Oxidation of methanol and formaldehyde to CO2 by a photocatalyst with an energy storage ability
Fei
Yang
,
Yukina
Takahashi
,
Nobuyuki
Sakai
and
Tetsu
Tatsuma
*
Institute of Industrial Science, University of Tokyo, Komaba, Meguro-ku, 153-8505, Tokyo, Japan. E-mail: tatsuma@iis.u-tokyo.ac.jp; Fax: +81-3-5452-6338; Tel: +81-3-5452-6336
First published on 1st April 2010
Abstract
A TiO2–Ni(OH)2 bilayer photocatalyst is known as a photocatalyst with energy storage abilities. Oxidative energy from the UV-irradiated TiO2 underlayer can be stored in the Ni(OH)2 overlayer. We investigated oxidation and mineralization of methanol and formaldehyde by the stored oxidative energy by mean of gas chromatography. When the methanol concentration in air is as low as 10 ppm, the mass conversion efficiency from methanol to CO2 is ∼86%. Formaldehyde can also be oxidized to CO2 by the stored energy.
1. Introduction
TiO2 photocatalyst,1 which drives oxidation and reduction reactions under UV light, has been applied to environmental remediation,2,3 self-cleaning coatings,2,3 self-sterilizing materials,3,4 anti-corrosion coatings5,6 and non-contact patterning (photocatalytic lithography).7 However, TiO2 photocatalyst functions only under light irradiation. To overcome this limitation, we have developed photocatalysts with reductive8–14 or oxidative15,16 energy storage abilities. The reductive energy of photocatalysts is stored in WO38–12 or MoO3,13 a redox-active n-type semiconductor, or H3PW12O40 (PWA),14 and the stored energy is utilized to keep anti-corrosion8 and bactericidal effects11 in the dark. The oxidative energy of photocatalysts is stored in Ni(OH)2,15,16 which is a redox-active p-type semiconductor.17,18Under UV light, electrons in the TiO2 valence band are excited to the conduction band, and holes generate in the valence band. In the case of the oxidative energy storage, Ni(OH)2 is oxidized to NiOx(OH)2−x (0 < x ≤ 1) by the holes, and it turns from colorless to brown.15 NiOx(OH)2−x is reduced back to Ni(OH)2 by some alcohols (methanol, ethanol and 2-propanol), aldehydes (formaldehyde and acetaldehyde), formate, acetone and phenol.15 The oxidation–reduction cycle is repeatable. Of those organic compounds, formaldehyde has been believed to be a cause of sick building syndrome.19 Therefore, its mineralization to CO2 and H2O is important to maintain indoor air quality. However, products of the formaldehyde oxidation by the stored energy are yet known.
In this work, therefore, we study oxidation of methanol and formaldehyde to CO2 by the oxidative energy stored in Ni(OH)2. Mass conversion and charge utilization efficiencies are also studied.
2. Experimental
2.1 Preparation of the bilayer film
An indium tin oxide (ITO) coated glass plate was cleaned by sonication in a detergent solution for 1 h, and further treated with 1 M aqueous NaOH for 1 h.A TiO2 underlayer of 4 cm × 4 cm was prepared on the ITO coated glass plate from a TiO2 sol (40 wt% sol, average particle size = 21 nm, STS-21, Ishihara Sangyo, Japan) that was diluted with water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume) by spin-coating at 1500 rpm for 10 s, and annealed at 400 °C for 1 h. It was kept under fluorescent black light (∼300–400 nm, ∼1 mW cm−2) before use to clean the surface for >12 h.
:1 by volume) by spin-coating at 1500 rpm for 10 s, and annealed at 400 °C for 1 h. It was kept under fluorescent black light (∼300–400 nm, ∼1 mW cm−2) before use to clean the surface for >12 h.
A Ni(OH)2 overlayer of 4 cm × 4 cm was prepared on the TiO2 coating by cathodic deposition from 0.02 M aqueous Ni(NO3)2 (pH 7) at 1 mA cm−2 for 210 s using a digital potentiostat (Model 263A, Princeton Applied Research, USA). A Ag|AgCl|sat. KCl electrode and a Pt wire were used as reference and counter electrodes, respectively.
2.2 Oxidative energy storage
Oxidative energy storage in Ni(OH)2 (i.e. oxidation of Ni(OH)2) was carried out in a 0.025 M carbonate buffer (pH 10). In the case of electrochemical storage, the TiO2–Ni(OH)2 bilayer-coated electrode was poised at +0.8 V vs. Ag|AgCl for 600 s. In the case of photoelectrochemical storage, the bilayer film was irradiated with UV light by using a Hg–Xe lamp (HB-25103BY, Ushio, Japan; light intensity, 30 mW cm−2).2.3 Characterization
Absorbance changes of the Ni(OH)2 films were measured by using a UV-Vis spectrophotometer (V-560, JASCO, Japan). For the present Ni(OH)2 films, the absorbance increase at 700 nm was in proportion to the stored charge (26.8 cm2 C−1), which was evaluated by a constant-current discharging at 0.5 μA cm−2 in the carbonate buffer. Therefore, the stored charge was calculated from the absorbance increase.Oxidation of methanol gas was carried out in the dark. After oxidative energy storage, the TiO2–Ni(OH)2 bilayer film was rinsed with water, dried and put into a 210 mL glass chamber, followed by flushing with clean air containing 39 ppm water for 5 min. Methanol was then injected into the chamber and mixed by a magnetic stirrer. The gas in the chamber was sampled (500 μL) every 30 min. The concentrations of methanol and CO2 were monitored by means of gas chromatography (GC) (GC-4000, GL Sciences, Japan) with a flame ionization detector (FID) equipped with a methanizer. N2 was employed as the carrier gas.
3. Results and discussion
3.1 Methanol oxidation by electrochemically stored energy
The TiO2–Ni(OH)2 bilayer film turned brown after electrochemical oxidation to NiOx(OH)2−x at +0.8 V vs. Ag|AgCl for 600 s (eqn (1)).| Ni(OH)2 + xOH− → NiOx(OH)2−x + xH2O + xe− | (1) |
| NiOx(OH)2−x + xH2O + xe− → Ni(OH)2 + xOH− | (2) |
| CH3OH + 6OH− → CO2 + 5H2O + 6e− | (3) |
 | ||
| Fig. 1 Time courses of (A) CO2 and methanol concentrations before (control) and after the electrochemical storage at +0.8 V vs. Ag|AgCl for 600 s and (B) the mass conversion efficiency from methanol to CO2 (=(ΔCCO2 − ΔCCont,CO2)/CIni,MeOH; see text for detail) calculated from (A). CIni,MeOH = 97 ppm. | ||
3.2 Photoelectrochemical storage
When the TiO2–Ni(OH)2 bilayer film was irradiated with UV light of 30 mW cm−2 in the carbonate buffer (pH 10), the film also turned from colorless to brown gradually, indicating that the film was charged.15,16 The increase of absorbance at 700 nm as a function of irradiation time is shown in Fig. 2. The absorbance reached a maximum value in 3 h. The quantum efficiency of oxidative energy storage (=the number of charges stored/the number of photons absorbed by TiO2 in the initial 1 h irradiation) was ∼0.086%. In our previous work,15 the efficiency was ∼0.40% at 10 mW cm−2 and ∼0.22% at 100 mW cm−2. The lower efficiency in the present work can be explained in terms of the greater Ni(OH)2 film thickness; the film was deposited at 1 mA cm−2 for 30 s in the previous work, whereas the deposition time was 210 s in the present work so as to maximize the storable charge. The maximum absorbance increase was about 50% of the absorbance increase for the 10 min electrochemical storage. There is a possibility that the rate of photoelectrochemical oxidation is balanced with the reduction by H2O2. It is known that H2O2 is one of the active oxygen species produced on UV-irradiated TiO2, and that H2O2 reduces oxidized Ni(OH)2.15,16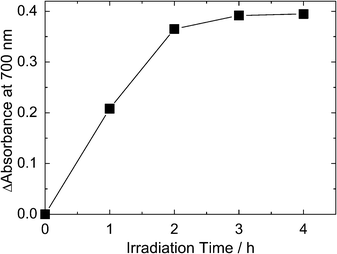 | ||
| Fig. 2 Oxidative coloration of the film by UV irradiation (light intensity: 30 mW cm−2) in the carbonate buffer (pH 10). | ||
3.3 Methanol oxidation by photoelectrochemically stored energy
After 4 h irradiation, the film was subjected to methanol oxidation (Fig. 3A). Again, the initial concentration was 97 ppm. The results were similar to those in the case of the electrochemical storage (Fig. 1A). The mass conversion efficiency to CO2 after 2 h reaction was 16 ± 1% (mean ± standard deviation, n = 4) (Fig. 3B). Charge used for the CO2 generation (QCO2), which was calculated from ΔCCO2 − ΔCCont,CO2, was 0.076 ± 0.006 C and the charge consumed during the reaction (QCons), which was evaluated from absorbance before and after the reaction, was 0.163 ± 0.013 C. Therefore, the charge utilization efficiency for CO2 generation (=QCO2/QCons) was 47 ± 3% or higher. The other 53% of the charge consumed should be used for oxidation of methanol to other products or adsorbed CO2/carbonate. If formaldehyde is assumed to be the other product, the mass conversion efficiency to it is 54%, and 30% of methanol is adsorbed on the film. If the other product is assumed to be formic acid, the mass conversion efficiency to it is 27%, and 57% of methanol is adsorbed on the film. | ||
| Fig. 3 Time courses of (A) CO2 and methanol concentrations before (control) and after the photoelectrochemical storage and (B) the mass conversion efficiency from methanol to CO2 (=(ΔCCO2 − ΔCCont,CO2)/CIni,MeOH; see text for detail) calculated from (A). CIni,MeOH = 97 ppm. | ||
When the initial concentration of methanol was decreased to 10 ppm, the mass conversion efficiency to CO2 increased to 86 ± 6% (n = 3) after 2 h reaction (Fig. 4). 14% of initial methanol is adsorbed on the film surface or oxidized to other products. The charge utilization efficiency (=QCO2/QCons) was 101 ± 27%. The large error was probably caused by small changes in the stored charges during the oxidation of methanol.
 | ||
| Fig. 4 Time courses of (A) CO2 and methanol concentrations before (control) and after the photoelectrochemical storage and (B) the mass conversion efficiency from methanol to CO2 (=(ΔCCO2 − ΔCCont,CO2)/CIni,MeOH; see text for detail) calculated from (A). CIni,MeOH = 10 ppm. | ||
Dependence of the mass conversion efficiency to CO2 on the initial methanol concentration was examined and plotted in Fig. 5 (filled circle). As the initial methanol concentration increases, the mass conversion efficiency decreases. On the other hand, the charge consumption ratio (=QCons/QStor; where QStor is the charge stored during the UV irradiation) increases with increasing initial methanol concentration. These results suggest that the mass conversion efficiency decreases as the charge consumption ratio increases. The potential of the TiO2–Ni(OH)2 bilayer film-coated electrode is about 0 V vs. Ag|AgCl in the pH 10 carbonate buffer before energy storage. It shifts positively to about +0.6 V after photoelectrochemical storage, so that methanol oxidation is possible. Although oxidation of methanol to CO2 is possible at a positive potential of +0.45 V vs. SCE,20 stored charges are consumed during the methanol oxidation and the potential shifts gradually in the negative direction, and eventually, the potential is not positive enough for methanol oxidation. Therefore, the mass conversion efficiency decreases if the stored charge is not sufficiently large in comparison with the amount of methanol. As described above, the charge utilization efficiency for CO2 generation also drops (i.e. reaction terminates before oxidation of intermediates to CO2) as the initial methanol concentration increases. This incomplete oxidation can also be explained in terms of the negative potential shift.
 | ||
| Fig. 5 Dependences of the mass conversion efficiency from methanol to CO2 (=(ΔCCO2 − ΔCCont,CO2)/CIni,MeOH; filled circle) and that from formaldehyde to CO2 (=(ΔCCO2 − ΔCCont,CO2 − CIni,MeOH)/CIni,HCHO; see text for detail; open circle) on the initial substrate (methanol or formaldehyde) concentration (CIni,Substrate). | ||
In the present system, no intermediary product21–24 such as formaldehyde, formic acid or methylformate was detected in the gas phase by GC-FID coupled with a methanizer even in the case where the initial methanol concentration was 97 ppm and the mass conversion efficiency from methanol to CO2 was only 20%. Here the lower detection limits for formaldehyde, formic acid and methylformate were <15, <16 and <10 ppm, respectively. It is possible that methanol, CO2/carbonate and/or intermediates stay on the surface or in the nanopores of the film. Hence, in a practical application, it is expected that the compounds entrapped on the film in the dark (e.g. in the night) are decomposed to CO2 when the film is irradiated again with UV light (e.g. in the morning).
3.4 Oxidation of formaldehyde
From a practical point of view, removal of methanol as well as formaldehyde is important because the latter is known to cause sick building syndrome.19 Since formaldehyde is easier to oxidize than methanol in general, it is reasonable to anticipate that formaldehyde is also oxidized to CO2 by the photoelectrochemically stored oxidative energy. So we examined oxidation of formaldehyde by vaporizing formalin containing methanol as a stabilizer for formaldehyde. The results obtained after 1 h reaction are summarized in Table 1. The mass conversion efficiencies from formaldehyde to CO2 are estimated on the assumption that the mass conversion efficiencies from the stabilizer (methanol) to CO2 are 100% and plotted in Fig. 5 (open circle). Although the values are slightly underestimated because the actual efficiencies from the stabilizer are less than 100%, the datapoints are in good agreement with the curve for methanol oxidation (Fig. 5, filled circle). Thus, we conclude that removal of formaldehyde is also possible by the oxidative energy stored in the TiO2–Ni(OH)2 bilayer photocatalyst (eqn (2) and (4)).| HCHO + 4OH− → CO2 + 3H2O + 4e− | (4) |
| C Ini,HCHO (ppm) | C Ini,MeOH (ppm) | ΔCCO2 − ΔCCont,CO2 (ppm) | C Fin,HCHO (ppm)b | C Fin,MeOH (ppm)c | Mass conversion efficiency from formaldehyde to CO2 (%)d |
|---|---|---|---|---|---|
| a Initial formaldehyde concentration. b Final formaldehyde concentration. c Final methanol concentration. d (ΔCCO2 − ΔCCont,CO2 − CIni,MeOH)/CIni,HCHO. | |||||
| 71 | 14 | 27 | 0 | 0 | 18 |
| 35 | 7 | 22 | 0 | 0 | 41 |
4. Conclusions
Methanol is oxidized to CO2 by oxidative energy stored in the TiO2–Ni(OH)2 bilayer photocatalyst. The mass conversion efficiency is ∼86% and the charge utilization efficiency for mineralization is ∼100% when the initial methanol concentration is 10 ppm. Formaldehyde is also oxidized to CO2 by the stored energy.Acknowledgements
We are grateful to Dr K. Kamata and Mr K. Yonehara for useful discussion regarding GC measurements. This work was supported in part by New Energy and Industrial Technology Development Organization (NEDO), Japan as a part of “Project to Create Photocatalyst Industry for Recycling-oriented Society” and Global COE Program “Chemistry Innovation through Cooperation of Science and Engineering”, MEXT, Japan.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CAS.
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, Electrochim. Acta, 2000, 45, 4683 CrossRef CAS.
- Y. Kikuchi, K. Sunada, T. Iyoda, K. Hashimoto and A. Fujishima, J. Photochem. Photobiol., A, 1997, 106, 51 CrossRef CAS.
- J. N. Yuan and S. Tsujikawa, J. Electrochem. Soc., 1995, 142, 3444 CAS.
- Y. Ohko, S. Saitoh, T. Tatsuma and A. Fujishima, J. Electrochem. Soc., 2001, 148, B24 CrossRef CAS.
- W. Kubo, T. Tatsuma, A. Fujishima and H. Kobayashi, J. Phys. Chem. B, 2004, 108, 3005 CrossRef CAS.
- T. Tatsuma, S. Saitoh, Y. Ohko and A. Fujishima, Chem. Mater., 2001, 13, 2838 CrossRef CAS.
- T. Tatsuma, S. Saitoh, P. Ngaotrakanwiwat, Y. Ohko and A. Fujishima, Langmuir, 2002, 18, 7777 CrossRef CAS.
- P. Ngaotrakanwiwat, T. Tatsuma, S. Saitoh, Y. Ohko and A. Fujishima, Phys. Chem. Chem. Phys., 2003, 5, 3234 RSC.
- T. Tatsuma, S. Takeda, S. Saitoh, Y. Ohko and A. Fujishima, Electrochem. Commun., 2003, 5, 793 CrossRef CAS.
- Y. Takahashi and T. Tatsuma, Electrochem. Commun., 2008, 10, 1404 CrossRef CAS.
- Y. Takahashi, P. Ngaotrakanwiwat and T. Tatsuma, Electrochim. Acta, 2004, 49, 2025 CrossRef CAS.
- P. Ngaotrakanwiwat, S. Saitoh, Y. Ohko, T. Tatsuma and A. Fujishima, J. Electrochem. Soc., 2003, 150, A1405 CrossRef CAS.
- Y. Takahashi and T. Tatsuma, Langmuir, 2005, 21, 12357 CrossRef CAS.
- Y. Takahashi and T. Tatsuma, Phys. Chem. Chem. Phys., 2006, 8, 2716 RSC.
- M. K. Carpenter and D. A. Corrigan, J. Electrochem. Soc., 1989, 136, 1022 CAS.
- C. J. Zhao, Y. Gu, H. Chen and Z. Y. Jiang, Electrochim. Acta, 2002, 47, 1801 CrossRef CAS.
- T. Godish, Sick Building: Definition, Diagnosis and Mitigation, Lewis Publishers-CRC Press, Boca Raton, 1994 Search PubMed.
- A. Kowal, S. N. Port and R. J. Nichols, Catal. Today, 1997, 38, 483 CrossRef CAS.
- M. W. Breiter, Electrochim. Acta, 1967, 12, 1213 CrossRef CAS.
- T. Iwasita, Electrochim. Acta, 2002, 47, 3663 CrossRef CAS.
- G. T. Burstein, C. J. Barnett, A. R. Kucernak and K. R. Williams, Catal. Today, 1997, 38, 425 CrossRef CAS.
- K. Otsuka, I. Yamanaka and K. Suga, Chem. Lett., 1987, 1087 Search PubMed.
| This journal is © the Owner Societies 2010 |