman and A. L. Spek, Organometallics, 2003, 22, 722 CrossRef CAS .
- A. J. Canty, H. Jin, B. W. Skelton and A. H. White, Inorg. Chem., 1998, 37, 3975 CrossRef CAS .
- A. J. Canty, H. Jin, A. S. Roberts, B. W. Skelton and A. H. White, Organometallics, 1996, 15, 5713 CrossRef CAS .
- P. K. Byers, A. J. Canty, M. Crespo, R. J. Puddephatt and J. D. Scott, Organometallics, 1988, 7, 1363 CrossRef CAS .
- A. Moravskiy and J. K. Stille, J. Am. Chem. Soc., 1981, 103, 4182 CrossRef CAS .
- A. Bayler, A. J. Canty, P. G. Edwards, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 2000, 3325 RSC .
- A. Bayler, A. J. Canty, B. W. Skelton and A. H. White, J. Organomet. Chem., 2000, 595, 296 CrossRef CAS .
- P. K. Byers and A. J. Canty, J. Chem. Soc., Chem. Commun., 1988, 639 RSC .
- R. Guo, J. L. Portscheller, V. W. Day and H. C. Malinakova, Organometallics, 2007, 26, 3874 CrossRef CAS .
- N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924 CrossRef CAS .
- M.-C. Lagunas, R. A. Gossage, A. L. Spek and G. van Koten, Organometallics, 1998, 17, 731 CrossRef CAS .
- A. R. Dick, J. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 12790 CrossRef CAS .
- S. R. Whitfield and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 15142 CrossRef CAS .
- R. Bhawmick, H. Biswas and P. Bandyopadhyay, J. Organomet. Chem., 1995, 498, 81 CrossRef CAS .
- K. Kamaraj and D. Bandyopadhyay, Organometallics, 1999, 18, 438 CrossRef CAS .
- A. R. Dick, M. S. Remy, J. W. Kampf and M. S. Sanford, Organometallics, 2007, 26, 1365 CrossRef CAS .
- A. J. Canty, J. Patel, T. Rodemann, J. H. Ryan, B. W. Skelton and A. H. White, Organometallics, 2004, 23, 3466 CrossRef CAS .
- A. J. Canty, T. Rodemann, B. W. Skelton and A. H. White, Organometallics, 2006, 25, 3996 CrossRef CAS .
- P. D. Chaudhuri, R. Guo and H. C. Malinakova, J. Organomet. Chem., 2008, 693, 567 CrossRef CAS .
- S. Shimada, M. Tanaka and M. Shiro, Angew. Chem., Int. Ed. Engl., 1996, 35, 1856 CrossRef CAS .
- H. Sakurai, Y. Kamiyama and Y. Nakadaira, J. Organomet. Chem., 1977, 131, 147 CrossRef CAS .
- M. Suginome, H. Oike and Y. Ito, J. Am. Chem. Soc., 1995, 117, 1665 CrossRef CAS .
- M. Suginome, Y. Kato, N. Takeda, H. Oike and Y. Ito, Organometallics, 1998, 17, 495 CrossRef CAS .
- Y. Yamamoto, T. Ohno and K. Itoh, Angew. Chem., Int. Ed., 2002, 41, 3662 CrossRef CAS .
- J. Cámpora, P. Palma, D. del Rio and E. Carmona, Organometallics, 2003, 22, 3345 CrossRef CAS .
- J. Cámpora, P. Palma, D. del Rio, J. A. Lopez, E. Alvarez and N. G. Connelly, Organometallics, 2005, 24, 3624 CrossRef CAS .
- T. Furuya, H. M. Kaiser and T. Ritter, Angew. Chem., Int. Ed., 2008, 47, 5993 CAS .
- T. Furuya and T. Ritter, J. Am. Chem. Soc., 2008, 130, 10060 CrossRef CAS .
- A. W. Kaspi, A. Yahav-Levi, I. Goldberg and A. Vigalok, Inorg. Chem., 2008, 47, 5 CrossRef CAS .
- For another example after submission of the manuscript: N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796 Search PubMed .
- Y. Fu, Z. Li, S. Liang, Q.-X. Guo and L. Liu, Organometallics, 2008, 27, 3736 CrossRef CAS .
- M. Uchiyama, T. Suzuki and Y. Yamazaki, Nippon Kagaku Kaishi, 1982, 236 CAS .
- T. Okuyama, Acc. Chem. Res., 2002, 35, 12 CrossRef CAS .
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523 CrossRef CAS .
- S. Ma and Z. Gu, Angew. Chem., Int. Ed., 2005, 44, 7512 CrossRef CAS .
- M. A. Campo, H. Zhang, T. Yao, A. Ibdah, R. D. McCulla, Q. Huang, J. Zhao, W. S. Jenks and R. C. Larock, J. Am. Chem. Soc., 2007, 129, 6298 CrossRef CAS , and references therein.
- A. E. Shilov and G. B. Shul’pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS ; G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1698 CrossRef ; J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS ; F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077 CrossRef CAS ; A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439 CrossRef CAS .
- K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS ; H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS ; J.-Q. Yu, R. Giri and S. Chen, Org. Biomol. Chem., 2006, 4, 4041 RSC ; B.-J. Li, S.-D. Yang and Z.-J. Shi, Synlett, 2008, 949 CAS .
- M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119 CrossRef CAS .
- M. Catellani, E. Motti and M. Minari, Chem. Commun., 2000, 157 RSC .
- M. Catellani, Synlett, 2003, 298 CrossRef CAS .
- M. Lautens and S. Piguel, Angew. Chem., Int. Ed., 2000, 39, 1045 CrossRef CAS .
- M. Lautens, J.-F. Paquin, S. Piguel and M. Dahlmann, J. Org. Chem., 2001, 66, 8127 CrossRef CAS .
- M. Lautens, J.-F. Paquin and S. Piguel, J. Org. Chem., 2002, 67, 3972 CrossRef CAS .
- S. Pache and M. Lautens, Org. Lett., 2003, 5, 4827 CrossRef CAS .
- A. Rudolph, N. Rackelmann and M. Lautens, Angew. Chem., Int. Ed., 2007, 46, 1485 CrossRef CAS .
- C. Bressy, D. Alberico and M. Lautens, J. Am. Chem. Soc., 2005, 127, 13148 CrossRef CAS .
- B. Mariampillai, D. Alberico, V. Bidau and M. Lautens, J. Am. Chem. Soc., 2006, 128, 14436 CrossRef CAS .
- B. Mariampillai, J. Alliot, M. Li and M. Lautens, J. Am. Chem. Soc., 2007, 129, 15372 CrossRef CAS .
- D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS .
- N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972 CrossRef CAS .
- O. Daugulis and V. G. Zaitsev, Angew. Chem., Int. Ed., 2005, 44, 4046 CrossRef CAS .
- K. L. Hull, E. L. Lanni and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 14047 CrossRef CAS .
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS ; I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC ; I. J. S. Fairlamb, Chem. Soc. Rev., 2007, 36, 1036 RSC ; L. Ackermann, Synlett, 2007, 507 CrossRef CAS .
- D. García-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066 CrossRef CAS .
- M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754 CrossRef .
- L.-C. Campeau, D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35 ; S. Pascual, P. de Mendoza and A. M. Echavarren, Org. Biomol. Chem., 2007, 5, 2727 RSC .
- M. Catellani and E. Motti, New J. Chem., 1998, 22, 759 RSC .
- M. Catellani, E. Motti and S. Ghelli, Chem. Commun., 2000, 2003 RSC .
- O. Reiser, M. Weber and A. de Meijere, Angew. Chem., Int. Ed. Engl., 1989, 28, 1037 CrossRef .
- M. Catellani, E. Motti and S. Baratta, Org. Lett., 2001, 3, 3611 CrossRef CAS .
- E. Motti, A. Mignozzi and M. Catellani, J. Mol. Catal. A: Chem., 2003, 204, 115 CrossRef .
- F. Faccini, E. Motti and M. Catellani, J. Am. Chem. Soc., 2004, 126, 78 CrossRef CAS .
- R. Ferraccioli, D. Carenzi, O. Rombola and M. Catellani, Org. Lett., 2004, 6, 4759 CrossRef CAS .
- G. Dyker, Angew. Chem., Int. Ed. Engl., 1992, 31, 1023 CrossRef .
- G. Dyker, J. Org. Chem., 1993, 58, 6426 CrossRef CAS .
- G. Dyker, Angew. Chem., Int. Ed. Engl., 1994, 33, 103 CrossRef .
- P. Mauleón, I. Alonso and J. C. Carretero, Angew. Chem., Int. Ed., 2001, 40, 1291 CrossRef CAS .
- A. Lazareva and O. Daugulis, Org. Lett., 2006, 8, 5211 CrossRef CAS .
- H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879 CrossRef CAS .
- O. Daugulis, V. G. Zaitsev, D. Shabashov, Q. N. Pham and A. Lazareva, Synlett, 2006, 3382 CrossRef CAS .
- T. Yoneyama and R. H. Crabtree, J. Mol. Catal. A: Chem., 1996, 108, 35 CrossRef CAS .
- A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2006, 126, 2300 .
- D. Kalyani and M. S. Sanford, Org. Lett., 2005, 7, 4149 CrossRef CAS .
- L. V. Desai, K. J. Stowers and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 13285 CrossRef CAS .
- L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141 CrossRef CAS .
- T. Jintoku, H. Taniguchi and Y. Fujiwara, Chem. Lett., 1987, 1865 CrossRef CAS .
- X. Wan, Z. Ma, B. Li, K. Zhang, S. Cao, S. Zhang and Z. Shi, J. Am. Chem. Soc., 2006, 128, 7416 CrossRef CAS .
- D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Org. Lett., 2006, 8, 2523 CrossRef CAS .
- D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483 CrossRef CAS .
- K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS .
- T.-S. Mei, R. Giri, N. Maugel and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 5215 CrossRef CAS .
- J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS .
- K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342 CrossRef CAS .
- W. C. P. Tsang, N. Zheng and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560 CrossRef CAS .
- H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048 CrossRef CAS .
- K. Inamoto, T. Saito, M. Katsuno, T. Sakamoto and K. Hiroya, Org. Lett., 2007, 9, 2931 CrossRef CAS .
- S. A. Reed and M. C. White, J. Am. Chem. Soc., 2008, 130, 3316 CrossRef CAS .
- M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14058 CrossRef CAS .
- J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184 CrossRef CAS .
- L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542 CrossRef CAS .
- R. Giri, J. Liang, J.-G. Lei, J.-J. Li, D.-H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 7420 CrossRef CAS .
- D.-H. Wang, X.-S. Hao, D.-F. Wu and J.-Q. Yu, Org. Lett., 2006, 8, 3387 CrossRef CAS .
- B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS .
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS .
- D. Shabashov and O. Daugulis, Org. Lett., 2005, 7, 3657 CrossRef CAS .
- R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS .
- For another similar work which may not involve a Pd(IV) intermediate, see: M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 9886 Search PubMed .
- R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef .
- R. Giri, X. Chen, X.-S. Hao, J.-J. Li, J. Liang, Z.-P. Fan and J.-Q. Yu, Tetrahedron: Asymmetry, 2005, 16, 3502 CrossRef CAS .
- R. Giri, M. Wasa, S. P. Breazzano and J.-Q. Yu, Org. Lett., 2006, 8, 5685 CrossRef CAS .
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974 CrossRef CAS .
- E. N. Jacobsen, I. Marko, W. S. Mungall, G. Schroder and K. B. Sharpless, J. Am. Chem. Soc., 1988, 110, 1968 CrossRef CAS .
- J. E. Backvall, B. Akermark and S. O. Ljunggren, J. Am. Chem. Soc., 1979, 101, 2411 CrossRef .
- X. Lu, G. Zhu and Z. Wang, Synlett, 1998, 115 CrossRef .
- A. K. El-Qisairi, H. A. Qaseer, G. Katsigras, P. Lorenzi, U. Trivedi, S. Tracz, A. Hartman, J. A. Miller and P. M. Henry, Org. Lett., 2003, 5, 439 CrossRef CAS .
- M. R. Manzoni, T. P. Zabawa, D. Kasi and S. R. Chemler, Organometallics, 2004, 23, 5618 CrossRef CAS .
- A. Lei, X. Lu and G. Liu, Tetrahedron Lett., 2004, 45, 1785 CrossRef CAS .
- E. J. Alexanian, C. Lee and E. J. Sorensen, J. Am. Chem. Soc., 2005, 127, 7690 CrossRef CAS .
- J. Streuff, C. H. Hovelmann, M. Nieger and K. Muñiz, J. Am. Chem. Soc., 2005, 127, 14586 CrossRef CAS .
- K. Muñiz, J. Am. Chem. Soc., 2007, 129, 14542 CrossRef CAS .
- P. A. Sibbald and F. E. Michael, Org. Lett., 2009, 11, 1147 CrossRef CAS .
- K. Muñiz, C. H. Hovelmann and J. Streuff, J. Am. Chem. Soc., 2008, 130, 763 CrossRef CAS .
- G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2006, 128, 7179 CrossRef CAS .
- L. V. Desai and M. S. Sanford, Angew. Chem., Int. Ed., 2007, 46, 5737 CrossRef CAS .
- Y. Li, D. Song and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 2962 CrossRef CAS .
- A. M. M. Antunes, S. J. L. Marto, P. S. Branco, S. Prabhakar and A. M. Lobo, Chem. Commun., 2001, 405 RSC .
- A. M. M. Antunes, V. D. B. Bonifácio, S. C. C. Nascimento, A. M. Lobo, P. S. Branco and S. Prabhakar, Tetrahedron, 2007, 63, 7009 CrossRef CAS .
- L. L. Welbes, T. W. Lyons, K. A. Cychosz and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 5836 CrossRef CAS .
- X. Tong, M. Beller and M. K. Tse, J. Am. Chem. Soc., 2007, 129, 4906 CrossRef CAS .
- G. Yin and G. Liu, Angew. Chem., Int. Ed., 2008, 47, 5442 CrossRef CAS .
- D. Kalyani and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 2150 CrossRef CAS .
- For another carbon–heteroatom difunctionalization of alkenes appearing after submission of the manuscript, see: C. F. Rosewall, P. A. Sibbald, D. V. Liskin and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 9488 Search PubMed .
- S. Tang, P. Peng, S.-F. Pi, Y. Liang, N.-X. Wang and J.-H. Li, Org. Lett., 2008, 10, 1179 CrossRef CAS .
- S. Tang, P. Peng, Z.-Q. Wang, B.-X. Tang, C.-L. Deng, J.-H. Li, P. Zhong and N.-X. Wang, Org. Lett., 2008, 10, 1875 CrossRef CAS .
- After submission of the manuscript, the first example of asymmetric reaction through a Pd(IV) intermediate has appeared: T. Tsujihara, K. Takenaka, K. Onitsuka, M. Hatanaka and H. Sasai, J. Am. Chem. Soc., 2009, 131, 3452 Search PubMed .
- After submission of the manuscript, an interesting bimetallic Pd(III) catalysis for carbon–heteroatom bond formation was disclosed: D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 Search PubMed .
Footnote |
| † These authors equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.