Complexation of apigenin and luteolin in weld lake: a DFT/TDDFT investigation†
Anna
Amat
b,
Catia
Clementi
b,
Costanza
Miliani
a,
Aldo
Romani
b,
Antonio
Sgamellotti
ab and
Simona
Fantacci
*a
aIstituto di Scienze e Tecnologie Molecolari del CNR (CNR-ISTM), c/o Dipartimento di Chimica, Università degli Studi di Perugia, via Elce di Sotto 8, I-06123 Perugia, Italy. E-mail: simona@thch.unipg.it
bDipartimento di Chimica, Università degli Studi di Perugia, via Elce di Sotto 8, I-06123 Perugia, Italy
First published on 23rd April 2010
Abstract
A DFT-TDDFT investigation on the aluminium complexation of apigenin and luteolin has been carried out. We have focused our attention on these hydroxyflavonoids, which are the main components of weld, one of the earliest natural dyestuff used in art. In particular, weld, upon complexation with Al(III) forms a highly prized lake which has been widely used in medieval manuscripts and easel paintings for its rich yellow colour and transparency. The experimental spectra of apigenin and luteolin upon addition of increasing [Al3+] show a general red-shift of the lowest absorption bands of both flavonoids spectra, associated with the presence of two and three isosbestic points for apigenin and luteolin, respectively. The molecular geometries of all the Al-apigenin and -luteolin complexes have been optimized, followed by calculation of the formation Gibbs free energies and UV–vis absorption spectra. The comparison between the computed absorption spectra of the Al-flavonoid complexes and the experimental ones corresponding to various limit [Al3+] concentrations has been used to discriminate between the possible complexation modes as well as the stoichiometry ratio. We have thus been able to associate specific Al-apigenin (-luteolin) complexes with the experimental absorption spectra as a function of the [Al3+] concentration, thus providing insights into the aluminium complexation of these hydroxyflavonoids and most importantly into the weld lake composition.
Introduction
In recent years, chemistry has acquired a central role in the field of painting restoration and conservation. Thus, along with restorers and art historians, chemists have investigated the art materials which can be specific, depending on the painting techniques, on the historical preparation recipes and on degradation occurring throughout the ages.From the chemical point of view, organic pigments (also called lakes) represent a very interesting class among the various coloured materials used in the past. Lakes are synthetic metal–organic complexes obtained in antiquity by adding metal salts to dyestuffs solutions. Different combinations of natural dyes (extracted either from plants or insects) and metal cations [i.e. Al(III), Fe(II) and Sn(II)] offered to ancient masters a wide array of lake pigments that were highly prized for their rich colour and transparency even though more prone to degradation than inorganic pigments. As a consequence, many topics of interest within paintings conservation are related to an in-depth comprehension of the nature and composition of organic pigments, and the changes that have occurred over the centuries, which might have affected both the lake composition and chromatic properties. In this respect, physical chemistry offers a wide spectrum of experimental techniques, which go from steady-state1–3 and time-resolved4 fluorescence to SERS5,6 and advanced NMR methods,7 capable of analysing the painting materials thus providing information which was inaccessible only a few years ago.
Such experimental methods may benefit from the interplay with theoretical and computational chemistry, which in recent decades has had a substantial impact in many research fields thanks to higher performing computers and more efficient numerical algorithms. In particular, quantum chemical methods based on Density Functional Theory (DFT) and its Time-Dependent extension (TDDFT) have changed the scale of the investigated systems, allowing to accurately describe complex molecular systems or materials composed by up to a few hundred atoms.
In this paper, we exploit DFT/TDDFT methods along with the experimental physical chemical data, to find a relationship between the structural and colour properties of weld lake. The main objective of our investigation is to provide a picture of Al complexation that can help in comprehending the weld lake system in real conditions.
Weld, extracted from Reseda LuteolaL., is one of the oldest natural dyestuffs known in
Europe and it has been used since the beginning of the Christian Era to dye textiles, and later on as an organic pigment in medieval
manuscripts as well as in easel paintings. Its dyeing properties depend on the two main
components luteolin (Lu) and apigenin (Ap) hydroxyflavonoids (Scheme
1), that are generally present in a ratio of about
9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.8 According to ancient treaties weld
lake was prepared adding potash alum (aluminium potassium double sulfate) to an alkaline solution of the dyestuff until to reach the
neutrality.2 After precipitation of the Al-dye complex together with hydrated alumina, the
resulting yellow organic pigment was filtered, washed with distilled water, dried and finely ground in a mortar.
:1.8 According to ancient treaties weld
lake was prepared adding potash alum (aluminium potassium double sulfate) to an alkaline solution of the dyestuff until to reach the
neutrality.2 After precipitation of the Al-dye complex together with hydrated alumina, the
resulting yellow organic pigment was filtered, washed with distilled water, dried and finely ground in a mortar.
In a recent paper by Favaro et al.8 a study on the spectral changes which occur upon Al(III) chelation of apigenin and luteolin was reported. The experimental spectrum of apigenin upon addition of [Al3+] up to 1 × 10−3 mol dm−3 shows the formation and increase of a band at 382 nm leading to a more intense yellow colour. Minor spectral changes were detected in the high-energy region concerning the increase of the band at 275 nm. Luteolin, differing from apigenin only by the presence of a further hydroxyl group situated in position 3′ of phenyl ring B, presents a more complex evolution of the absorption spectrum with the increase of aluminium concentration revealing the appearance of a third band. Limit Al3+ concentrations have been determined to be 8 × 10−6, 5 × 10−5 and 3 × 10−4 mol dm−3 pointing to the formation of three different complexed species.
Based on these experimental results, the formation of 2(3) apigenin(luteolin) aluminium complexes has been detected even though the nature of these species in terms of chelating modes, involved sites and stoichiometry has not been determined. Therefore we investigate the complexation of both weld component dyes with Al(III) with Density Functional Theory (DFT) and Time-Dependent Functional Theory (TDDFT).
Several experimental8–17 and theoretical11–17 studies have been reported on flavonoid complexation with aluminium and with other metals,18–20 but to our knowledge, no theoretical investigation has been performed on the spectroscopic properties of Al(III)–Ap/Lu complexes. To shed light on the metal–dye complexation processes, all the possible Al(III)–apigenin(luteolin) complexes have been taken into consideration and scrutinized through their computed complexation Gibbs energies and by comparison between simulated and experimental spectra. While the thermodynamic analysis does not lead to clear conclusions, the simulation of the UV–vis spectra has revealed itself to be an efficient tool to discriminate between the possible complexation modes.
Computational methodology
Several Al3+–Ap/Lu complexation modes were investigated, considering different molecular sites with different Al:Ap/Lu stoichiometric ratios, i.e. 1:1,
1:2 and 2:1. The Al3+ cation can bind to both the keto and deprotonated hydroxy
oxygen atoms in a monodentate fashion; moreover the
presence of the geminal keto O4 and hydroxyl O5 opens the possibility of a bi-dentate chelation mode
for both flavonoids. Furthermore, for luteolin the
presence of an additional hydroxyl
group in the 3′ position allows for a further
bi-dentate complexation mode involving the geminal hydroxyl O3′ and O4′.
We have considered all the possible monodentate Al–Ap, Lu complexes with stoichiometry 1:1 and the bi-dentate
Al–Ap,Lu ones involving the O4
and O5 atoms with stoichiometries of 1:2 and
1:1. Moreover, for luteolin we have
taken into account: (i) the bi-dentate complexes involving the O3′ and
O4′ atoms with stoichiometric ratios 1:2 and
1:1; (ii) a mixed 1:2 Al:Lu bi-dentate complex
in which Al(III) binds to the
O4, O5 atoms of one luteolin and
O3′, O4′ of the other luteolin unit; and (iii) a binuclear complex in which two Al(III) cations chelate in a bi-dentate mode to the
O4, O5 and O3′, O4′ sites of
the same luteolin. Since our calculations on Al–Ap,Lu monodentate and bidentate complexes with
stoichiometry 1:1 pointed out the major stability of the bidentate
complexes with respect to the monodentate ones, see below, only the bidentate complexes
were considered for the stoichiometric ratios 1:2 and
2:1.
Given the large number of Al(III) complexes to be investigated, we labelled them according to the following nomenclature: AlnXyFkm, explanatory of the chelation mode. In particular, Al corresponds to the Al(H2O)x species, with x being the number of water molecules necessary to reach the octahedral geometry; F = Ap|Lu stands for the type of flavonoid considered; n and m are the number of metal and flavonoid units, respectively; X = M|B is the mono-|bi- dentate complexation modes, y refers to the site positions involved in the complexation and k is the charge of the complex.
Since we consider reactions in which a different number of metal centres, flavonoid units, water molecules, and protons can be involved, we computed Gibbs formation energies for the various possible complexes, to gain insight into the leading thermodynamically stable species in solution. In particular, we computed the Gibbs free energies of the complexation reactions in water using a procedure analogous to that employed for pKa calculations.21–22 We consider Al3+ as binding six water molecules to reach the octahedral geometry, Al(H2O)63+; this hypothesis is supported by the fact that the experimental spectra were measured in non-anhydrous methanol, and moreover the Gibbs energies of complexation reactions are computed in water.
The Gibbs free energy for the formation of one mole of the considered complexes was
estimated using the following relation:
| ΔGsolv = ΣGiPsolv − ΣGiRsolv |
| Gisolv = Givac + ΔGisolv |
Two different possible thermodynamical cycles which are commonly employed in the calculation of free energy differences involving deprotonation reactions were considered for the complexes formation energies. The two cycles consider different reaction products, i.e. dissociated proton and water, H+ and H2O, or associated H3O+, namely:
| nAl(H2O)63+ + mF → AlnXyFkm + (l − (3n − k))H2O + (3n − k)H3O+ | (1) |
| nAl(H2O)63+ + mF → AlnXyFkm + lH2O + (3n − k)H+ | (2) |
In cycle (i) the experimental value of −ΔGvap(H2O) is used as ΔGsolv(H2O) while the ΔGsolv of the other species is computed in water. In cycle (ii) the experimental values of −ΔGvap(H2O) as ΔGsolv(H2O) and Gsolv(H+) were used while the ΔGisolv of the other species is computed in water. The experimental ΔGsolv(H2O) was substituted by the experimental value of −ΔGvap(H2O) because it has been demonstrated to perform much better for reactions in water.23 We note that for a more detailed picture, reactions kinetics, evaluating the transition states along the reaction pathway, should be investigated,24 but this is beyond the aim of the present investigation.
DFT25 and its Time-Dependent extension TDDFT26–28 within the Gaussian03 (G03) program package29 were used to study the structural and optical properties of the Al complexes and their formation Gibbs energies. Calculations were performed using the hybrid functional B3LYP30−32 and the 6–31g** basis set.33–34 Solvation effects were included by means of the conductor-like polarisable continuum model CPCM35–38 using the UAHF solvation radii.39
Geometry optimizations with no symmetry constraints were performed both in vacuo, in methanol and in water solution and frequency calculations were carried out on the optimized structures in vacuo to check the nature of the stationary points and to evaluate the Gibbs free energy (Gvac). ΔGsolv values were computed on the optimized structures in water using as a reference the optimization in vacuo.
TDDFT calculations were performed on the optimized structures in methanol using the CPCM non-equilibrium version38 as implemented in G03. The lowest 40 or 60 singlet–singlet excitations were computed and transition energies and oscillator strengths were interpolated using a Gaussian convolution, with a σ = 0.20 or 0.15 depending on the investigated system. The computed absorption spectra of each complex were compared to the experimental spectra at the limit aluminium concentrations. These comparisons were used to discriminate the different investigated Al–Ap(Lu) complexes.
Results and discussion
Apigenin and luteolin absorption spectra
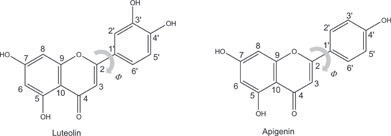
The calculated apigenin and luteolin absorption spectra in methanol solution are reported in Fig. 1, together with the frontier orbitals energy levels and the electronic distribution isodensity plots.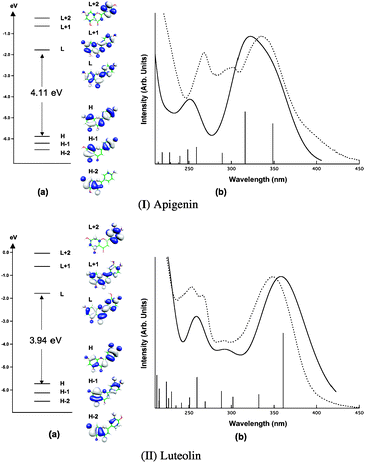 | ||
| Fig. 1 (a) Apigenin (top) and luteolin (bottom) frontier molecular orbitals energies and isodensity plots. (b) Comparison between the experimental (from ref. 8, dotted line) and the computed (solid line) absorption spectra of apigenin and luteolin. | ||
Both absorption spectra are very similar to those computed in water previously reported40 and will be therefore only be briefly discussed. The agreement between the computed UV–vis spectra and those experimentally measured in methanol solution is excellent for both species, reproducing the experimental red-shift of the luteolin spectrum with respect to the apigenin one. We have demonstrated that the inclusion of solvation effects and the use of hybrid xc functionals, i.e. B3LYP and PBE0, are mandatory for the quantitative reproduction of the experimental absorption spectra of these systems.40 The TDDFT methodology is therefore able to accurately describe the excited states of these compounds despite the known limitations of TDDFT in describing long-range charge-transfer excited states,41–43 or the systematic errors found within certain families of organic dyes.44
We are able to assign the lowest energy band for both apigenin and luteolin
absorption spectra as the result of the two π
→ π* HOMO(H) → LUMO(L) and H-1 → L transitions. Moreover, the
different spectral shape of the two experimental spectra
can be explained in terms of the different oscillator strengths ratio of the component
transitions, 0.34:0.44 for apigenin
and 0.49:0.12 for luteolin. The dim
signal of both the experimental spectra around 290 nm is
composed of an H-2 → L transition for apigenin and an H-2 →
L and a H-3 → L for luteolin, showing partial n → π* character.
Aluminium complexation of apigenin
The optimized molecular geometries of all the investigated Al3+–Ap complexes are reported in Fig. 2.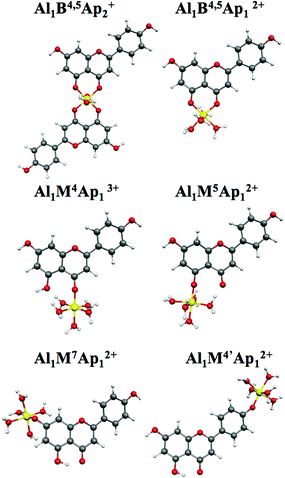 | ||
| Fig. 2 Optimized geometries of Al–Ap complexes | ||
The molecular geometries optimized in water and methanol are almost coincident and show only slight differences with respect to that computed in vacuo, similar to what was found for the non-complexed flavonoids.45 Therefore, we will refer only to the optimized geometries in methanol. For the Al1B4,5Ap2+ complex, we optimized the two apigenin units coplanar, with the four oxygen atoms in equatorial positions and two water molecules in the axial sites of the octahedral arrangement, see Fig. 2. On the other hand, for a similar complex with only the Al(III) centre without water molecules, the two apigenins were optimized orthogonally, assuming the metal a pseudo-tetrahedral coordination.10
Planarity of flavonoids has been extensively discussed in the literature11–15,45–48 and has been often related to the presence of a
hydroxyl
group in position 3, see Scheme
1.47
Apigenin and luteolin were shown to be non-planar, with dihedral angles between ring
B and the condensed A–C rings
(Φ) computed to be ca. 15° both in vacuo and in
methanol–water solution. The optimized geometries of the Al–Ap complexes involving the 4
and 5 positions show a decrease of this dihedral angle. In particular, for complexes
Al1M4Ap13+ and
Al1M5Ap12+, Φ is computed
to be 1.1° and 5.5°, respectively. For the bi-dentate complexes, the
Al1B4,5Ap2+ (stoichiometry
1:2) shows a Φ angle of 9.6°, larger than the
corresponding one in Al1B4,5Ap12+
(stoichiometry 1:1) which is 2.6°. On the other hand, the metal
complexation involving 7 and 4′ oxygens only slightly affects the
Φ angle, which is computed to be 15.5° and 12.2° for
Al1M7Ap12+ and
Al1M4′Ap12+, respectively.
Incidentally, the planarization of the Al–Ap complexes does not follow
the behaviour shown by the mono-deprotonated species in a previous study,45 where only deprotonation at position 4′ lead to a
planar species at the B3LYP/6-31+g* level of
calculation. Planarization is representative of an increase in conjugation and is therefore accompanied by a
C2C1′ bond decrease. Φ and the
C2C1′ bond seem to be the only parameters substantially
affected by the presence of the aluminium, not even the CyO distance, where y, the position where
the metal centre is bound, is changed after complexation. For all the monodentate
complexes the Al–O length is computed to be in the range 1.756–1.800
Å while for the Al1B4,5Ap2+ and the Al1B4,5Ap2+ complexes the distances Al–O5(O4) are computed to be
1.805(1.840) and 1.850(1.890) Å, respectively.
Some difficulties arose in the Al1M4Ap13+ and Al1M5Ap12+ complex optimization in vacuo. For Al1M4Ap13+ two of the five water molecules used to reach octahedral coordination get close to the oxygen in position 5. On the other hand, a similar but stronger effect is retrieved for Al1M5Ap12+ where the ketonic oxygen O4 leads to the detachment of one proton belonging to one of the water molecules coordinated by the aluminium centre. All the attempts to avoid this detachment were unsuccessful for in vacuo calculations and therefore we have not been able to compute Gvac for this species nor Gsolv.
In Table 1 the formation reactions together with the
computed ΔG values in water
following the procedures (i) and (ii) described above and ΔΔG, relative to the lower free energy reaction,
are reported for all the investigated Al–Ap species. The
Givac and
ΔGis computed
for all the reactants and products can be found in the ESI.†
Tunega et al.49 reported the difficulties in
obtaining realistic reaction enthalpies in solution for the interaction between the
acetate anion and
Al(H2O)63+. The same kind of conclusions can be
inferred from our calculations whatever procedure is used. In fact, reaction energies
are often endothermic and quite dependent on the employed methodology. However, it has
to be noted that complex multi-step reactions could occur in solution, and thus our
formation reactions are extremely simplified with a single-step picture and the
inclusion of the solvent by means of a continuum model.
We also evaluated ΔGin vacuo using the computed
Gvac, finding that the formation reactions are extremely
exothermic in these conditions (from −213 kcal mol−1 to
−135 kcal mol−1) and differ from the solvent case by up to 200 kcal mol−1, in line with the
enthalpy differences found for the acetate
complexes.49 Looking at Table
1, we retrieve a preference for the bi-dentate modes, with
Al1B4,5Ap2+ being the favoured species for
procedure (i), while procedure (ii) favours the stoichiometry 1:1 with
respect to 1:2. Overall, the results obtained seem to point to a
preference for a bi-dentate complexation, although the preferred stoichiometry ratio
cannot be discriminated so far. Moreover the small differences between the mono-dentate
complexes ΔΔGsolv computed with procedures (i) and
(ii) do not allow us to rule out the formation of these complexes. We have therefore
simulated the absorption spectra of all the considered
Al–Ap
complexes in an attempt to further discriminate between the various possible
complexation modes.
| Complex | Reaction | ΔGsolv | ΔΔGsolv |
|---|---|---|---|
| Al1B4,5Ap2+ | Al(H2O)63+ + 2Ap → Al1(H2O)2Ap2+ + 2H2O + 2H3O + (i) | −4.95 | 0.00 |
| Al(H2O)63+ + 2Ap → Al1(H2O)2Ap2+ + 4H2O + 2H +(ii) | 19.17 | 5.07 | |
| Al1B4,5Ap12+ | Al(H2O)63+ + Ap → Al1(H2O)4Ap12+ + H2O + H3O + (i) | 2.04 | 6.99 |
| Al(H2O)63+ + Ap → Al1(H2O)4Ap12+ + 2H2O + H +(ii) | 14.10 | 0.00 | |
| Al1M4Ap13+ | Al(H2O)63+ + Ap → Al1(H2O)5Ap13+ + H2O (i) | 23.93 | 28.88 |
| Al(H2O)63+ + Ap → Al1(H2O)5Ap13+ + H2O (ii) | 23.93 | 18.98 | |
| Al1M5Ap12+ | Al(H2O)63+ + Ap → Al1(H2O)5Ap12+ + H3O + (i) | — | — |
| Al(H2O)63+ + Ap → Al1(H2O)5Ap12+ + H2O + H +(ii) | — | — | |
| Al1M7Ap12+ | Al(H2O)63+ + Ap → Al1(H2O)5Ap12+ + H3O + (i) | 12.25 | 17.20 |
| Al(H2O)63+ + Ap → Al1(H2O)5Ap12+ + H2O + H +(ii) | 24.31 | 10.21 | |
| Al1M4′Ap12+ | Al(H2O)63+ + Ap → Al1(H2O)5Ap12+ + H3O + (i) | 7.47 | 12.42 |
| Al(H2O)63+ + Ap → Al1(H2O)5Ap12+ + H2O + H +(ii) | 19.53 | 5.43 |
The computed absorption spectra of the six complexes were compared to the experimental spectra at concentrations of 8 × 10−5 and 1 × 10−3 mol dm−3 for the first and second experimentally determined complexation steps, respectively, as reported in Fig. 3. The comparison has shown that the spectra of the mono-dentate complexes Al1M5Ap12+, Al1M7Ap12+ and Al1M4′Ap12+ are strongly blue-shifted and do not satisfactorily reproduce the experimental spectral features, while the computed spectrum of Al1M4Ap13+, showing only an intense narrow transition in the low-energy region, does not qualitatively reproduce the two-shoulder shape of the main experimental band, see Fig. 3.
![Experimental spectra at [Al3+] = 8 × 10−5 and 1 ×
10−3 and [Ap] = 1 ×
10−5 mol dm−3 from ref. 8 (dotted line). Theoretical spectra (solid line) of
Al1M4Ap13+,
Al1M5Ap12+,
Al1M7Ap12+ and
Al1M4′Ap12+.](/image/article/2010/CP/b925700d/b925700d-f3.gif) | ||
| Fig. 3 Experimental spectra at [Al3+] = 8 × 10−5 and 1 × 10−3 and [Ap] = 1 × 10−5 mol dm−3 from ref. 8 (dotted line). Theoretical spectra (solid line) of Al1M4Ap13+, Al1M5Ap12+, Al1M7Ap12+ and Al1M4′Ap12+. | ||
In contrast to the mono-dentate cases, the theoretical spectra of the two bi-dentate complexes are both in fair agreement with experiment, see Fig. 4, in particular the computed spectra of Al1B4,5Ap2+ and Al1B4,5Ap12+ agree well with those measured at [Al3+] = 8 × 10−5 mol dm−3 and 1 × 10−3 mol dm−3, respectively, suggesting the initial formation of a Al1B4,5Ap2+ complex at low [Al3+] concentrations followed by the formation of Al1B4,5Ap12+ at higher metal concentrations. The bi-dentate complexes show similar spectral features, both in terms of shape and absorption maxima. The main differences going from Al1B4,5Ap2+ to Al1B4,5Ap12+ spectra are: (a) the two overlapping lowest energy bands assume comparable intensity; (b) the intensity increase of the band computed at ca. 260 nm with respect to the feature at 300 nm, consistent with the changes observed experimentally with the increase of [Al3+] concentration, see Fig. 4.
![(a) Al1B4,5Ap2+ (top) and
Al1B4,5Ap12+ (bottom) frontier molecular
orbitals energies and isodensity plots in methanol. (b) Al1B4,5Ap2+
(top) and Al1B4,5Ap12+ (bottom) computed
(solid line) vs. experimental (dotted line) spectra at 1 × 10−5 mol dm−3
[Ap] and 8 × 10−5 (top)
and 1 × 10−3 (bottom) mol dm−3 [Al3+], from ref. 8.](/image/article/2010/CP/b925700d/b925700d-f4.gif) | ||
| Fig. 4 (a) Al1B4,5Ap2+ (top) and Al1B4,5Ap12+ (bottom) frontier molecular orbitals energies and isodensity plots in methanol. (b) Al1B4,5Ap2+ (top) and Al1B4,5Ap12+ (bottom) computed (solid line) vs. experimental (dotted line) spectra at 1 × 10−5 mol dm−3 [Ap] and 8 × 10−5 (top) and 1 × 10−3 (bottom) mol dm−3 [Al3+], from ref. 8. | ||
In Table 2 computed transitions, oscillator
strengths and involved molecular orbitals of
Al1B4,5Ap2+ and
Al1B4,5Ap12+ are reported. From the
analysis of the computed TDDFT eigenvectors we
found that the main computed transition energies are very similar for both complexes and
main differences are found in their oscillator strengths. In
Al1B4,5Ap12+ the two lowest
singlet–singlet excitations show comparable relative oscillator strengths
(0.39:0.45), while for
Al1B4,5Ap2+ the computed transition at 393
nm is significantly less intense than the one at 343 nm (0.45:1.11).
Moreover, a very small red-shift, of the second transition is computed for
Al1B4,5Ap12+(1:1) with
respect to Al1B4,5Ap2+(1:2),
353 nm vs. 343 nm, implying a slightly larger separation between the two lowest
bands. These changes are reflected in the absolute and relative intensity of the two
lowest bands, with a decrease of the second band which assumes a similar intensity to
the first one. In the higher energy region both experimental absorption spectra show two bands at 300 nm and 275 nm, computed at
ca. 300 nm and 260 nm: the former is constituted by a single transition of
oscillator strength 0.23 (0.08) for
Al1B4,5Ap2+
(Al1B4,5Ap12+), while the latter is
originated by two transitions which for
Al1B4,5Ap2+ are more separated in energy
and show comparable oscillator strengths with respect to the
Al1B4,5Ap12+ complex, see Table 2. All these changes translate into an increase of the
band at 260 nm in comparison with those at lower energies on going from stoichiometry
1:2 to 1:1, showing the same trend observed in the
experimental spectra, even though we are not able to
quantitatively reproduce the relative band intensity over the entire investigated
spectral range.
| Al1B4,5Ap2+ // [Al3+] = 8 × 10−5 mol dm−3; [Ap] = 1 × 10−5 mol dm−3 | ||||
|---|---|---|---|---|
| Exp.a max. (nm/eV) | Comp. max. (nm/eV) | Comp. trans. (nm/eV) | f | MO |
| a Experimental spectra from ref. 8. | ||||
| 382/3.25 | 393/3.15 | 393/3.15 (S1) | 0.54 | (61%) H→L |
| (27%) H-1→L + 1 | ||||
| 344/3.60 | 343/3.61 | 343/3.62 (S5) | 1.11 | (59%) H-3→L |
| (26%) H-2→L + 1 | ||||
| 301/4.12 | 299/4.15 | 299/4.15 (S9) | 0.23 | (58%) H-4→L |
| (17%) H-5→L+1 | ||||
| (10%) H→L + 2 | ||||
| (9%) H-1→L + 3 | ||||
| 275/4.51 | 261/4.75 | 268/4.63 (S18) | 0.29 | (40%) H→L + 2 |
| (10%) H-5→L+1 | ||||
| (10%) H-1→L + 3 | ||||
| (9%) H-3→L + 2 | ||||
| 256/4.85 (S21) | 0.29 | (36%) H-3→L + 2 | ||
| (30%) H-2→ L + 3 | ||||
| (8%) H-1→L + 3 | ||||
| Al1B4,5Ap12+ // [Al3+] = 1 × 10−3 mol dm−3; [Ap] = 1 × −5 mol dm−3 | ||||
|---|---|---|---|---|
| Exp.a max. (nm/eV) | Comp. max. (nm/eV) | Comp. trans. (nm/eV) | f | MO |
| 382/3.25 | 391/3.17 | 391/3.17 (S1) | 0.39 | (74%) H→L |
| (17%) H-1→L | ||||
| 347/3.52 | 353/3.51 | 353/3.51 (S2) | 0.45 | (75%) H-1→L |
| (14%) H→L | ||||
| 300/4.13 | 300/4.13 | 300/4.13 (S3) | 0.08 | (76%) H-2→L |
| (15%) H→ L + 1 | ||||
| 275/4.51 | 260/4.77 | 264/4.70 (S5) | 0.14 | (47%) H→L + 1 |
| (20%) H-1→L + 1 | ||||
| (12%) H-2→L | ||||
| 261/4.75 (S6) | 0.24 | (65%) H-1→L + 1 | ||
| (24%) H→L + 1 | ||||
To investigate the relation between the spectral modifications and the changes in the
electronic transitions, we analysed the involved molecular orbitals, see Table 2 and Fig. 4. The
Al1B4,5Ap2+ molecular orbitals are
degenerate pairs, with energy differences within 0.07 eV. These orbitals are completely
delocalized on both apigenin units, see Fig. 4. It is worth noting that the character of the frontier
molecular orbitals of both Al complexes is very
similar to those computed for isolated apigenin in
methanol, suggesting that the Al centre only slightly affects the electron density
distribution of the organic ligand. On the other hand. the H–L gap decreases from
4.11 eV, to 3.73 eV, and to 3.59 eV passing from Ap
to the Al1B4,5Ap2+ and e
Al1B4,5Ap12+ complexes, respectively,
rationalizing the spectral red-shift shown by the Al
complexes with respect to the apigenin case. Looking at
the composition of the two lowest transitions, we noticed that for
Al1B4,5Ap2+ only orbitals of one
degenerate pair are involved both in the starting and final states of each transition,
see Table 2; for
Al1B4,5Ap12+ these transitions originate
from orbitals of different character and energy, H
and H-1, while the L is maintained as final state. These changes in the character of the
electronic transition are probably responsible for the oscillator strength differences
discussed above going from a Al:Ap stoichiometry of 1:2 to 1:1.
We can therefore conclude that overall a good agreement with the experiment is
retrieved for both stoichiometries, transition energies are computed in excellent
agreement with the experimental bands while the relative intensities are partly
underestimated in the high-energy region. Moreover, we are also able to follow the
spectral changes upon Al3+ addition. The
lowest band of the Ap is red-shifted in the spectra of both Al–Ap complexes and arises as
a two-peak band, since the two transitions are more separated in energy. Our theoretical
findings which associate the Al1B4,5Ap2+
complex to low [Al3+] and the
Al1B4,5Ap12+ complex to high metal
concentrations are consistent with the experimental
data. Moreover, the hypothesis of the formation of the
Al1B4,5Ap2+ complex is supported by an
experimental study of mass spectrometry, where the
complexation of flavonoids by Al(III) is
used for their characterization, finding the Al–flavonoid 1:2
stoichiometry favoured with minimal competing products.10 In another experimental study, the Al–flavonoid complexes (with
flavonoids similar to apigenin and luteolin) were investigated by electrospray mass spectrometry and UV–vis spectrophotometry techniques, demonstrating that the complex formation is
dependent on the Al3+–flavonoid concentration ratio, on the solvent and on the flavonoids. In particular, it was found that for both
quercetin and kaempferol the dimeric Al–flavonoid
complexes predominantly form at low [Al3+] leaving the scene in favour of the monomeric species at
very high [Al3+], and that methanol favours the formation of dimers with respect to
other solvents.9
Aluminium complexation of luteolin
The same computational approach used for Al(III) complexation of apigenin was used to investigate luteolin complexation. For luteolin, the presence of the 3′,4′-dihydroxyl together with the 5-hydroxy-4-keto groups as chelating sites, allow the formation of six bi-dentate complexes, two for each complexation site with Al:Lu
1:2 and 1:1 stoichiometry, a 1:2
complexation involving the two different sites, 5-hydroxy-4-keto and 3′,4′-dihydroxyl, in each luteolin unit, and a bi-nuclear one involving both carboxylic and cathecolic
functionalities of the same luteolin. The optimized
geometries for all the investigated complexes are shown in Fig.
5, showing very similar results to the corresponding Al–Ap
complexes. In general, the bond distance Al–O is very similar in all complexes,
with a slight increase in the bi-dentate cases. Moreover, for
Al1B4,5Lu12+ and
Al1B4,5Lu2+ we computed longer
Al–O4 distances than Al–O5 ones, while for
Al1B3′,4′Lu2−
and Al1B3′,4′Lu1+
complexes both Al–O distances are very similar. Dihedral angles also reproduce
the behaviour observed for the corresponding Al–Ap complexes. The
bi-nuclear complex is retrieved almost planar with Φ = 1.2°, while
Al1B3′,4′Lu2−,
Al1B3′,4′Lu1+,
and Al1B4,5−3′,4′Lu1+
maintain the original non-planarity of luteolin,
with computed averaged angles of 12.3°, 14.4°, and 11.3°,
respectively.
 | ||
| Fig. 5 Optimized geometries of Al–Lu complexes. | ||
The same problem found for apigenin in optimizing the mono-dentate complex in position 5 was obersved also for luteolin, therefore ΔGs for this species was not computed. In Table 3 the formation reactions together with the computed ΔGsolv and ΔΔGsolv (relative to the favoured species) following the procedures (i) and (ii) are reported for all the Al–Lu species except for Al1M5Lu12+. The terms Gvac, ΔGsolv and Gsolv computed for all the reactants and products are reported in the ESI.†
| Complex | Reaction | ΔGsolv | ΔΔGsolv |
|---|---|---|---|
| Al1B4,5Lu2+ | Al(H2O)63+ + 2Lu → Al1(H2O)2Lu2+ + 2H2O + 2H3O+ (i) | −6.78 | 0.00 |
| Al(H2O)63+ + 2Lu → Al1(H2O)2Lu2+ + 4H2O + 2H+ (ii) | 17.34 | 2.83 | |
| Al1B3′,4′Lu2− | Al(H2O)63+ + 2Lu → Al1(H2O)2Lu2− + 4H3O+ (i) | −1.11 | 5.67 |
| Al(H2O)63+ + 2Lu → Al1(H2O)2Lu2− + 4H2O + 4H+ (ii) | 47.14 | 32.63 | |
| Al1B4,5−3′,4′Lu2 | Al(H2O)63+ + 2Lu → Al1(H2O)2Lu2 + H2O + 3H3O+ (i) | −4.27 | 2.51 |
| Al(H2O)63+ + 2Lu → Al1(H2O)2Lu2 + 4H2O + 3H+ (ii) | 31.92 | 17.40 | |
| Al1B4,5Lu12+ | Al(H2O)63+ + Lu → Al1(H2O)4Lu22+ + H2O + H3O+ (i) | 2.45 | 9.23 |
| Al(H2O)63+ + Lu → Al1(H2O)4Lu22+ + 2H2O + H+ (ii) | 14.51 | 0.00 | |
| Al1B3′,4′Lu1+ | Al(H2O)63+ + Lu → Al1(H2O)4Lu2+ + 2 H3O+ (i) | −1.52 | 5.26 |
| Al(H2O)63+ + Lu → Al1(H2O)4Lu2+ + 2H2O+2H+ (ii) | 22.60 | 8.09 | |
| Al2B4,5−3′,4′Lu13+ | 2Al(H2O)63+ + Lu → Al2(H2O)8Lu23+ + H2O + 3H3O+ (i) | 6.42 | 13.20 |
| 2Al(H2O)63+ + Lu → Al2(H2O)8Lu23+ + 4H2O + 3H+ (ii) | 42.61 | 28.10 | |
| Al1M4Lu13+ | Al(H2O)63+ + Lu → Al1(H2O)5Lu13+ + H2O (i) | 21.40 | 28.18 |
| Al(H2O)63+ + Lu → Al1(H2O)5Lu13+ + H2O (ii) | 21.40 | 6.88 | |
| Al1M5Lu12+ | Al(H2O)63+ + Lu → Al1(H2O)5Lu12+ + H3O+ (i) | — | — |
| Al(H2O)63+ + Lu → Al1(H2O)5Lu12+ + H2O + H+(ii) | — | — | |
| Al1M7Lu12+ | Al(H2O)63+ + Lu → Al1(H2O)5Lu12+ + H3O+ (i) | 12.92 | 19.70 |
| Al(H2O)63+ + Lu → Al1(H2O)5Lu12+ + H2O + H+(ii) | 24.98 | 10.47 | |
| Al1M3′Lu12+ | Al(H2O)63+ + Lu → Al1(H2O)5Lu12+ + H3O+ (i) | 15.18 | 21.97 |
| Al(H2O)63+ + Lu → Al1(H2O)5Lu12+ + H2O + H+ (ii) | 27.25 | 12.73 | |
| Al1M4′Lu12+ | Al(H2O)63+ + Lu → Al1(H2O)5Lu12+ + H3O+ (i) | 7.78 | 14.57 |
| Al(H2O)63+ + Lu → Al1(H2O)5Lu12+ + H2O + H+ (ii) | 19.84 | 5.33 |
We find for the Al–Lu complexes a similar situation to the apigenin case, where the bi-dentate complexes involving the 4-keto-5-hydroxy site are thermodynamically favoured. In
particular, the Al–Lu 1:2 or 1:1 stoichiometry is
preferred, depending on whether the (i) or (ii) procedure is employed, analogously to
the corresponding Al–Ap complexes.
For Al–Lu complexes the thermodynamics analysis is further complicated by the
presence of the bidentate complexes involving the 3′ and 4′ sites with
both 1:2 and 1:1 Al–Lu stoichiometry. Looking at
the ΔG values obtained within procedure (i) we found the bi-dentate
complexes, independently from the involved cathecolic or pseudo-carboxylic
functionalities, were thermodynamically favoured with respect to the mono-dentate
complexes. On the other hand, procedure (ii) provides discordant values, with all the
bi-dentate Al–Lu complexes chelating the O3′ and
O4′ atoms showing very high formation ΔG values. The
calculation of the formation Gibbs energies for the investigated Al–Lu complexes
does not allow us to delineate any definitive conclusion on which complexes are formed
upon [Al3+] addition, although
mono-dentate complexes are found again to be thermodynamically disfavoured with respect
to the bi-dentate ones.
We therefore resorted to the computed absorption spectra in methanol for all the studied Al–Lu complexes, and compared them to the experimental spectra at the following limit concentrations: 8 × 10−6, 5 × 10−5 and 3 × 10−4 mol dm−3, see Figs. 6 and 7. These concentrations indicate three different situations upon addition of aluminium, delimited by two isosbestic points, a fact that points out the formation of three possible complexes. The experimental spectrum at 8 × 10−6 mol dm−3 resembles that of the apigenin case, being only slightly red-shifted and with two overlapping bands at 402 and 355 nm. The second considered spectrum at [Al3+] = 5 × 10−5 mol dm−3 reproduces the same trend observed for apigenin, a decrease of the relative intensity of the peak at 355 nm with respect to the lowest energy one. In luteolin this change in relative intensity is more pronounced and leads to the almost complete disappearance of the second peak, although it remains as a dim shoulder at ca. 352 nm, to completely disappear upon further Al3+ addition. Indeed, at high aluminium concentrations the UV–vis spectra exhibit a main band which is red-shifted up to 426 nm pointing out the formation of a new species.
![Experimental spectra at [Al3+] = 8 ×10−6, 5 ×
10−5 and 3 × 10−4 and [Lu] = 2.7 × 10−5 mol
dm−3 (dotted line) from ref. 8. Theoretical spectra (solid line) of
Al1M4Lu13+,
Al1M5Lu12+,
Al1M7Lu12+,
Al1M3′Lu12+,
Al1M4′Lu12+,
Al1B3′,4′Lu2−,
Al1B3′,4′Lu1+
and Al1B4.5−3′,4′Lu2
complexes.](/image/article/2010/CP/b925700d/b925700d-f6.gif) | ||
| Fig. 6 Experimental spectra at [Al3+] = 8 ×10−6, 5 × 10−5 and 3 × 10−4 and [Lu] = 2.7 × 10−5 mol dm−3 (dotted line) from ref. 8. Theoretical spectra (solid line) of Al1M4Lu13+, Al1M5Lu12+, Al1M7Lu12+, Al1M3′Lu12+, Al1M4′Lu12+, Al1B3′,4′Lu2−, Al1B3′,4′Lu1+ and Al1B4.5−3′,4′Lu2 complexes. | ||
![(a) Al1B4,5Lu2+ (i),
Al1B4,5Lu12+ (ii) and
Al2B4,5−3′4′Lu13+
(iii) frontier molecular orbital energies and isodensity plots in
methanol. (b)
Al1B4,5Lu2+ (i),
Al1B4,5Lu12+ (ii), and
Al2B4,5−3′4′Lu13+
(iii) computed (solid line) vs. experimental (dotted line)
spectra from ref. 8 at 8 × 10−6 (i), 5 × 10−5 (ii), and 3 ×
10−4 (iii) mol
dm−3 [Al3+].](/image/article/2010/CP/b925700d/b925700d-f7.gif) | ||
| Fig. 7 (a) Al1B4,5Lu2+ (I), Al1B4,5Lu12+ (II) and Al2B4,5−3′4′Lu13+ (III) frontier molecular orbital energies and isodensity plots in methanol. (b) Al1B4,5Lu2+ (I), Al1B4,5Lu12+ (II), and Al2B4,5−3′4′Lu13+ (III) computed (solid line) vs. experimental (dotted line) spectra from ref. 8 at 8 × 10−6 (I), 5 × 10−5 (II), and 3 × 10−4 (III) mol dm−3 [Al3+]. | ||
Analogously to apigenin, almost all the computed
spectra of the mono-dentate complexes do not reproduce
the main experimental spectral features. The
Al1M5Lu12+,
Al1M7Lu12+,
Al1M3′Lu12+ and
Al1M4′Lu12+
spectra are similar, showing a single intense band at
ca. 360 nm, strongly blue-shifted with respect to experiment, see Fig. 6. On the other hand, the
Al1M4Lu13+ computed spectrum shows a main band at 412 nm and higher energy spectral features
compatible with the experimental spectra at high
[Al3+]. This resemblance, however,
seems a coincidence when considering that the reaction is strongly thermodynamically
disfavoured with procedure (i) and above all this complex seems not compatible with the
two formed complexes at low concentrations, as will be discussed below. Regarding the
bi-dentate complexes, on the one hand the computed spectrum of complex
Al1B3′,4′Lu1+ has
a single band in the low-energy region strongly red-shifted compared to experiment. On
the other hand, the
Al1B3′,4′Lu2−
and the
Al1B4,5−3′,4′Lu2
(Al:Lu stoichiometry 1:2) are consistent in the lower energy
range with the experimental spectra at medium and high
concentrations, respectively. We discard however the formation of these complexes since
it implies the loss of four and three protons,
respectively and because in Al3+ excess
the 1:2 complex stoichiometry is disfavoured.14
The main features of the experimental spectra at [Al3+] concentrations of 8 × 10−6, 5 × 10−5 and 3 × 10−4 mol dm−3, are fairly reproduced by the computed spectra of Al1B4,5Lu2+, Al1B4,5Lu12+ and Al2B4,5−3′,4′Lu13+ complexes, respectively, as shown in Fig. 7 and the maxima of the main experimental absorption bands8 together with the computed vertical excitation energies, oscillator strengths and composition in terms of the main orbitals are collected in Table 4.
| Al1B4,5Lu2+ // [Al3+] = 8 × 10−6 mol dm−3; [Lu] = 2.7 × 10−5 mol dm−3 | ||||
|---|---|---|---|---|
| Exp.a max. (nm/eV) | Comp. max. (nm/eV) | Comp. trans. (nm/eV) | f | MO |
| a Experimental spectra from ref. 8. | ||||
| 402/3.08 | 400/3.10 | 400/3.10 (S1) | 0.75 | (43%) H → L |
| (18%) H → L + 1 | ||||
| (8%) H-1 → L | ||||
| (7%) H-1 → L + 1 | ||||
| 355/3.49 | 360/3.44 | 360/3.44 (S6) | 0.55 | (26%) H-2 → L + 1 |
| (26%) H-2 → L | ||||
| (19%) H-3 → L | ||||
| (10%) H-1 → L + 1 | ||||
| 296/4.19 (sh) | 296/4.19 (S12) | 0.19 | (34%) H-7 → L | |
| (8%) H-5 → L | ||||
| (8%) H-4 → L + 1 | ||||
| 267/4.64 | 266/4.67 | 266/4.67 (S23) | 0.40 | (40%) H-2 → L + 3 |
| (38%) H-3 → L + 2 | ||||
| Al1B4,5Lu12+ // [Al3+] = 5 × 10−5 mol dm−3; [Lu] = 2.7 × 10−5 mol dm−3 | ||||
|---|---|---|---|---|
| Exp.a max. (nm/eV) | Comp. max. (nm/eV) | Comp. trans. (nm/eV) | f | MO |
| 402/3.08 | 404/3.07 | 404/3.07 (S1) | 0.55 | (75%) H → L |
| (13%) H-1 → L | ||||
| 352/4.92 (sh) | — | 370/3.35 (S2) | 0.11 | (80%) H-1 → L |
| (11%) H → L | ||||
| 328/3.78 (S3) | 0.14 | (90%) H-2 → L | ||
| 298/4.16 (sh) | — | 300/4.13 (S4) | 0.08 | (75%) H-3 → L |
| (12%) H → L + 1 | ||||
| 277/4.48 (S5) | 0.08 | (76%) H → L + 1 | ||
| (9%) H-1 → L + 1 | ||||
| 271/457 | 264/4.70 | 264/4.70 (S6) | 0.25 | (70%) H-1→L+1 |
| (7%) H-3 → L | ||||
| Al2B4,5−3′4′Lu13+ // [Al3+] = 3 × 10−4 mol dm−3; [Lu] = 2.7 × 10−5 mol dm−3 | ||||
|---|---|---|---|---|
| Exp.a max. (nm/eV) | Comp. max. (nm/eV) | Comp. trans. (nm/eV) | f | MO |
| 426/2.91 | 436/2.85 | 436/2.85 (S1) | 0.63 | (83%) H → L |
| 333/3.72 | 342/3.62 | 342/3.62 (S3) | 0.26 | (92%) H-2 → L |
| 296/4.19 (sh) | — | 295/4.20 (S5) | 0.09 | (56%) H-3 → L |
| (21%) H → L + 1 | ||||
| (14%) H-1 → L + 1 | ||||
| 273/4.54 | 265/4.68 (sh) | 265/4.68 (S6) | 0.23 | (60%) H-1 → L + 1 |
| (14%) H → L + 2 | ||||
| (7%) H-3 → L | ||||
For the first two concentrations a similar behaviour as in apigenin is retrieved except for a general spectral red-shift. The
experimental absorption spectrum at an Al3+ concentration of 8 ×
10−6 mol dm−3 agrees well with the one computed
for the Al1B4,5Lu2+ complex in terms of
bands position, in fact the first two experimental bands at 402 and 355 nm are computed
at 400 and 360 nm. The intensity of these two computed absorption bands is inverted with
respect to the experiment. This reversed intensity distribution has been observed also
when comparing the spectrum of
Al1B4,5Lu2+ with that of
Al1B4,5Ap2+. The spectrum at [Al3+] = 5 ×
10−5 mol dm−3 is consistent with the computed one
for Al1B4,5Lu12+, the bi-dentate complex
involving the same chelation site evidenced in apigenin and showing an Al:Lu stoichiometry
1:1. The computed spectrum is in good
agreement with the experimental one, in particular looking at the single transitions,
they reproduce the main spectral features even though their oscillator strength
distribution quantitatively does not fit the absorption band intensity. Compared to the
corresponding apigenin
absorption spectrum the two overlapping bands, see
Fig. 2, become a single band computed at 404 nm in
Al1B4,5Lu12+ reproducing the experimental
spectral differences observed for the two complexes. This change is due to a different
relative intensity and to an approach of the two first transitions computed for
Al1B4,5Lu12+ (0.55:0.11;
404:370 nm) with respect to
Al1B4,5Ap2+ (0.75:0.55;
400:360 nm). For an analogous flavonoid,
isoquercitrin (IsQ), upon Al3+ addition
the formation of Al complexes involving the
bi-dentate 4-keto-5-hydroxyl site has been found,
evidencing that at low [Al3+] the Al:IsQ 1:2
stoichiometry ratio is dominant, and is substituted by the corresponding complex with an
Al:IsQ
1:1 stoichiometry ratio upon increasing the [Al3+].9
We then compared the experimental spectrum at high
concentrations with that computed for the binuclear complex. In fact, at high [Al3+], apigenin and luteolin experimental
absorption spectra show a different shape bringing us
to think about a participation of a hydroxyl
group in position 3′ which is the only difference
between the two flavonoids. The computed
Al2B4,5−3′,4′Lu13+
spectrum is consistent with the experiment, with a
red-shift of the band at 426 nm (333 nm) by 0.10 eV (0.09 eV), within the accuracy of
the employed method. The absorption bands at higher energies are well reproduced by the
computed transitions, see Table 4, and differences in
the spectra are probably due to the oscillator strength
distribution and to the Gaussian convolution used being inadequate for this spectral
region. However, the experimental spectrum might not be
due to a single species but to an equilibrium between two complexes. If we admit the
presence of both Al1B4,5Lu12+and
Al2B4,5−3′,4′Lu13+
at high [Al3+], the resulting spectrum should be a superposition of the spectra computed for the two complexes with a weight depending on their
concentration. We simulate an average of the spectra
calculated for the mono- and bi-nuclear complexes assuming a 50![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) :50
distribution. Under this assumption, the calculated spectrum agrees well with the experimental spectrum at 3 × 10−4 mol dm−3,
showing the absorption maximum at 424 nm, compared to the experimental value of 426 nm
(see the ESI†). The presence of such an equilibrium would be
consistent with a previous study by Cornard et al.14 that proved for the similar IsQ the
presence of both the 1:1 and 2:1 complexes even at high
metal concentration. The only other computed spectra that
might be consistent with the experimental spectra at high
concentration are: the mono-dentate
Al1M4Ap12+ and the bi-dentate with
stoichiometry 1:2
Al1B3′,4′Lu2−,
but both complexes are anyway related to
Al1B4,5Lu12+, which should be the species
present in solution before a further amount of metal is added.
:50
distribution. Under this assumption, the calculated spectrum agrees well with the experimental spectrum at 3 × 10−4 mol dm−3,
showing the absorption maximum at 424 nm, compared to the experimental value of 426 nm
(see the ESI†). The presence of such an equilibrium would be
consistent with a previous study by Cornard et al.14 that proved for the similar IsQ the
presence of both the 1:1 and 2:1 complexes even at high
metal concentration. The only other computed spectra that
might be consistent with the experimental spectra at high
concentration are: the mono-dentate
Al1M4Ap12+ and the bi-dentate with
stoichiometry 1:2
Al1B3′,4′Lu2−,
but both complexes are anyway related to
Al1B4,5Lu12+, which should be the species
present in solution before a further amount of metal is added.
To find a rationale for the spectral changes going from Lu to Al–Lu complexes, we analyzed the character of the computed
transitions that give rise to the absorption bands in terms of involved molecular
orbitals as starting and arriving states. In particular, we notice for
Al1B4,5Lu2+ that the frontier orbitals
appear as almost degenerate pairs, whose single orbital electronic distribution is
mainly delocalized on each Lu unit, see Fig. 7. This situation is apparently quite different from the
apigenin case, but from an in depth analysis we find
that even though the frontier orbitals (see Figs. 4 and
7) are the result of different flavonoid
orbital combinations, the electronic transitions have very similar character for
corresponding Ap and Lu complexes. Regarding the stoichiometry ratio for the Al:Lu
1:1 complex, we found that the computed frontier molecular orbitals look
like those of uncomplexed luteolin, while the energy
pattern is changed, with the H and H-1 inverted with
respect to the luteolin. This has a strong impact on the
excitation character, in fact for
Al1B4,5Lu12+ the lowest computed
absorption band originates from two transitions mainly having as starting states
H and H-1, respectively, analogously to the
uncomplexed luteolin case even though the character
of the two lowest transitions is different. The Al
luteolin computed spectrum is red-shifted so
as to become the lowest transition in the
Al1B4,5Lu12+
spectrum. Interestingly, this transition can be
considered the spectral signature of the Al chelation of
the 4-keto-5-hydroxy functionality, being the lowest
transition of the three computed spectra of Fig. 7, showing essentially the same character even though
increasingly red-shifted going from
Al1B4,5Lu2+ to
Al2B4,5−3′,4′Lu13+.
The effect of the second Al complexation on the
site 3′,4′-dihydroxy has a further
stabilization effect on the original luteolin H-1,
that in the Al complexes has become the H. While the lowest absorption bands of the
Al1B4,5Lu2+ and
Al1B4,5Lu12+ complexes are broadened and
sub-structured, being originated by two or three transitions, in the binuclear complex
the lowest absorption band at 426 nm is the result of only one intense transition, and
it shows a narrow shape, consistent with experiment. The experimental feature at 333 nm,
computed at 342 nm whose intensity might be overestimated, is characteristic of the
Al chelation in the 3′,4′-dihydroxy site and involves as starting orbital the
H-3 of luteolin,
which is stabilized by the Al complexation.
Conclusions
Our DFT/TDDFT results, in a complementary way and integrated with the experimental data, provide an interpretative picture of the interaction mode between Al(III) and apigenin and luteolin, underlying the difference between the two flavonoids. Several possible Al–Ap and Al–Lu complexes have been taken into account, the thermodynamics of their formation reactions have been analysed and their UV–vis absorption spectra have been simulated with the aim to follow the changes of the spectral features associated with the formation of different complexes. The calculated thermodynamics do not allow us to draw definitive conclusions but only general trends on the preference for the bi-dentate mode involving the 4-keto-5-hydroxy site, probably due to the fact that also the kinetics and mechanistic aspects have a relevant role in the discrimination between the various Al complexes. On the other hand, the comparison between the simulated and experimental spectra allow us to associate different Al–flavonoid complexes with experimental spectra measured at different Al concentrations, suggesting which complexes form in solution and qualitatively at which Al:flavonoid concentration ratios.The information obtained from the comparison between the experimental8 and computed spectra, allow us to follow the Al(III) complexation of apigenin and luteolin. For apigenin, the favoured chelating mode is the bi-dentate one on
the 4-keto-5-hydroxy
group, providing two Al–Ap complexes,
Al1B4,5Ap2+ and
Al1B4,5Ap12+, whose spectra are consistent with those measured at [Al3+] = 8 × 10−5 mol
dm−3 and 1 × 10−3 mol dm−3,
respectively, suggesting the initial formation of the complex Al–Ap with 1:2
Al:Ap
stoichiometric ratio at low [Al3+] followed
by the formation of the 1:1 Al:Ap complex at higher metal
concentrations. For luteolin complexation three main
steps are indicated: (i) the formation of an Al complex
with luteolin chelated through the bi-dentate
4-keto-5-hydroxy site with a 1:2
Al:Lu stoichiometry at low [Al3+]; (ii) upon increasing the [Al3+], the 1:1 Al:Lu stoichiometry
becomes dominant and the formation of the
Al1B4,5Lu12+ complex is observed; (iii) at
high [Al3+] we suppose the chelation of the
Al1B4,5Lu12+ complex occurring on the
luteolin
3′,4′-dihydroxyl site providing a
binuclear complex, which might be in equilibrium with the
Al1B4,5Lu12+ complex.
The provided information on the complexation of apigenin and luteolin can be transferred to the weld lake. As already mentioned, weld lake was prepared by adding potash alum to an alkaline solution of the dyestuff. When alum is added to water, it dissociates to give trivalent Al3+ ions, which hydrate to form the hexa-aquo-aluminium(III) ion, Al(H2O)63+. In alkaline solution, Al(H2O)63+ undergoes a series of rapid hydrolytic reactions to form soluble monomeric and polymeric species, as well as Al(OH)3 in a solid phase. The dye molecules may be involved in the precipitation of hydrate alumina as co-precipitated Al complexes or may be adsorbed on the surface of the amorphous solid.2
The UV–vis reflectance spectrum of weld lake
exhibits a strong absorption in the visible spectral region with a single maximum at 410
nm.2 By comparing the spectral behavior of a flavonoid-based lake with that observed in solution for
luteolin and apigenin in the presence of Al(III) cations, it is clear that the absorption properties of
the pigment are determined by luteolin, which is the
main colored component of Reseda luteola L. Considering the computed spectra of the various Al–Lu complexes it can be argued
that during the precipitation of hydrate alumina, luteolin is preferentially co-precipitated or absorbed in a
bi-dentate mode involving the 4-keto-5-hydroxy site and
with a Al:Lu 1:1 stoichiometry.
The results of this Al complexation study, obtained by an alternative approach for the typical cultural heritage field investigations, are crucial in understanding several aspects not completely accessible to the experimental work, even though this investigation represents only the first step towards the comprehension of very complex materials.
Acknowledgements
We thank F. De Angelis for helpful discussions. This work was carried out within the joint research activities of CHARISMA (Cultural Heritage Advanced Research Infrastructures: Synergy for a Multidisciplinary Approach to Conservation–Restoration project No. 228330) a Combination of Collaborative Project and Coordination and Support Action for Integrating Activities supported by the 7th F.P. of the European Commission.References
- G. Favaro, C. Miliani, G. Romani and M. Vagnini, J. Chem. Soc., Perkin Trans. 2, 2002, 192 RSC.
- C. Clementi, B. Doherty, P. L. Gentili, C. Miliani, A. Romani, B. G. Brunetti and A. Sgamellotti, Appl. Phys. A: Mater. Sci. Process., 2008, 92, 25 CrossRef CAS.
- A. Claro, M. J. Melo, S. Schafer, J. S. Seixas de Melo, F. Pina, K. J. van den Berg and A. Burnstock, Talanta, 2008, 74, 922 CrossRef CAS.
- A. Romani, C. Clementi, C. Miliani, G. Favaro, B. Brunetti and A. Sgamellotti, Appl. Spectrosc., 2008, 62, 1395 CrossRef CAS.
- K. L. Wustholz, C. L. Brosseau, F. Casadio and R. P. Van Duyne, Phys. Chem. Chem. Phys., 2009, 11, 7350 RSC.
- Z. Jurasekova, C. Domingo, J. V. Garcia-Ramos and S. Sanchez-Cortes, J. Raman Spectrosc., 2008, 39, 1309 CrossRef CAS.
- P. Soubayrol, G. Dana and P. P. Man, Magn. Reson. Chem., 1996, 34, 638 CrossRef CAS.
- G. Favaro, C. Clementi, A. Romani and V. Vickackaite, J. Fluoresc., 2007, 17, 707 CrossRef CAS.
- H. T. Deng and G. J. Van Berkel, J. Mass Spectrom., 1998, 33, 1080 CrossRef CAS.
- J. Zhang, J. Wang and J. S. Brodbelt, J. Mass Spectrom., 2005, 40, 350 CrossRef CAS.
- J. P. Cornard, A. C. Boudet and J. C. Merlin, Spectrochim. Acta, Part A, 2001, 57, 591 CrossRef CAS.
- J. P. Cornard and J. C. Merlin, J. Mol. Struct., 2001, 569, 129 CrossRef CAS.
- J. P. Cornard and J. C. Merlin, J. Inorg. Biochem., 2002, 92, 19 CrossRef CAS.
- J. P. Cornard and J. C. Merlin, Polyhedron, 2002, 21, 2801 CrossRef CAS.
- J. P. Cornard and J. C. Merlin, J. Mol. Struct., 2003, 651–653, 381 CrossRef CAS.
- L. Dangleterre, J. P. Cornard and C. Lapouge, Polyhedron, 2008, 27, 1581 CrossRef CAS.
- A. C. Boudet, J. P. Cornard and J. C. Merlin, Spectrochim. Acta, Part A, 2000, 56, 829 CrossRef CAS.
- J. P. Cornard, L. Dangleterre and C. Lapouge, J. Phys. Chem. A, 2005, 109, 10044 CrossRef CAS.
- M. Leopoldini, N. Russo, S. Chiodo and M. Toscano, J. Agric. Food Chem., 2006, 54, 6343 CrossRef CAS.
- J. Ren, S. Meng, C. E. Lekka and E. Kaxiras, J. Phys. Chem. B, 2008, 112, 1845 CrossRef CAS.
- G. A. A. Saracino, R. Improta and V. Barone, Chem. Phys. Lett., 2003, 373, 411 CrossRef CAS.
- A. Amat, F. De Angelis, A. Sgamellotti and S. Fantacci, Chem. Phys. Lett., 2008, 462, 313 CrossRef CAS.
- M. D. Liptak, K. C. Gross, P. G. Seybold, S. Feldgus and G. C. Shields, J. Am. Chem. Soc., 2002, 124, 6421 CrossRef CAS.
- C. Lapouge and J. P. Cornard, ChemPhysChem, 2007, 8, 473 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr.T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS.
- M. Cossi and V. Barone, J. Phys. Chem. A, 2000, 104, 10614 CrossRef CAS.
- M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Chem. Phys., 1997, 107, 3210 CrossRef CAS.
- A. Amat, C. Clementi, F. De Angelis, A. Sgamellotti and S. Fantacci, J. Phys. Chem. A, 2009, 113, 15118 CrossRef CAS.
- R. J. Magyar and S. Tretiak, J. Chem. Theory Comput., 2007, 3, 976 CrossRef CAS.
- S. Grimme and M. Parac, ChemPhysChem, 2003, 4, 292 CrossRef CAS.
- A. Dreuw, J. L. Weisman and M. Head-Gordon, J. Chem. Phys., 2003, 119, 2943 CrossRef CAS.
- M. Guillaume, B. Champagne and F. Zutterman, J. Phys. Chem. A, 2006, 110, 13007 CrossRef CAS.
- A. Amat, F. De Angelis, A. Sgamellotti and S. Fantacci, THEOCHEM, 2008, 868, 12 CrossRef CAS.
- M. Leopoldini, I. Pitarch, N. Russo and M. Toscano, J. Phys. Chem. A, 2004, 108, 92 CrossRef CAS.
- S. van Acker, M. de Groot, D. J. van den Berg, M. Tromp, G. Donne-OpdenKelder, W. van der Vijgh and A. Bast, Chem. Res. Toxicol., 1996, 9, 1305 CrossRef CAS.
- J. C. del Valle, J. Chem. Phys., 2006, 124, 104506 CrossRef.
- D. Tunega, G. Haberhauer, M. Gerzabek and H. Lischka, J. Phys. Chem. A, 2000, 104, 6824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electronic supplementary information (ESI) available: Computed Givac and ΔGis for all the reactants and products. Experimental spectra at [Al3+] = 3 × 10−4 and [Lu] = 2.7 × 10−5 mol dm−3vs. theoretical spectra of Al1B3′,4′Lu1++Al2B4,5–3′,4′Lu13+ (50%–50%) complexes. See DOI: 10.1039/b925700d |
| This journal is © the Owner Societies 2010 |