Postsynthetic diazeniumdiolate formation and NO release from MOFs†
Joseph G.
Nguyen
,
Kristine K.
Tanabe
and
Seth M.
Cohen
*
Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093-0358, USA. E-mail: scohen@ucsd.edu; Fax: +1 858 822 5598; Tel: +1 858 822 5596
First published on 10th April 2010
Abstract
Two different metal–organic frameworks (MOFs) have been modified with nitric oxide (NO) through covalent postsynthetic modification (PSM) to form diazeniumdiolate-functionalized and releasing MOFs.
Metal–organic frameworks (MOFs) represent a relatively new class of hybrid inorganic–organic porous materials that have garnered an immense amount of attention because of their chemical tunability, high porosities, and good thermal stability.1 Because of the unique combination of these properties, these materials are appealing for applications in gas adsorption,2 separations,3 and catalysis.4,5 The application of MOFs in the biotechnology and biomedical arena has only recently been explored.6–15 Traditional MOF synthetic methods have limited the ability to produce materials with suitable stability and desired functionality for biotechnology uses. Postsynthetic modification (PSM) of MOFs is a general, practical approach for incorporating a wide range of functional groups into MOFs.16 We17–20 and others21–27 have shown that a diverse set of MOFs can be customized by PSM with many different functional groups; thus, PSM provides a unique avenue to produce MOFs relevant for biomedical applications.
One function that is not easily available through traditional MOF syntheses is the preparation of nitric oxide (NO) donors, which have the capacity to release NO for therapeutic applications. Nitric oxide is an important physiological mediator involved in cellular signaling,28–30 and its delivery from a storage material is attractive for many anti-bacterial, anti-thrombic, and wound healing applications.31–34 Currently, all the NO-releasing porous materials, including zeolites and MOFs, are prepared using non-covalent interactions (e.g. NO coordination to a metal ion).9,15,35–37 Unfortunately, these materials show limited control over the rate of NO release. One way to circumvent this issue is to incorporate diazeniumdiolate (NONOate) functional groups within the MOF. NONOate functional groups (Fig. 1) are attractive for NO release because: (i) they decompose at physiological pH and temperature, generating up to two molar equivalents of NO and (ii) the rate of NO release can be controlled, which can facilitate the targeting of NO to specific sites of interest.38,39
 | ||
| Fig. 1 The NONOate functional group and mechanism of release. Through variation of the R-groups the rate of NO release can be controlled. | ||
In an attempt to exploit these advantageous features, Rosseinsky and coworkers synthesized a NONOate-functionalized MOF through a combination of non-covalent and covalent PSM.8 In this report, 4-(methylamino)-pyridine (4-map) was coordinated to the open metal sites in the MOF HKUST-1 (non-covalent PSM), which was then subsequently converted to a NONOate functional group upon exposure to NO (covalent PSM). Significant leaching of 4-map occurred when the NONOate-functionalized MOF was suspended in water, thus presenting one shortcoming of the non-covalent approach, and thereby curtailing some of the advantages of NONOates in this platform.
A better way to control NO release from NONOate-modified MOFs is to functionalize them through only covalent PSM because R1 and R2 can be chosen appropriately to be part of the framework ligand. Two different MOFs containing pendant amines, IRMOF-340 and UMCM-1–NH2,20 were successfully transformed with NO (100 psi, 24 h) into two new MOFs designated IRMOF-3–NONO and UMCM-1–NONO, respectively (Scheme 1). The NONOate functionalities were confirmed utilizing electronic and Fourier transform infrared (FT-IR) spectroscopy. Electronic spectroscopy is the most widely used method for identification of NONOates;39 diffuse reflectance solid-state spectroscopy on crystalline samples of IRMOF-3–NONO provides a characteristic signal for the formation of the NONOate functionality. Aromatic NONOates generally display absorption maxima between 285 and 320 nm.41,42 The diffuse reflectance solid-state UV-Vis spectrum for IRMOF-3–NONO shows a broad absorption between 270 and 290 nm, in contrast to the same spectral region for IRMOF-3 (Fig. 2). A similar difference in features is observed in the diffuse reflectance solid-state UV-Vis spectra for the respective ligands, NONO–BDC and NH2–BDC (NH2–BDC = 2-amino-1,4-benzenedicarboxylic acid, Fig. S1A†). This same feature is not apparent in UMCM-1–NONO perhaps because it has fewer pendant amine groups per secondary building unit (Fig. S1B†). There is also a broad absorption above 400 nm unique to both NO-modified MOFs, but the origin of this band is presently unknown.
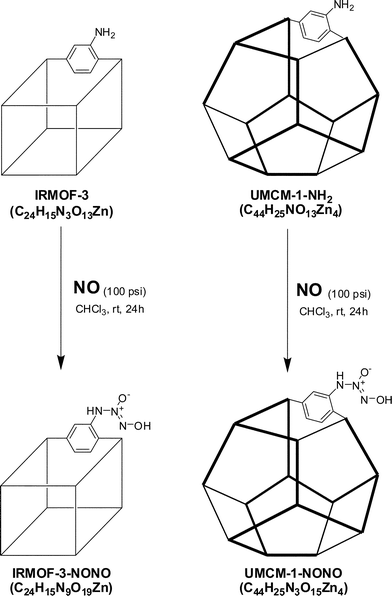 | ||
| Scheme 1 Synthesis of IRMOF-3–NONO (left) and UMCM-1–NONO (right). | ||
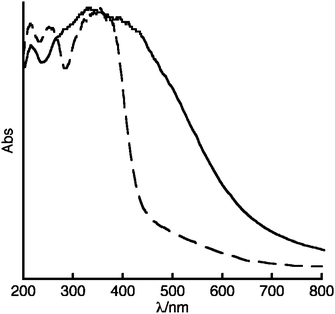 | ||
Fig. 2 Diffuse reflectance solid-state UV-Vis spectra of IRMOF-3–NONO (—) and IRMOF-3 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ). ). | ||
Further evidence for formation of the NONOate species was provided by the appearance of new stretching frequencies in the FT-IR spectra for both modified MOFs. The stretching frequencies at 1040–1043 cm−1 (N–O), 1310–1320 cm−1 (N–O), and 1493–1505 cm−1 (N–N) are characteristic of NONOates (Fig. 3 and S2†).39 There is also an additional stretching band at 2270–2275 cm−1 present in all NO-modified compounds, which is most likely due to an N–N stretch. NO release from these materials (vide infra) only occurs when this new band is present. While it is known that amines can react with NO+ to form azides,43 the 2270–2275 cm−1 stretching frequency is too high for aromatic azides.44 It was also observed that after NO-modification the resulting MOFs are much more readily dissolved in phosphate buffer solution (pH ≈ 7.4), resulting in an orange-colored solution with a small amount of white precipitate. When both IRMOF-3 and UMCM-1–NH2 are placed in phosphate buffer solution, a large amount of white precipitate is formed in a colorless solution. Thus, this change in the solubility property suggests that MOF modification has occurred.
 | ||
| Fig. 3 FT-IR spectra of IRMOF-3 () and IRMOF-3–NONO (—). | ||
The modified MOFs visually show no signs of degradation after treatment with NO (Fig. S3†). Powder X-ray diffraction (PXRD) patterns also show that the crystallinity of the bulk materials is generally preserved (Fig. S4†). Crystallinity was further confirmed by using single crystal X-ray diffraction analysis, which showed that for both IRMOF-3–NONO and UMCM-1–NONO cell parameters identical to those of the parent material are obtained (Table 1). The MOFs were also examined by thermal gravimetric analysis (TGA) to determine the thermal and structural stability of the materials. The modified MOFs showed good thermal stability, with decomposition temperatures around 450 °C (Fig. S5†). A weight loss observed between 270 and 360 °C is likely due to the NONOate functionality (Fig. S6†). IRMOF-3–NONO displays an 8% weight loss (theoretical 18% for complete conversion of all amines to NONOates), which suggests that 44% of the free amines have been modified. UMCM-1–NONO displays a 5% weight loss (6% theoretical weight loss), which implies modification of 83% of the amines. The Brunauer–Emmett–Teller (BET) surface areas of the modified samples were measured via dinitrogen adsorption at 77 K. The modified samples showed BET surface areas of 1294 m2 g−1 and 2560 m2 g−1 for IRMOF-3–NONO and UMCM-1–NONO, respectively (Fig. S7†). The surface areas are significantly reduced relative to the parent MOF (∼2400 m2 g−1 for IRMOF-3 and ∼4000 m2 g−1 for UMCM-1–NH2). While the BET surface areas are still far in excess of typical zeolites, the reduction is larger than expected for this modification, and is likely due to some degradation of the MOFs during PSM.
| MOF | Cell setting | a = b = c/Å | α = β = γ/° | Volume/Å3 |
|---|---|---|---|---|
| IRMOF-3 | Cubic F | 25.7465 | 90 | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 066 066 |
| IRMOF-3–NONO | Cubic F | 25.76 | 90 | 17092 |
| UMCM-1–NH2 | Hexagonal | 41.2555 | 90 | 25808.2 |
| 17.5091 | 120 | |||
| UMCM-1–NONO | Hexagonal | 41.49 | 90 | 26172 |
| 17.56 | 120 |
NO release studies in aqueous media showed that the observed NONOate-modification is unique to MOFs containing pendant amines. Approximately 10 mg of the NONOate MOF samples were placed in a 0.01 M phosphate buffer (pH ≈ 7.4). In this buffer the MOFs rapidly decompose (almost complete dissolution in 5 min), and the amount of NO released in these studies was monitored by determining the nitrite concentration with the commonly used Griess assay.45,46 IRMOF-3–NONO releases 0.51 ± 0.11 mmol NO per g of material, and UMCM-1–NONO releases 0.10 ± 0.01 mmol NO per g of material (Fig. 4). The release data are equivalent to only 8% of the amine sites releasing NO in IRMOF-3–NONO and 6% for UMCM-1–NONO. These values are much lower than the degree of conversion and potential amounts of NO for release estimated by TGA analysis. Part of the discrepancy between the TGA and NO release may be due to limitations in quantifying the TGA data, making it difficult to accurately estimate the extent of modification. In addition, complete release of the NONOate functionality might not be achieved or the Griess assay conditions may be contributing towards the loss of NO before it can be detected. Although the extent of the modification and maximum release is unclear, our findings demonstrate that NO release only occurs from amine-containing MOFs. Structurally identical MOFs, IRMOF-140 and UMCM-1,47 which lack the pendant amine groups, show no release of NO when treated under identical conditions. IRMOF-3–NONO and UMCM-1–NONO release a relatively high amount of NO compared to some NO-releasing materials. IRMOF-3–NONO releases 250 times more NO than HKUST-1 (0.002 mmol NO per g of material),15 but 3.5 times less than the highest NO-releasing MOF or zeolite porous materials (7 mmol NO per g of material).9 The NONOate functionality in IRMOF-3–NONO and UMCM-1–NONO shows some instability, as a 20% decrease in NO release is observed when the samples are tested 10 days after the initial release study (Fig. S8†). This instability is not surprising as the NONOate functionality within the MOF may be in its protonated form, which is more hygroscopic and less stable than the sodium salt analogs.38,39 Despite the limited stability of the NONOate functionality in these materials, our findings are significant because they show that amine-containing MOFs can be covalently modified to form NO-releasing materials.
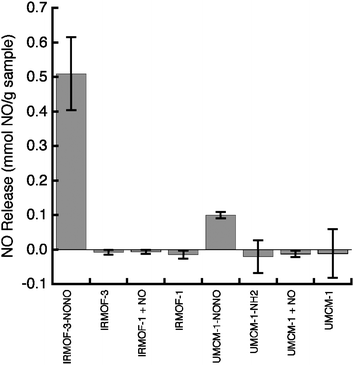 | ||
| Fig. 4 Release of NO from IRMOF-3–NONO, UMCM-1–NONO, and different control systems using the Griess assay. The amount of NO release (with standard deviations) is an average of at least three independent experiments. | ||
In conclusion, we have successfully developed two new NONOate-functionalized MOFs using covalent PSM. The NONOate functional group would not be attainable in a MOF by traditional solvothermal synthesis. More importantly, NO-release studies show that NONOate modification is specific to MOFs containing the NH2–BDC ligand and that large amounts of NO can be liberated. Overall, these results show that covalent PSM provides an excellent avenue for the production of NO-releasing MOFs.
Acknowledgements
This work was supported by the National Science Foundation (instrumentation grants CHE-9709183, CHE-0116662, and CHE-0741968) and the Department of Energy (DE-FG02-08ER46519). Joseph G. Nguyen was supported by an NIH Training Grant (5T32DK007233).References
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS.
- M. J. Ingleson, R. Heck, J. A. Gould and M. J. Rosseinsky, Inorg. Chem., 2009, 48, 9986–9988 CrossRef CAS.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440–10444 CrossRef CAS.
- W. J. Rieter, K. M. Pott, K. M. L. Taylor and W. Lin, J. Am. Chem. Soc., 2008, 130, 11584–11585 CrossRef CAS.
- W. J. Rieter, K. M. L. Taylor, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2006, 128, 9024–9025 CrossRef CAS.
- W. J. Rieter, K. M. L. Taylor and W. Lin, J. Am. Chem. Soc., 2007, 129, 9852–9853 CrossRef CAS.
- K. M. L. Taylor, A. Jin and W. Lin, Angew. Chem., Int. Ed., 2008, 47, 7722–7725 CrossRef CAS.
- K. M. L. Taylor, W. J. Rieter and W. Lin, J. Am. Chem. Soc., 2008, 130, 14358–14359 CrossRef CAS.
- B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203–1209 CrossRef CAS.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- E. Dugan, Z. Wang, M. Okamura, A. Medina and S. M. Cohen, Chem. Commun., 2008, 3366–3368 RSC.
- S. J. Garibay, Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 7341–7349 CrossRef CAS.
- K. K. Tanabe, Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2008, 130, 8508–8517 CrossRef CAS.
- Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 296–306 CrossRef CAS.
- T. Ahnfeldt, D. Gunzelmann, T. Loiseau, D. Hirsemann, J. Senker, G. Ferey and N. Stock, Inorg. Chem., 2009, 48, 3057–3064 CrossRef CAS.
- J. S. Costa, P. Gamez, C. A. Black, O. Roubeau, S. J. Teat and J. Reedijk, Eur. J. Inorg. Chem., 2008, 1551–1554 CrossRef CAS.
- C. J. Doonan, W. Morris, H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 9492–9493 CrossRef CAS.
- T. Gadzikwa, O. K. Farha, K. L. Mulfort, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2009, 3720–3722 RSC.
- M. J. Ingleson, J. P. Barrio, J.-B. Guilbaud, Y. Z. Khimyak and M. J. Rosseinsky, Chem. Commun., 2008, 2680–2682 RSC.
- T. Kawamichi, T. Kodama, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2008, 47, 8030–8032 CrossRef CAS.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS.
- Nitric Oxide: Biology and Pathobiology, ed. L. J. Ignarro, Academic Press, San Diego, 2000 Search PubMed.
- S. Y. Low, Mol. Aspects Med., 2005, 26, 97–138 CrossRef CAS.
- S. Moncada and E. A. Higgs, Br. J. Pharmacol., 2006, 147, S193–S201 CrossRef CAS.
- M. C. Frost, M. M. Reynolds and M. E. Meyerhoff, Biomaterials, 2005, 26, 1685–1693 CrossRef CAS.
- E. M. Hetrick, J. H. Shin, N. A. Stasko, C. B. Johnson, D. A. Wespe, E. Holmuhamedov and M. H. Schoenfisch, ACS Nano, 2008, 2, 235–246 CrossRef CAS.
- M. R. Miller and I. L. Megson, Br. J. Pharmacol., 2007, 151, 305–321 CrossRef CAS.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS.
- F. Bonino, S. Chavan, J. G. Vitillo, E. Groppo, G. Agostini, C. Lamberti, P. D. C. Dietzel, C. Prestipino and S. Bordiga, Chem. Mater., 2008, 20, 4957–4968 CrossRef CAS.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef.
- P. S. Wheatley, A. R. Butler, M. S. Crane, S. Fox, B. Xiao, A. G. Rossi, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2006, 128, 502–509 CrossRef CAS.
- L. K. Keefer, Annu. Rev. Pharmacol. Toxicol., 2003, 43, 585–607 CrossRef CAS.
- J. A. Hrabie and L. K. Keefer, Chem. Rev., 2002, 102, 1135–1154 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef.
- L. K. Keefer, J. L. Flippen-Anderson, C. George, A. P. Shanklin, T. M. Dunams, D. Christodoulou, J. E. Saavedra, E. S. Sagan and D. S. Bohle, Nitric Oxide, 2001, 5, 377–394 CrossRef CAS.
- J. E. Saavedra, A. Srinivasan, C. L. Bonifant, J. Chu, A. P. Shanklin, J. L. Flippen-Anderson, W. G. Rice, J. A. Turpin, K. M. Davies and L. K. Keefer, J. Org. Chem., 2001, 66, 3090–3098 CrossRef CAS.
- I. Gusarov, K. Shatalin, M. Starodubtseva and E. Nudler, Science, 2009, 325, 1380–1384 CrossRef CAS.
- Azides and Nitrenes: Reactivity and Utility, ed. E. F. V. Scriven, Academic Press, Inc., Orlando, 1984 Search PubMed.
- L. J. Ignarro, J. M. Fukuto, J. M. Griscavage, N. E. Rogers and R. E. Byrns, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 8103–8107 CAS.
- H. H. H. W. Schmidt and M. Kelm, in Methods in Nitric Oxide Research, ed. M. Feelish and J. S. Stamler, Wiley & Sons, New York, 1996 Search PubMed.
- K. Koh, A. G. Wong-Foy and A. J. Matzger, Angew. Chem., Int. Ed., 2008, 47, 677–680 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and crystallographic details, Fig. S1–S8. See DOI: 10.1039/c000154f |
| This journal is © The Royal Society of Chemistry 2010 |