DOI:
10.1039/B914419F
(Paper)
CrystEngComm, 2010,
12, 122-133
[Cr(dmbipy)(ox)2]−: a new bis-oxalato building block for metal assembling. Crystal structures and magnetic properties of XPh4[Cr(dmbipy)(ox)2]·5H2O (X = P and As), {Ba(H2O)2[Cr(dmbipy)(ox)2]2}n·17/2nH2O and {Ag(H2O)[Cr(dmbipy)(ox)2]}n·3nH2O†
Received
17th July 2009
, Accepted 11th September 2009
First published on
22nd September 2009
Abstract
The synthesis, X-ray structure and variable-temperature magnetic study of new compounds of formula PPh4[Cr(dmbipy)(ox)2]·5H2O (1), AsPh4[Cr(dmbipy)(ox)2]·5H2O (2), {Ba(H2O)2[Cr(dmbipy)(ox)2]2}n·17/2nH2O (3) and {Ag(H2O)[Cr(dmbipy)(ox)2]}n·3nH2O (4) (PPh4+ = tetraphenylphosphonium cation; AsPh4+ = tetraphenylarsonium cation; dmbipy = 4,4′-dimethyl-2,2′-bipyridine; ox2− = oxalate dianion) are reported herein. The isomorphous compounds 1 and 2 are made up of discrete [Cr(dmbipy)(ox)2]− anions, XPh4+ cations [X = P (1) and As (2)] and uncoordinated water molecules. The chromium environment in 1 and 2 is distorted octahedral with Cr–O and Cr–N bond distances varying in the ranges 1.950(2)–1.9782(12) and 2.047(3)–2.0567(14) Å, respectively. The angles subtended at the chromium atom by the two bidentate oxalate ligands cover the range 82.58(10)–83.11(5)°, and they are somewhat greater than those concerning the chelating dmbipy [79.04(10) (1) and 79.24(5)° (2)]. The [Cr(dmbipy)(ox)2]− unit of 1 and 2 also occurs in 3 and 4 but it adopts different coordination modes. It acts as a chelating ligand through its two oxalate groups towards the divalent barium cations in 3 affording neutral chains with diamond-shaped units sharing the barium atoms, while the two other corners are occupied by two crystallographically independent chromium atoms. The barium atom in 3 is coordinated by eight oxygen atoms from four oxalate groups and two aqua ligands. The structure of 4 consists of neutral bimetallic layers where the [Cr(dmbipy)(ox)2]− unit acts as a ligand towards the univalent silver(I) cation through its two oxalate groups, one of them being bidentate and the other bidentate/monodentate (outer). Each silver atom is six-coordinated with a water molecule and five oxygen atoms from three oxalate groups building a highly distorted octahedral environment. Magnetic susceptibility measurements for 1–4 in the temperature range 1.9–300 K show the occurrence of weak ferro- (1 and 2) and antiferromagnetic (3 and 4) interactions which are mediated by π–π stacking between dmbipy ligands through the spin polarization mechanism. A comparative study of the potentiality of the [Cr(AA)(ox)2]− unit (AA = bidentate nitrogen donor) as a building block for designing heterometallic species is carried out in the light of the available structural information.
Introduction
The tris(oxalato)metallate complex [M(ox)3]3− (MIII = trivalent metal ion; ox2− = oxalate dianion) has been widely used as a ligand towards fully solvated metal ions in the preparation of extended multifunctional magnetic systems in the last fifteen years.1 The main virtues of this building block are the following: (a) its high negative charge; (ii) the ability of the oxalate group to act as a bridge between metal ions through the bis-bidentate coordination mode; (iii) the remarkable ability of the oxalate ligand to mediate magnetic interactions between the paramagnetic centers through the previous bridging mode; (iv) and finally, the inert character towards the ligand exchange of the tris-oxalato precursor for certain metal ions such as Cr(III). This last point is very important because it precludes the presence of free oxalate in solution that would lead to an undesired mixture of products. These advantages account for the impressive success of this preparative route of oxalate-bridged heterobimetallic compounds with controlled nuclearity and dimensionality.2–12 In the light of these results, it is clear now that their dimensionality is controlled by the templating counterion. So, layered honeycomb structures with the formula cat[MIIMIII(ox)3] (cat+ = univalent cation) are obtained when the cation is of the type XR4+ (X = N and P; R = aryl and alkyl)2–7,8a,9 or even the decamethylmetallocenium unit [A(Cp*)2]+ (A = Fe and Co; Cp* = pentamethylcyclopentadienyl);8c,10 whereas the tris-chelated [M(bipy)3]2+ (bipy = 2,2′-bipyridine) entity affords chiral anionic 3D networks of formula [MnII2(ox)3]−, [MIMIII(ox)3]3− and [MIIMIII(ox)3]−.8b,11,12 All these two- and three-dimensional systems are examples of polyfunctional molecular-based materials in which the magnetic properties are often complemented by additional properties such as electrical conductivity,10c prefixed chirality8 and non-linear optical activity.9
In attempt to direct these studies towards the rational design of lower dimensionality compounds, several mononuclear chromium(III) complexes of general formula [Cr(AA)ox)2]− were synthesized where one of the oxalate groups has been replaced by a bidentate13–15 or potentially bis-bidentate16 nitrogen donor (AA). The self-assembly of these species with uni- or divalent metal ions has afforded a good number of oxalato-bridged heterometallic complexes with different structures, depending on the assembling cation.13,14a,b,15,16a,17–21
The new precursor [Cr(dmbipy)(ox)2]− (dmbipy = 4,4′-dimethyl-2,2′-bipyridine) was prepared in an attempt to extend this preparative route with the [Cr(AA)(ox)2]− building block to both polar and non polar solvents and also to check the influence of the peripheral AA ligand on the magnetic properties. Moreover, its coordinating ability towards some metal ions was foreseen. In this work, we present the syntheses and the magnetic properties of the mononuclear species XPh4[Cr(dmbipy)(ox)2]·5H2O [X = P (1) and As (2)] as well as those of the heterometallic one- {Ba(H2O)2[Cr(dmbipy)(ox)2]2}n·17/2nH2O (3) and two-dimensional {Ag(H2O)[Cr(dmbipy)(ox)2]}n·3nH2O (4) compounds.
Experimental
Materials and methods
All reagents and solvents were purchased from commercial sources and used as received. Elemental analyses (C, H and N) of 1–4 were performed at the Centro de Microanálisis from the Universidad Complutense de Madrid. The values of the P:Cr (1), As:Cr (2), Ba:Cr (3) and Ag:Cr (4) molar ratios [1:1 (1, 2 and 4) and 1:2 (3)] were determined by electron microscopy at the Servicio Interdepartamental de Investigación de la Universitat de València. Infrared spectra (4000–300 cm−1) were recorded on a Bruker IF S55 spectrophotometer on samples of 1–4 prepared as KBr pellets. Variable-temperature (1.9–300 K) magnetic susceptibility measurements on polycrystalline samples of 1–4 were carried out with a SQUID susceptometer using applied magnetic fields of 1 T (T ≥ 50 K) and 250 G (T < 50 K). Diamagnetic corrections of the constituents atoms were estimated from Pascal's constants22 as −480 × 10−6 (1) −485 × 10−6 (2), −274 × 10−6 (3) and −266 × 10−6 cm3 mol−1 (4) [per mol of Cr(III) ion]. The magnetic data were also corrected for the magnetization of the sample holder.
Synthesis
XPh4[Cr(dmbipy)(ox)2]·5H2O [X = P (1) and As (2)].
Chromium(III) chloride hexahydrate (1 mmol), 4,4′-dimethyl-2,2′-bipyridine (dmbipy) (1 mmol) and sodium oxalate (2 mmol) were dissolved in a 1:1 mixed water/ethanol solution. The initial green reaction mixture was refluxed for 5 h turning into a deep violet solution. After the addition of tetraphenylphosphonium (1) or tetraphenylarsonium chloride (2) (1 mmol) dissolved in a minimum amount of water, the resulting solution was allowed to evaporate at room temperature. After a few days, violet parallelepipeds (1)/prisms (2) were obtained in different crops. Yield: 40 (1) and 50% (2). Anal. Calcd for C40H42PCrN2O13 (1): C, 57.10; H, 4.99; N, 3.33. Found: C, 56.93; H, 4.90; N, 3.27%. Anal. Calcd for C40H42AsCrN2O13 (2): C, 54.26; H, 4.74; N, 3.16. Found: C, 54.10; H, 4.63; N, 3.19%.
{Ba(H2O)2[Cr(dmbipy)(ox)2]2}n·17/2nH2O (3).
An aqueous solution of barium(II) perchlorate (1 mmol) was added to a concentrated aqueous suspension of 2 (1 mmol) and the resulting mixture was kept under continuous stirring for 2 h. The white solid (tetraphenylarsonium perchlorate) formed was removed by filtration and the resulting violet solution allowed to evaporate at room temperature. X-Ray quality prisms of 3 were obtained by slow evaporation. The yield is practically quantitative. Anal. Calcd for C32H45BaCr2N4O26.5 (3): C, 33.40; H, 3.91; N, 4.87. Found: C, 33.35; H, 3.85; N, 4.83%.
{Ag(H2O)[Cr(dmbipy)(ox)2] }n·3nH2O (4).
The synthesis of 4 is analogous to that of 3, except for the addition of silver(I) perchlorate (1 mmol) instead of barium perchlorate. The experiment was performed in the darkness to avoid the silver(I) reduction by the daylight. Violet rectangles of 4 were grown from the mother liquor by slow evaporation at room temperature. The yield is practically quantitative. Anal. Calcd for C16H20AgCrN2O12 (4): C, 32.46; H, 3.38; N, 4.73. Found: C, 32.40; H, 3.34; N, 4.69%.
Crystal data collection and refinement
The data collection on single crystals of 1, 3 and 4 was performed on a Bruker Smart CCD diffractometer at 293 (1 and 4) and 213 K (3) by using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) with a nominal crystal-to-detector distance of 4 cm. A hemisphere of data was collected based on three ω-scans runs (starting ω = −28°) at values ϕ = 0, 90 and 180° with the detector at 2θ = 28°. At each of these runs, frames (606, 435 and 230) were collected at 0.3° intervals and 35 (1) and 25 s (3 and 4) per frame. The data collection on a single crystal of 2 was carried out on a Bruker-Nonius X8APEXII CCD area detector diffractometer at 100 K by using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The diffraction frames for 1–4 were integrated using the SAINT package [versions 5.0 (1, 3 and 4) and 6.45 (2)]23 and corrected for absorption with SADABS.24 A summary of the crystallographic data and structure refinement of 1–4 is given in Table 1.
Table 1 Crystallographic data for (PPh4)[Cr(ox)2(dmbipy)]·5H2O (1), (AsPh4)[Cr(ox)2(dmbipy)]·5H2O (2), {Ba(H2O)2[Cr(dmbipy)(ox)2]2·17/2nH2O}n (3) and {Ag(H2O)[Cr(dmbipy)(ox)2]·3nH2O}n (4)
| Compound |
1
|
2
|
3
|
4
|
|
R
1
= ∑||Fo| − |Fc||/∑|Fo|.
wR
2
= {∑[w(Fo2 − Fc2)2]/∑w(Fo2)2}1/2.
|
| Formula |
C40H42CrN2O13P |
C40H42AsCrN2O13 |
C64H90Ba2Cr4N8O53 |
C16H20AgCrN2O12 |
| Formula weight |
841.73 |
885.68 |
2302.12 |
592.21 |
| Crystal system |
Triclinic |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P-1 |
P-1 |
P2/c |
P2(1)/n |
|
a/Å |
9.804(4) |
9.7453(8) |
19.493(8) |
10.106(4) |
|
b/Å |
11.001(4) |
10.9615(9) |
9.687(4) |
12.278(5) |
|
c/Å |
20.153(8) |
20.060(2) |
24.200(9) |
17.285(7) |
|
α/° |
92.799(9) |
93.031(2) |
90 |
90 |
|
β/° |
98.239(9) |
99.433(2) |
98.736(10) |
92.851(9) |
|
γ/° |
109.087(8) |
108.355(2) |
90 |
90 |
|
V/Å3 |
2022.1(13) |
1993.8(3) |
4517(3) |
2142.1(14) |
|
T/K |
293(2) |
100(2) |
213(2) |
293(2) |
|
Z
|
2 |
2 |
2 |
4 |
|
µ(Mo Kα)/mm−1 |
0.389 |
1.178 |
1.430 |
1.487 |
| Reflections collected |
16274 |
17168 |
30406 |
11790 |
|
ϕ range for data collection |
1.03 to 30.00 |
1.04 to 26.37 |
1.06 to 27.50 |
2.04 to 27.50 |
| Unique reflections/Rint |
11369/0.0296 |
8045/0.0187 |
10393/0.0819 |
4920/0.0785 |
| Goodness-of-fit on F2 |
1.014 |
1.056 |
1.053 |
1.045 |
|
R1a/wR2b |
0.0619/0.1596 |
0.0263/0.0711 |
0.0553/0.1241 |
0.0681/0.1790 |
|
R1a/wR2b (all data) |
0.1289/0.1996 |
0.0298/0.0735 |
0.1149/0.1483 |
0.1417/0.2153 |
| Residual ρ/e A−3 |
0.573 and −0.507 |
0.539 and −0.483 |
1.212 and −1.068 |
0.704 and −1.013 |
The structures of 1–4 were solved by direct methods through the SHELXTL 5.10 program package25 and subsequently refined by Fourier recycling. All non-hydrogen atoms of 1–4 were refined anisotropically. The analysis of the residual electronic density in the final difference Fourier maps of compounds 1–4 revealed several peaks which were attributed to crystallization water molecules. Five (1 and 2) and three (4) distinct peaks in general positions [O(1W), O(2W), O(3W), O(4W) and O(5W) for 1 and 2, and O(1W), O(2W) and O(3W) for 4] were found which were refined anisotropically. For compound 3, eleven peaks in general positions, six of them [O(1W) to O(6W)] and the other five [O(7W) to O(11W)] with full and half occupancies respectively, were found and refined anisotropically. In the case of 2, the hydrogen atoms of the water molecules were located on a ΔF map and refined with 15 restraints, the thermal factors being fixed to 0.06 Å2, whereas in the case of 1, 3 and 4 they were neither found nor fixed at idealized positions. All hydrogen atoms of the AsPh4+/PPh4+ cations and those of the dmbipy ligand were generally set in calculated positions and refined as riding atoms. The final full-matrix least-squares refinement on F2, minimizing the function ∑w|Fo| − |Fc|)2, reached convergence at the values of the discrepancy indices listed in Table 1. The final geometrical calculations for 1, 2 and 4 were carried out with the XP utility of the SHELXTL 5.10 system and the PARST 9726 program, whereas the graphical manipulations were performed with the DIAMOND27 program. The main interatomic bond distances and angles are listed in Tables 2 (1 and 2), 3 (2), 4 (3), 5 (4) and S1–S3 (1, 3 and 4). CCDC reference numbers 740221 (1), 740222 (2), 740223 (3) and 740224 (4). See http://www.rsc.org/suppdata for crystallographic data in CIF or other electronic format.
Table 2 Selected bond lengths (Å) and angles (°) for compounds 1 and 2
| Compound |
1
|
2
|
| Cr(1)–O(1) |
1.952(2) |
1.9554(12) |
| Cr(1)–O(2) |
1.967(2) |
1.9559(12) |
| Cr(1)–O(5) |
1.964(2) |
1.9782(12) |
| Cr(1)–O(6) |
1.950(2) |
1.9673(12) |
| Cr(1)–N(1) |
2.047(3) |
2.0567(14) |
| Cr(1)–N(2) |
2.056(3) |
2.0492(14) |
| O(1)–Cr(1)–O(2) |
82.96(9) |
83.11(5) |
| O(1)–Cr(1)–O(5) |
172.44(9) |
171.43(5) |
| O(1)–Cr(1)–O(6) |
93.24(10) |
92.41(5) |
| O(1)–Cr(1)–N(1) |
91.35(10) |
91.84(5) |
| O(1)–Cr(1)–N(2) |
92.94(9) |
93.87(5) |
| O(2)–Cr(1)–O(5) |
90.92(9) |
90.09(5) |
| O(2)–Cr(1)–O(6) |
92.75(10) |
93.06(5) |
| O(2)–Cr(1)–N(1) |
95.75(10) |
95.48(5) |
| O(2)–Cr(1)–N(2) |
173.33(10) |
173.86(5) |
| O(5)–Cr(1)–O(6) |
82.58(10) |
82.73(5) |
| O(5)–Cr(1)–N(1) |
93.68(10) |
93.99(5) |
| O(5)–Cr(1)–N(2) |
93.55(9) |
93.38(5) |
| O(6)–Cr(1)–N(2) |
92.73(10) |
92.41(5) |
| O(6)–Cr(1)–N(1) |
170.77(10) |
170.86(5) |
| N(2)–Cr(1)–N(1) |
79.04(10) |
79.24(5) |
Results and discussion
It has been shown in previous works that the heteroleptic [Cr(AA)(ox)2]− species [AA = 2,2′-bipy (2,2′-bipyridine), phen (1,10-phenanthroline), dpa (2,2′-dipyridylamine) and bpym (2,2′-bipyrimidine)] are highly versatile ligands in designing heterometallic compounds.13–21 Their relative stability, the possibility that they have to act as a ligand towards a second metal ion through the oxalate groups and the supramolecular interactions involving the AA ligand [π–π stacking between the aromatic rings or hydrogen bonds (when AA = dpa)] make them very appropriate tools for metal assembling in crystal engineering. In the present work, we have enlarged this chemistry by preparing the new precursor [Cr(dmbipy)(ox)2]− as tetraphenylphosphonium (1) and tetraphenylarsonium (2) salts and testing its coordinating ability towards the univalent Ag(I) (4) and divalent Ba(II) (3) cations. The metathetic reaction of (1)/(2) with the perchlorate salts of these cations in aqueous solution, which is based on the insolubility in water of the XPh4ClO4 (X = P or As) ionic salt, affords the bimetallic compounds 3 and 4 in a practically quantitative yields.
The high frequency region of the infrared spectra of 1–4 has in common the occurrence of a broad and strong absorption in the range 3504–3449 cm−1 [ν(OH)] which is due to water molecules linked by hydrogen bonds.28 The stretching vibrations of the oxalate are observed at 1714s and 1680s (1 and 2), 1713s and 1678s cm−1 (3), 1709s and 1671s cm−1 (4) [νas(CO)] and at 807m (1), 806m (2), 810m (3) and 804m cm−1 (4) [δ(OCO)], suggesting the presence of coordinated oxalate in them. The [νs(CO)] stretching peaks are obscured by the vibrations of the methyl groups of the dmbipy ligand which appear at 1382s (1), 1381s (2), 1389s (3) and 1396s cm−1 (4). The out-of-plane bending vibrations of the phenyl rings of the XPh4+ [X = P (1) and As (2)] are located at 764m, 723s and 692s cm−1 (1) and at 747m and 691m cm−1 (2).
Description of the structures
PPh4[Cr(ox)2(dmbipy)]·5H2O (1) and AsPh4[Cr(ox)2(dmbipy)]·5H2O (2).
Compounds 1 and 2 crystallize in the triclinic P(−1) space group and they are isomorphous. Their crystal structure consists of discrete [Cr(dmbipy)(ox)2]− anions (Fig. 1 and S1), tetraphenylphosphonium (1) or tetraphenylarsonium (2) cations and uncoordinated water molecules, held together by electrostatic forces, hydrogen bonds (see Tables 3 and S1,† for 2 and 1, respectively) and van der Waals interactions (see below).
Table 3 Hydrogen-bonding interactions in compound 2a,b
| D–H⋯A |
D–H/Å |
H⋯A/Å |
(DHA)/° |
D⋯A/Å |
|
D = Donor, A = Acceptor.
Symmetry code: (g) = x, y + 1, z; (f) = 2 − x, 2 − y, 1 − z; (e) = 1 − x, 2 − y, 1 − z; (h) = −x + 1, −y + 1, −z + 1; (i) = x, −1 + y, z.
|
| O(1W)–H(1W)⋯O(7i) |
0.946(13) |
2.29(2) |
141(2) |
3.078(2) |
| O(1W)–H(1W)⋯O(8g) |
0.944(9) |
2.37(2) |
143(2) |
3.179(2) |
| O(1W)–H(2W)⋯O(3) |
0.950(9) |
1.95(1) |
175(2) |
2.901(2) |
| O(2W)–H(4W)⋯O(7g) |
0.926(9) |
2.02(1) |
176(2) |
2.943(2) |
| O(2W)–H(3W)⋯O(3) |
0.939(9) |
1.93(1) |
162(2) |
2.844(2) |
| O(3W)–H(6W)⋯O(4W) |
0.944(9) |
1.96(1) |
166(2) |
2.882(2) |
| O(3W)–H(5W)⋯O(1W) |
0.947(9) |
1.97(1) |
169(2) |
2.902(2) |
| O(4W)–H(8W)⋯O(5W) |
0.937(9) |
1.82(1) |
175(2) |
2.755(3) |
| O(4W)–H(7W)⋯O(8f) |
0.948(9) |
2.04(1) |
170(2) |
2.978(2) |
| O(5W)–H(10W)⋯O(7e) |
0.944(9) |
1.91(1) |
171(2) |
2.849(2) |
| O(5W)–H(9W)⋯O(3Wh) |
0.946(9) |
1.92(1) |
163(2) |
2.835(2) |
![Perspective view of the [Cr(dmbpy)(ox)2]− anion of 2 showing the atom numbering (the same labelling was adopted for the corresponding anion in 1). The hydrogen atoms other than those of the methyl groups were omitted for clarity.](/image/article/2010/CE/b914419f/b914419f-f1.gif) |
| | Fig. 1 Perspective view of the [Cr(dmbpy)(ox)2]− anion of 2 showing the atom numbering (the same labelling was adopted for the corresponding anion in 1). The hydrogen atoms other than those of the methyl groups were omitted for clarity. | |
Each chromium atom in 1 and 2 is six-coordinated with the two dmbipy-nitrogen atoms and four oxygen atoms from two oxalate molecules building a somewhat distorted octahedral environment. The reduced value of the bite angle of the bidentate dmbipy [79.04(10)° (1) and 79.24(5)° (2)] and oxalate ligands [82.58(10) and 82.96(9)° (1) and 83.11(5) and 82.73(5)° (2)] is the main factor accounting for the distortion of the metal environment from the ideal octahedral geometry. The Cr–N(dmbipy) [2.047(3) and 2.056(3) Å (1) and 2.0492(14) and 2.0567(14) Å (2)] and Cr–O(ox) [values varying in the ranges 1.950(2)–1.967(2) (1) and 1.9554(12)–1.9782(12) Å (2)] bond lengths compare well with the corresponding mean values reported in the literature for the parent [Cr(bipy)(ox)2]− (2.06 and 1.96 Å for Cr–N and Cr–O, respectively),13 [Cr(phen)(ox)2]− (2.08 and 1.95 Å),14 [Cr(bpym)(ox)2]− (2.08 and 1.96 Å),16 and [Cr(dpa)(ox)2]− (2.07 and 1.96 Å)15 complexes as AsPh4+/PPh4+ salts.
The dmbipy and ox ligands do not show significant deviations from planarity. The carbon-carbon bond distance in the ox ligands is as expected for a single C – C bond [average values 1.55 (1) and 1.54 Å (2)]. Two sets of oxalate carbon-oxygen bond distances are observed, the shortest values belonging to the peripheral C–O bonds, in agreement with their greater double bond character [the ranges of the values for the outer and inner C–O bonds are 1.215(4)–1.226(4) and 1.263(4)–1.291(4) Å for 1 and 1.217(2)–1.232(2) and 1.283(2)–1.292(2) Å for 2]. The values of the dihedral angle between the two oxalate ligands are 85.8(9)° in 1 and 88.1(4)° in 2 while those of the dmbipy molecule with the two oxalate ligands are 80.5(8) and 86.4(5)° (1) and 86.3(4) and 86.0(2)° (2). The values of the inter-ring carbon-carbon bond length of the dmbipy ligand in 1 and 2 are identical [1.474(4) and 1.475(2) Å] and they are very close to that observed for the free molecule [1.4931(12) Å].29 The trans conformation observed in the crystal structure of the uncoordinated dmbipy molecule turns into the cis one in 1 and 2 because of its coordination to the chromium(III) ion as a bidentate ligand. The bulky PPh4+ (1) and AsPh4+ (2) cations exhibit the typical tetrahedral shape and their bond lengths and angles do not exhibit any unusual feature.
In the overall crystal packing of 1 and 2, different domains occur for the anions and cations. Focusing on the XPh4+ cations, they are involved in two types of supramolecular interactions:30 the sextuple phenyl embrace (SPE) along the b axis in which three phenyl rings from each unit give rise to six edge-to-face local phenyl⋯phenyl interactions, and the double phenyl embrace (DPE) along the [210] direction where two phenyl rings with an offset face-to-face (off) relationship are in the interaction domain [Fig. 2 (2) and S2† (1)]. As far as the anions are concerned, in- and off-set π–π type interactions between dmbipy ligands of adjacent [Cr(dmbipy)(ox)2]− units generate anionic chains running along the a axis [Fig. 3 (2)]. The values of the interplanar distance occurring between neighbouring dmbipy molecules in 1 [3.43(1) and 3.31(1) Å for the in- and the off-set interactions, respectively] are identical to that of 2 [3.43(1) and 3.28(1) Å] and they compare well with those observed in the parent AsPh4[Cr(bpym)(ox)2]·H2O16a and AsPh4[Cr(phen)(ox)2]·H2O14b compounds [3.48(3) and 3.53(9) Å for the bpym⋯bpym and phen⋯phen interplanar separations, respectively] but they are somewhat shorter than the reported one for AsPh4[Cr(bipy)(ox)2]·H2O13a [3.7(1) Å]. This is quite surprising in particular when comparing the complexes with bipy and dmbipy because of the lack of substituents on the bipy ligand would preclude the occurrence of the steric effects which could issue from the presence of the methyl groups in dmbipy. Most likely, the somewhat stronger π–π interaction when passing from bipy to dmbipy has to be associated to the donor effect of the methyl groups and it is the origin of the weak intermolecular ferromagnetic coupling observed in 1 and 2 (see the magnetic discussion).
![A view showing the SPE and DPE supramolecular interactions between the AsPh4+ cations in 2 (the same information is given for 1 in Fig. S2). Regular alternating SPE interactions occur along b with As(1)⋯As(1k) = 5.912(2) Å and As(1)⋯As(1j) = 6.618(2) Å whereas two DPE interactions develop along the [210] direction with As(1)⋯As(1l) = 9.273(2) Å and As(1j)⋯As(1m) = 10.744(2) Å [symmetry code: (j) = −x, 1 − y, −z; (k) = −x, 2 − y, −z; (l) = 1 − x, 2 − y, −z; (m) = 1 + x, y, z].](/image/article/2010/CE/b914419f/b914419f-f2.gif) |
| | Fig. 2 A view showing the SPE and DPE supramolecular interactions between the AsPh4+ cations in 2 (the same information is given for 1 in Fig. S2†). Regular alternating SPE interactions occur along b with As(1)⋯As(1k) = 5.912(2) Å and As(1)⋯As(1j) = 6.618(2) Å whereas two DPE interactions develop along the [210] direction with As(1)⋯As(1l) = 9.273(2) Å and As(1j)⋯As(1m) = 10.744(2) Å [symmetry code: (j) = −x, 1 − y, −z; (k) = −x, 2 − y, −z; (l) = 1 − x, 2 − y, −z; (m) = 1 + x, y, z]. | |
![View of the stacking of the mononuclear [Cr(dmbipy)(ox)2]− anions of 2 through π–π type interactions.](/image/article/2010/CE/b914419f/b914419f-f3.gif) |
| | Fig. 3 View of the stacking of the mononuclear [Cr(dmbipy)(ox)2]− anions of 2 through π–π type interactions. | |
Finally, an extensive network of hydrogen bonds involving all the crystallization water molecules and the peripheral O(3), O(7) and O(8) oxalate-oxygens [Fig. 4 (2); see Tables 3 (2) and S1 (1)] contributes to the stabilization of the structures of 1 and 2. Remarkably, an hexameric ring of water molecules with two dangling waters which are anchored by four [Cr(dmbipy](ox)2]− anions occurs in these compounds. The hydrogen bonds together with the π–π type interactions between adjacent dmbipy ligands lead to anionic layers growing in the ab plane [(Fig. 4 (2)] which are separated from each other by the layers of the XPh4+ cations.
 |
| | Fig. 4 Hydrogen bonds (dashed blue lines) involving the water molecules and some of the peripheral oxalate-oxygen atoms in 2 (dashed grey lines refer to π–π interactions). | |
{Ba(H2O)2[Cr(dmbipy)(ox)2]2}n·17/2nH2O (3).
Compound 3 crystallizes in the monoclinic P2/c space group. Its structure is made up of crystallographically independent heterotrinucler units of formula {Ba(H2O)2[Cr(dmbipy)(ox)2]2} (Fig. 5) that grow along the b axis through oxalate bridges to afford neutral double zigzag chains (Fig. 6). Each chain is formed of diamond shaped units sharing the barium atoms, while the other two corners are occupied by chromium atoms having opposite chirality. Adjacent double chains are related by an inversion center leading to an achiral structure. The same topology of the metallic centers was reported for the oxalato-bridged heterometallic compound of formula {Mn[Cr(bipy)(ox)2]2}n,17a {Sr[Cr(dpa)(ox)2]}n·8nH2O15 and {Ba(H2O)2[Cr(bipy)(ox)2]2}n·4nH2O.21a Some selected bond lengths and angles for compound 3 are presented in Table 4.
 |
| | Fig. 5 (left) Perspective view of the crystallographically independent heterotrinuclear BaCr2 unit of 3 showing the atom numbering. The hydrogen atoms other than those of the methyl groups were omitted for clarity. (right) Top view of the coordination environment of the barium atom. | |
 |
| | Fig. 6 View of the double chain arrangement of 3 along the b axis showing the interchain π–π interactions involving the dmbipy ligands. The crystallization water molecules have been omitted for clarity. | |
Table 4 Selected bond lengths (Å) and angles (°) for compound 3a
|
Symmetry code: (a) = x, y + 1, z; (b) = x, y − 1, z.
|
| Cr(1)–O(5) |
1.960(4) |
Cr(1)–O(6) |
1.962(4) |
Cr(1)–O(2) |
1.965(4) |
Cr(1)–O(1) |
1.977(4) |
| Cr(1)–N(1) |
2.044(5) |
Cr(1)–N(2) |
2.045(5) |
Cr(1)–N(2) |
2.045(5) |
Cr(2)–O(9) |
1.949(4) |
| Cr(2)–O(16) |
1.954(4) |
Cr(2)–O(15) |
1.973(4) |
Cr(2)–O(10) |
1.974(4) |
Cr(2)–N(3) |
2.045(5) |
| Cr(2)–N(4) |
2.056(5) |
|
|
|
|
|
|
| O(1)–Cr(1)–N(1) |
92.09(18) |
O(1)–Cr(1)–N(2) |
89.06(18) |
O(2)–Cr(1)–O(1) |
83.20(17) |
O(2)–Cr(1)–N(1) |
172.67(19) |
| O(2)–Cr(1)–N(2) |
95.35(18) |
O(5)–Cr(1)–O(1) |
173.27(18) |
O(5)–Cr(1)–O(6) |
82.91(16) |
O(5)–Cr(1)–O(2) |
90.08(17) |
| O(5)–Cr(1)–N(1) |
95.71(18) |
O(5)–Cr(1)–N(2) |
174.57(19) |
O(6)–Cr(1)–O(1) |
173.27(18) |
O(6)–Cr(1)–O(2) |
92.44(17) |
| O(6)–Cr(1)–N(1) |
92.74(18) |
O(6)–Cr(1)–N(2) |
96.47(18) |
N(1)–Cr(1)–N(2) |
78.91(19) |
O(9)–Cr(2)–O(16) |
92.88(18) |
| O(9)–Cr(2)–O(15) |
91.67(16) |
O(9)–Cr(2)–O(10) |
83.15(16) |
O(9)–Cr(2)–N(3) |
94.46(18) |
O(9)–Cr(2)–N(4) |
174.13(19) |
| O(10)–Cr(2)–N(3) |
93.47(18) |
O(10)–Cr(2)–N(4) |
96.49(17) |
O(15)–Cr(2)–O(10) |
172.05(17) |
O(15)–Cr(2)–N(3) |
92.94(18) |
| O(15)–Cr(2)–N(4) |
89.27(17) |
O(16)–Cr(2)–O(15) |
83.69(17) |
O(16)–Cr(2)–O(10) |
90.53(17) |
O(16)–Cr(2)–N(3) |
172.01(18) |
| O(16)–Cr(2)–N(4) |
92.98(18) |
N(3)–Cr(2)–N(4) |
79.70(18) |
|
|
|
|
| Ba(1)–O(11b) |
2.763(4) |
Ba(1)–O(14) |
2.773(4) |
Ba(1)–O(7) |
2.799(4) |
Ba(1)–O(8) |
2.816(4) |
| Ba(1)–O(3a) |
2.822(5) |
Ba(1)–O(18) |
2.833(4) |
Ba(1)–O(4a) |
2.854(4) |
Ba(1)–O(13) |
2.858(4) |
| Ba(1)–O(12b) |
2.869(4) |
Ba(1)–O(17) |
2.967(5) |
|
|
|
|
| O(3a)–Ba(1)–O(18) |
111.63(13) |
O(3a)–Ba(1)–O(4a) |
58.40(13) |
O(3A)–Ba(1)–O(12b) |
137.87(12) |
O(3a)–Ba(1)–O(17) |
68.40(13) |
| O(3b)–Ba(1)–O(13) |
125.25(12) |
O(4A)–Ba(1)–O(12b) |
145.18(14) |
O(4a)–Ba(1)–O(17) |
114.67(13) |
O(4b)–Ba(1)–O(13) |
75.72(14) |
| O(7)–Ba(1)–O(3a) |
91.74(13) |
O(7)–Ba(1)–O(8) |
59.15(11) |
O(7)–Ba(1)–O(18) |
113.58(12) |
O(7)–Ba(1)–O(4a) |
142.32(14) |
| O(7)–Ba(1)–O(13) |
140.91(13) |
O(7)–Ba(1)–O(12b) |
72.30(12) |
O(7)–Ba(1)–O(17) |
66.60(13) |
O(8)–Ba(1)–O(3a) |
68.37(13) |
| O(8)–Ba(1)–O(18) |
73.23(12) |
O(8)–Ba(1)–O(4a) |
86.79(13) |
O(8)–Ba(1)–O(13) |
140.85(12) |
O(8)–Ba(1)–O(12b) |
126.38(12) |
| O(8)–Ba(1)–O(17) |
106.94(13) |
O(11b)–Ba(1)–O(14) |
133.89(13) |
O(11b)–Ba(1)–O(7) |
68.92(13) |
O(11b)–Ba(1)–O(8) |
82.27(12) |
| O(11b)–Ba(1)–O(3a) |
150.37(13) |
O(11b)–Ba(1)–O(18) |
60.82(12) |
O(11b)–Ba(1)–O(4a) |
125.88(12) |
O(11b)–Ba(1)–O(13) |
80.37(13) |
| O(11b)–Ba(1)–O(12b) |
58.91(11) |
O(11b)–Ba(1)–O(17) |
119.25(13) |
O(12b)–Ba(1)–O(17) |
69.48(13) |
O(13)–Ba(1)–O(12b) |
71.30(12) |
| O(13)–Ba(1)–O(17) |
112.17(13) |
O(14)–Ba(1)–O(7) |
131.99(12) |
O(14)–Ba(1)–O(8) |
143.09(13) |
O(14)–Ba(1)–O(3a) |
75.74(13) |
| O(14)–Ba(1)–O(18) |
114.13(13) |
O(14)–Ba(1)–O(4a) |
66.79(14) |
O(14)–Ba(1)–O(13) |
58.77(12) |
O(14)–Ba(1)–O(12b) |
86.45(12) |
| O(14)–Ba(1)–O(17) |
65.69(13) |
O(18)–Ba(1)–O(4a) |
65.24(12) |
O(18)–Ba(1)–O(13) |
67.65(12) |
O(18)–Ba(1)–O(12b) |
110.49(12) |
| O(18)–Ba(1)–O(17) |
179.81(14) |
|
|
|
|
|
|
Three crystallographically independent metal atoms, namely Cr(1), Cr(2) and Ba(1) are present in 3. The two chromium atoms are six-coordinated in a somewhat distorted CrN2O4 octahedral surrounding as in 1 and 2, the Cr–N(dmbipy) and Cr–O(ox) bond distances being similar to those observed in those compounds. The angles subtended by the bidentate dmbipy [78.91(19) and 79.70(18)° at Cr(1) and Cr(2), respectively] and the bis-bidentate oxalate [83.20(17) and 82.91(16)° at Cr(1) and 83.15(16) and 83.69(17)° at Cr(2)] are the main sources of the distortion of the ideal octahedron. Each barium atom is ten-fold coordinated by eight oxygen atoms of four bis-bidentate oxalate ligands [values of the Ba–O(ox) bond distances varying in the range 2.763(3)–2.869(4) Å] and two water molecules [values of the Ba–O(water) bond lengths of 2.833(4) and 2.967(5) Å]. The O–Ba–O bite angles of the bridging oxalate are very acute [values varying in the range 58.40(13)–59.15(11)°]. The barium atom has a bicapped flattened square antiprism geometry (Fig. 5), the distance between the basal [O(3)O(7)O(12)O(14)] and upper [O(4)O(8)O(11)O(13)] mean planes being 2.118(6) Å.
The two dmbipy ligands in 3 are quasi planar, the values of the dihedral angle between the two pyridyl rings being 5.4(2)° [N(1)/N(2)] and 11.1(2)° [N(3)/N(4)]. The values of inter-ring carbon-carbon bond length of dmbipy [1.492(8) and 1.475(8) Å for C(10)–C(11) and C(26)–C(27)] agree with those observed for this molecule in 1 and 2. The four oxalate ligands are also planar and the values of their carbon-carbon bond are as expected for a single a single C–C bond [values covering the range 1.552(8)–1.563(8) Å]. The C–O bonds within each oxalate group are significantly different [the shortest and longest values being 1.219(7) and 1.288(7) Å]. The different nature and charge of the two metal ions bridged by the oxalate [Cr(III) and Ba(II)] account for asymmetry in the carbonyl-oxalate bonds. The values of the dihedral angle between the mean plane of the dmbipy molecule and those of the oxalate ligands at the two chromium atoms vary in the range 79.2(1)–88.0(1)° whereas those between adjacent oxalate groups at each barium atoms lie in the range 54.9(1)–85.2(1)°.
The neighbouring double chains in 3 interact through π–π contacts between dmbipy along the b axis in quasi eclipsed trans conformation [the shortest separation between the two mean dmbipy planes being ca. 3.566(7) Å] (Fig. 6). An extensive network of hydrogen bonds involving all the water molecules and some oxalate-oxygens (Table S2†) leads to a supramolecular three-dimensional structure for 3. The Cr⋯Ba separation through the oxalate bridge are 6.125(2) [Cr(1)⋯Ba(1)] and 6.142(2) Å [Cr(2)⋯Ba(1)] whereas the shortest interchain Cr⋯Ba separations are 8.454(3) [Ba(1)⋯Cr(2g)] and 8.499(3)Å [Ba(1)⋯Cr(2d)] for the two closest neighbouring chain generated through the symmetry operations (g) = 1 − x, y − 1, 1/2 −z and (d) = 1 − x, y, 1/2 − z, respectively.
{Ag(H2O)[Cr(dmbipy)(ox)2]}n·3nH2O (4).
Compound 4 crystallizes in the monoclinic P21/n space group. It exhibits a two-dimensional structure which is made up by bimetallic oxalato-bridged Cr(III)–Ag(I) helical chains (Fig. 7) that are linked through centrosymmetric Ag2O2 units (Fig. 8). Because of this inversion center, the chromium complexes from adjacent chains have opposite chirality. The resulting neutral layers grow in the bc plane and they eclipsed to each other with the shortest interlayer distances of 7.461(3) [Ag(1)⋯Ag(1c)] and 8.749(3) Å [Cr(1)⋯Cr(1e)] [symmetry code: (c) = 1 − x, 1 − y, −z; (e) = 1 − x, 2 − y, −z]. Hydrogen bonds involving four oxalate-oxygens [O(2), O(3), O(4) and O(8)] and the coordinated [O(9)] and free [O(1W), O(2W) and O(3W)] water molecules [O⋯O values varying in the range 2.668(11)–3.169(9) Å, Table S3†] provide an additional stabilization to the crystal packing energy and lead to a three-dimensional supramolecular structure. Within each layer, four octanuclear Cr4Ag4 rings and four tetranuclear Cr2Ag2 rhombuses alternate regularly around each Cr4Ag4 unit (Fig. 9) with a four-fold axis being perpendicular to the layer and passing through the middle point between two opposite chromium atoms. This type of topology was previously observed in the complexes of formula {Na(H2O)[Cr(bipy)(ox)2]}n·2nH2O13a and {Na(H2O)[Cr(phen)(ox)2]}n·2nH2O.14b
 |
| | Fig. 7 Perspective view of a fragment of the helical bimetallic chain of 4 with the atom numbering. The hydrogen atoms other than those of the methyl groups were omitted for clarity. | |
 |
| | Fig. 8 A view of a fragment of the bimetallic layer of 4 showing the CrIII4AgI4 holes partially filled by the dmbipy ligands. The hydrogen atoms and crystallization water molecules were omitted for the sake of clarity. | |
 |
| | Fig. 9 A topological drawing of a fragment of a layer of 4 down the a axis visualizing the alternating arrangement of Cr4Ag4 octogons and Ag2O2 rhombuses. Green and grey circles stand for chromium and silver atoms, respectively. | |
The crystallographically independent chromium atom [Cr(1)] in 4 exhibits the same distorted octahedral CrN2O4 environment occurring in 1–3, the bond lengths and angles around the chromium atoms comparing well with those of the previous structures. The two oxalate groups of the Cr(dmbipy(ox)2]− unit adopt different coordination modes: the bis-bidentate of C(1a)/C(2a) [towards Cr(1a) and Ag(1)] and bis-bidentate/monodentate outer of C(3)/C(4) [towards Cr(1) and Ag(1)/through O(7) at Ag(1b)]. The metal-metal separations through these bridges are 5.652(6) [Cr(1a)⋯Ag(1)], 5.824(6) [Cr(1)⋯Ag(1)] 3.731(6) Å [Ag(1)⋯Ag(1b)] [symmetry code: (a) = −x + 1/2, y − 1/2, −z + 1/2; (b) = −x, −y + 1, −z]. Each silver atom [Ag(1)] is also six-coordinated with a water molecule and five oxygen atoms from three oxalate groups building a highly distorted octahedron. The silver to water bond length [2.329(7) Å for Ag(1)–O(9)] is shorter that the silver to oxalate bond distances [values varying in the range 2.403(5)–2.626(6) Å]. The values of the angles subtended by the chelating oxalate ligands at the silver atom are very reduced [65.7(2) and 67.4(2)° for O(7)–Ag(1)–O(8) and O(3a)–Ag(1)–O(4a), respectively] and they agree with those previously observed in the sheetlike coordination polymer {Ag3(H2O)[Cr(dpa)(ox)2]}n·2nH2O.20
The dmbipy ligand in 4 is practically planar [the value of the dihedral angle between its two pyridyl rings being 1.4(3)°] and the value of the inter-ring carbon-carbon bond is 1.485(9) Å. Weak intrasheet π–π type interactions across the Cr4Ag4 cavity occur involving two dmbipy ligands in a practically eclipsed conformation with the methyl substituents in trans arrangement [the shortest separation between the two dmbipy mean planes being 3.500(9) Å] (see Fig. 8). One of the oxalate ligands, C(3)/C(4), is somewhat twisted [the value of the dihedral angle between the O(5)C(3)O(7) and O(6)C(4)O(8) planes is 8.6(1)°] whereas the other one, C(1)–C(2), is closer to planarity [dihedral angle between O(1)C(1)O(3) and O(2)C(2)O(4) is 1.7(1)°]. Their different bridging modes would account for this structural difference. The values of the C(1)–C(2) and C(3)–C(4) bond lengths are as expected for a single carbon-carbon bond [1.519(12) and 1.548(11) Å, respectively]. Finally, the C–O bonds within each oxalate group in 4 are significantly different as occurred in 3, with the longest values being on the side of the chromium atom [C–O values in the ranges 1.266(9)–1.271(9) and 1.214(9)–1.271(9) Å at the chromium and silver atoms, respectively]. Selected bond lengths and angles for compound 4 are presented in Table 5.
Table 5 Selected bond lengths (Å) and angles (°) for compound 4a
|
Symmetry codes: (a) = −x + 1/2, y − 1/2, −z + 1/2; (b) = −x, −y + 1, −z.
|
| Cr(1)–O(1) |
1.940(5) |
Cr(1)–O(2) |
1.964(5) |
Cr(1)–O(5) |
1.962(5) |
Cr(1)–O(6) |
1.965(5) |
| Cr(1)–N(1) |
2.051(6) |
Cr(1)–N(2) |
2.056(6) |
|
|
|
|
| O(1)–Cr(1)–O(5) |
173.1(2) |
O(5)–Cr(1)–N(1) |
93.6(2) |
O(1)–Cr(1)–O(2) |
83.1(2) |
O(2)–Cr(1)–N(1) |
94.4(2) |
| O(5)–Cr(1)–O(2) |
93.2(2) |
O(6)–Cr(1)–N(1) |
173.2(2) |
O(1)–Cr(1)–O(6) |
92.0(2) |
O(1)–Cr(1)–N(2) |
93.0(2) |
| O(5)–Cr(1)–O(6) |
82.2(2) |
O(5)–Cr(1)–N(2) |
91.4(2) |
O(2)–Cr(1)–O(6) |
91.2(2) |
O(2)–Cr(1)–N(2) |
171.9(2) |
| O(1)–Cr(1)–N(1) |
92.6(2) |
O(6)–Cr(1)–N(2) |
96.1(2) |
N(1)–Cr(1)–N(2) |
78.6(2) |
|
|
| Ag(1)–O(3a) |
2.525(6) |
Ag(1)–O(4a) |
2.498(6) |
Ag(1)–O(7) |
2.506(6) |
Ag(1)–O(8) |
2.626(6) |
| Ag(1)–O(9) |
2.330(7) |
Ag(1)–O(7b) |
2.403(6) |
Ag(1)⋯Ag(1b) |
3.731(2) |
|
|
| Ag(1b)–O(7)–Ag(1) |
98.92(19) |
O(4a)–Ag(1)–O(7) |
137.77(19) |
O(4a)–Ag(1)–O(3a) |
67.4(2) |
O(7)–Ag(1)–O(3a) |
78.65(19) |
| O(7b)–Ag(1)–O(3a) |
95.7(2) |
O(7b)–Ag(1)–O(4a) |
125.2(2) |
O(7b)–Ag(1)–O(7) |
81.1(2) |
O(8)–Ag(1)–O(3a) |
78.6(18) |
| O(8)–Ag(1)–O(4a) |
83.1(19) |
O(8)–Ag(1)–O(7) |
65.7(2) |
O(8)–Ag(1)–O(7a) |
146.8(18) |
O(8)–Ag(1)–O(9) |
100.8(2) |
| O(9)–Ag(1)–O(3a) |
171.8(2) |
O(9)–Ag(1)–O(4a) |
104.4(2) |
O(9)–Ag(1)–O(7) |
108.7(2) |
O(9)–Ag(1)–O(7b) |
89.2(2) |
Magnetic properties
The magnetic properties of 1–4 under the form of the χMTversusT plot [χM is the magnetic susceptibility per Cr(III) ion] at T ≤ 50 K are shown in Fig. 10. At 300 K, χMT is ca. 1.86 cm3 mol−1 K for 1–4, a value which is as expected for a magnetically isolated spin quartet (SCr = 3/2). This value remains constant upon cooling until ca. 20 K and it further increases (1 and 2) or decreases (3 and 4) to reach values of 1.91 (1), 2.40 (2), 1.78 (3) and 1.20 cm3 mol−1 K (4) at 1.9 K. These features can be ascribed to the occurrence of very weak ferro- (1 and 2) and antiferromagnetic (3 and 4) interactions (θ) between the local spin quartets, together with zero-field splitting effects (D).
 |
| | Fig. 10
χ
M
T versus T plots for 1–4 in the low temperature region. The solid lines are the best-fit curves to the experimental data (see text). | |
In order to evaluate these interactions in 1–4 and having in mind their crystal structures we have used a simple Curie-Weiss expression [eq (1)]
| | | χM = 15Nβ2gCr2/12(T − θ) | (1) |
where
θ accounts for the intermolecular magnetic interactions and
gCr is the average Landé factor of the
chromium(III) ion. Least-squares best-fit parameters are:
gCr = 1.98(1) (
1–4) and
θ = +0.020(2) (
1), +0.050(1) (
2), −0.050(1) (
3) and −0.020(1) K (
4). Given that
D values reported in the literature for six-coordinated mononuclear Cr(III) complexes may attain values up to 1.0 cm
−1,
31 it is clear that the computed positive values of
θ (complexes
1 and
2) are the minimum ones whereas the negative ones (complexes
3 and
4) are the maximum values. In an attempt to check if the slight decrease of
χMT for
3 and
4 can be reproduced with a reasonable value of
D instead of
θ, we have analyzed their magnetic data through eqns (2)−(4) which are derived from the Hamiltonian given by eqn (5) (case of an axial zero field splitting and S = 3/2):
32| |  | (2) |
| |  | (3) |
| | 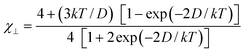 | (4) |
| | | Ĥ = D[Ŝz2 −1/3(S(S + 1)] | (5) |
Best-fit parameters through
eqns (2)–(4) lead to
gCr = 1.98(1) (
3 and
4) and |
D| = 0.80(2) (
3) and 0.50(2) cm
−1 (
4), the quality of the fit in both cases being similar to the previous ones with
gCr and
θ as variable parameters. The fact that the values of
D are in the upper range for six-coordinate Cr(III) complexes suggests that a very weak antiferromagnetic coupling could be involved in
3 and
4.
A careful examination of the structures of 1–4 shows that the weak π–π type interactions between the dmbipy of adjacent [Cr(dmbipy)(ox)2]− units could mediate these weak ferro- (1 and 2) and antiferromagnetic (3 and 4) interactions. In that respect, it deserves to be noted that weak either ferro- or antiferromagnetic interactions across π–π stacking have been observed in several other compounds.13b,17c,33,34 The spin polarization mechanism proposed by McConnell as early as 196335 can account for the nature of the magnetic couplings in 1–4. The McConnell's strategy requires molecular units showing noncompensating regions of positive (α) and negative (β) spin densities, a situation which may arise from the spin polarization effects. If the organization in the packing is such that the positive spin density of a unit interacts in an up-down fashion with the negative spin density of the adjacent unit, then the molecular spins are aligned along the same direction corresponding to intermolecular ferromagnetic interactions. This is the case for 1 and 2 which is illustrated by Scheme 1a whereas the opposite situation occurs in 3 and 4 (see Scheme 1b).
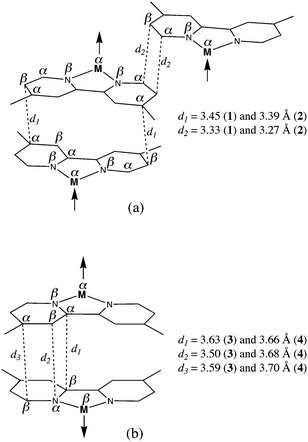 |
| | Scheme 1 | |
One can see therein how the π–π overlap between the dmbipy ligands leads to a parallel alignment of the positive spin densities of the Cr(III) ions (Scheme 1a), the interaction in 2 being predicted to be somewhat stronger than that of 1 because of the shorter values for the d1 and d2 distances. However, the π–π stacking in the case of 3 and 4 (Scheme 1b) favours the antiparallel alignment of the spin densities of the Cr(III) ions, the somewhat weaker antiferromagnetic coupling observed in 4 being as expected given the longer values of d1, d2 and d3 (carbon–carbon distances between adjacent dmbipy ligands). These examples show how subtle structural effects concerning weak supramolecular interactions can produce different magnetic effects.
The versatility of the [Cr(AA)(ox)2]− building block in designing heterometallic complexes
The [Cr(AA)(ox)2]− anion (AA= bidentate nitrogen donor) has been proved to be a suitable building block for the synthesis of heterometallic systems.13,14a,b,15,16a,17–21 The topology and dimensionality of the resulted product depends on type of [Cr(AA)(ox)2]− anion and also on the nature, charge, oxidation state and preferred stereochemistry of the cation which is used.
With alkaline earth ions (MII), the [Cr(AA)(ox)2]− unit (AA = bipy, dpa, dmbipy and phen) acts as bidentate/monodentate (MII = BaII), bis-bidentate/bis-monodentate (MII = BaII) and bis-bidentate (MII = SrII, BaII) (Scheme 2).15,17a,21a Apparently, the AA ligand determines the type of structure that is formed and the coordination modes of the oxalate ligand, as shown in Scheme 2. The coordination number of the alkaline earth cations is eight in the case of Sr(II) (eight oxalate-oxygens),15 whereas for Ba(II) is ten (nine oxalate-oxygens and one water molecule) for the complex with the bipy ligand17a and also (eight oxalate-oxygens and two water molecules) for complexes containing phen and bipy ligands21a and dmbipy (3). The self-assembly of the [Cr(AA)(ox)2]− building block with MII affords complexes with different dimensionalities, ranging from 3D (MII = BaII, AA = bipy),17a 2D (MII = BaII, AA = phen)21a to 1D [MII = SrII (AA = dpa),15 BaII (AA = bipy and dmbipy (4))].21a It should be noticed that the [Cr(AA)(ox)2]− building block with the same ligand, i.e. bipy affords two compounds with different dimensionality, the number of coordinated water molecules being the main factor to establish the dimensionality.17a,21a
 |
| | Scheme 2 | |
The reaction of the [Cr(AA)(ox)2]− building block with Ag(I) ion also affords different structures depending on the AA ligand (AA = bipy, dpa, bpym, dmbipy and phen) (Scheme 3).16b,17b, 20 In the case of AA = bipy, one of the oxalate groups acts as bis-bidentate whereas the other one adopts the bidentate/monodentate (inner) coordination mode resulting in a tetranuclear [(CrAg)2] metallocycle.17b However, when AA = bpym, the [Cr(AA)(ox)2]− unit loses the bpym ligand, which is replaced by two water molecules and the bpym ligand is coordinated to the Ag(I) in a bis-bidentate manner forming a cationic chain.16b When the dpa ligand is used, five different bridging modes of the oxalate ligand are displayed: bis-bidentate, bis-bidentate/monodentate (inner), bis-bidentate/monodentate (outer), bidentate/monodentate (outer) and bidentate/bis-monodentate (outer). The chromium and silver atoms are assembled in a layered structure with cycles comprised by decagons and rhombuses.20 In compound 4, the oxalate ligand exhibits the bis-bidentate and bis-bidentate/monodentate (outer) coordination modes. In this case, a layered motif is also formed but the cycles are octagons and rhombuses. This last topology was previously observed in the complexes with the Na(I) ion, such as {Na(H2O)[Cr(bipy)(ox)2]}n·2nH2O13a and {Na(H2O)[Cr(phen)(ox)2]}n·2nH2O.14b
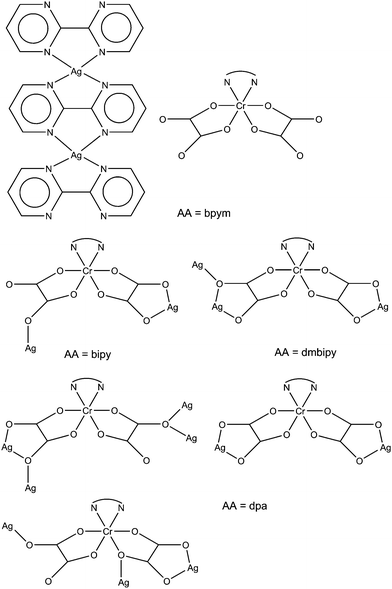 |
| | Scheme 3 | |
Conclusions
A new building block of formula XPh4[Cr(dmbipy)(ox)2]·5H2O [X = P (1), As (2)] has been synthesized and characterized and its complex ability tested versus Ba(II) (3) and Ag(I) (4) cations. The weak intermolecular ferromagnetic interactions occurring in 1 and 2 (which are lacking in other [Cr(AA)(ox)2]− containing compounds) are mediated by π–π stacking between the peripheral dmbipy ligands through the spin polarization mechanism. This work shows that the AA ligand can play a relevant role in mediating magnetic coupling across supramolecular interactions and also proves that [Cr(dmbipy)(ox)2]− is as good building block to afford heterobimetallic complexes. The topology and dimensionality of the resulting structures are not only determined by the nature of the outer metal ion but also on the peripheral AA ligand. An extensive use of the [Cr(dmbipy)(ox)2]− unit as a ligand towards fully solvated or partially blocked paramagnetic metal ions is in progress in order to explore the broad magneto-structural possibilities that it offers.
Acknowledgements
Financial support from the Ministerio Español de Ciencia e Innovación (Project CTQ2007-61690), the European Network MAGMANet (NMP-CT-2005-515767) and the Generalitat Valenciana (Project PROMETEO2009/108) is gratefully acknowledged. We thank C. Yuste for useful discussions. Authors are also grateful to the Servei Central d'Instrumentació Cientifica (SCIC) of the University Jaume I for providing us with X-ray facilities.
References
-
(a) O. Kahn, Adv. Inorg. Chem., 1995, 43, 179–259 CrossRef CAS;
(b)
S. Decurtins, R. Pellaux, M. Gross and H. W. Schmalle, Kluwer Academic Publishers, Dordrecht, 1999; p 175.
-
(a) H. Tamaki, M. Mitsumi, K. Nakamura, N. Matsumoto, S. Kida, H. Okawa and S. Iijima, Chem. Lett., 1992, 1975–1978 CAS;
(b) H. Tamaki, Z. J. Zhong, N. Matsumoto, S. Kida, M. Koikawa, N. Achiwa, Y. Hashimoto and H. Okawa, J. Am. Chem. Soc., 1992, 114, 6974–6979 CrossRef;
(c) M. Ohba, H. Tamaki, N. Matsumoto and H. Okawa, Inorg. Chem., 1993, 32, 5385–5390 CrossRef CAS.
- L. O. Atovmyan, G. V. Shilov, R. N. Lyubovskaya, E. I. Zhilyaeva, N. S. Ovanesyan, S. I. Pirumova, I. G.
Gusakovskaya and Y. G. Morozov, JETP Lett., 1993, 58, 766–769.
- R. P. Farrell, T. W. Hambley and P. A. Lay, Inorg. Chem., 1995, 34, 757–758 CrossRef CAS.
-
(a) S. Decurtins, H. W. Schmalle, H. R. Oswald, A. Linden, J. Ensling, P. Gutlich and A. Hauser, Inorg. Chim. Acta, 1994, 216, 65–73 CrossRef CAS;
(b) R. Pellaux, H. W. Schmalle, R. Huber, P. Fischer, T. Hauss, B. Ouladdiaf and S. Decurtins, Inorg. Chem., 1997, 36, 2301–2308 CrossRef CAS;
(c) S. Decurtins, M. Gross, H. W. Schmalle and S. Ferlay, Inorg. Chem., 1998, 37, 2443–2449 CrossRef CAS.
-
(a) S. G. Carling, C. Mathonière, P. Day, K. M. A. Malik, S. J. Coles and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1996, 1839–1843 RSC;
(b) C. Mathonière, C. J. Nuttall, S. G. Carling and P. Day, Inorg. Chem., 1996, 35, 1201–1206 CrossRef CAS;
(c) I. D. Watts, S. G. Carling and P. Day, J. Chem. Soc., Dalton Trans., 2002, 1429–1434 RSC.
- J. Larionova, B. Mombelli, J. Sanchiz and O. Kahn, Inorg. Chem., 1998, 37, 679–684 CrossRef CAS.
-
(a) R. Andrés, M. Gruselle, B. Malézieux, M. Verdaguer and J. Vaissermann, Inorg. Chem., 1999, 38, 4637–4646 CrossRef CAS;
(b) R. Andrés, M. Brissard, M. Gruselle, C. Train, J. Vaissermann, B. Malézieux, J. P. Jamet and M. Verdaguer, Inorg. Chem., 2001, 40, 4633–4640 CrossRef CAS;
(c) B. Malézieux, R. Andrés, M. Brissard, M. Gruselle, C. Train, P. Herson, L. L. Troitskaya, V. I. Sokolov, S. T. Ovseenko, T. V. Demeschik, N. S. Ovanesyan and I. A. Mamed'yarova, J. Organomet. Chem., 2001, 637–639, 182–190 CrossRef CAS;
(d) M. Gruselle, R. Andrés, B. Malézieux, M. Brissard, C. Train and M. Verdaguer, Chirality, 2001, 13, 712–714 CrossRef CAS.
-
(a) S. Benard, P. Yu, J. P. Audière, E. Rivière, R. Clement, J. Guilhem, L. Tchertanov and K. Nakatani, J. Am. Chem. Soc., 2000, 122, 9444–9454 CrossRef CAS;
(b) J. S. O. Evans, S. Benard, P. Yu and R. Clement, Chem. Mater., 2001, 13, 3813–3816 CrossRef CAS.
-
(a) M. Clemente-León, E. Coronado, J. R. Galán-Mascarós and C. J. Gomez-García, Chem. Commun., 1997, 1727–1728 RSC;
(b) E. Coronado, J. R. Galán-Mascarós, C. J. Gomez-García and J. M. Martínez-Agudo, Adv. Mater., 1999, 11, 558–561 CrossRef CAS;
(c) E. Coronado, J. R. Galán-Mascarós, C. J. Gomez-García and V. Laukhin, Nature, 2000, 408, 447–449 CrossRef CAS;
(d) E. Coronado, J. R. Galán-Mascarós, C. J. Gomez-García, J. Ensling and P. Gutlich, Chem.–Eur. J., 2000, 6, 552–563 CrossRef CAS;
(e) E. Coronado, J. R. Galán-Mascarós, C. J. Gomez-García and J. M. Martínez-Agudo, Inorg. Chem., 2001, 40, 113–120 CrossRef CAS.
-
(a) S. Decurtins, H. W. Schmalle, P. Schneuwly and H. R. Oswald, Inorg. Chem., 1993, 32, 1888–1892 CrossRef CAS;
(b) S. Decurtins, H. W. Schmalle, P. Schneuwly, J. Ensling and P. Gutlich, J. Am. Chem. Soc., 1994, 116, 9521–9528 CrossRef CAS;
(c) S. Decurtins, H. W. Schmalle, R. Pellaux, R. Huber, P. Fischer and B. Ouladdiaf, Adv. Mater., 1996, 8, 647–651 CrossRef CAS;
(d) S. Decurtins, H. W. Schmalle, R. Pellaux, P. Schneuwly and A. Hauser, Inorg. Chem., 1996, 35, 1451–1460 CrossRef CAS;
(e) R. Sieber, S. Decurtins, H. Stoeckli-Evans, C. Wilson, D. Yufit, J. A. K. Howard, S. C. Capelli and A. Hauser, Chem.–Eur. J., 2000, 6, 361–368 CrossRef CAS.
- M. Hernández-Molina, F. Lloret, C. Ruiz-Pérez and M. Julve, Inorg. Chem., 1998, 37, 4131–4135 CrossRef CAS.
-
(a) M. C. Muñoz, M. Julve, F. Lloret, J. Faus and M. Andruh, J. Chem. Soc., Dalton Trans., 1998, 3125–3131 RSC;
(b) R. Lescouëzec, G. Marinescu, J. Vaissermann, F. Lloret, J. Faus, M. Andruh and M. Julve, Inorg. Chim. Acta, 2003, 350, 131–142 CrossRef.
-
(a) F. D. Rochon and G. Massarweh, Can. J. Chem., 1999, 77, 2059–2068 CrossRef CAS;
(b) G. Marinescu, M. Andruh, R. Lescouëzec, M. C. Muñoz, J. Cano, F. Lloret and M. Julve, New J. Chem., 2000, 24, 527–536 RSC;
(c) V. Russell, D. Craig, M. Scudder and I. Dance, CrystEngComm, 2001, 3, 96–106 RSC.
- R. Lescouëzec, G. Marinescu, M. C. Muñoz, D. Luneau, M. Andruh, F. Lloret, J. Faus, M. Julve, J. A. Mata, R. Llusar and J. Cano, New J. Chem., 2001, 25, 1224–1235 RSC.
-
(a) G. De Munno, D. Armentano, M. Julve, F. Lloret, R. Lescouëzec and J. Faus, Inorg. Chem., 1999, 38, 2234–2237 CrossRef;
(b) G. Marinescu, R. Lescouëzec, D. Armentano, G. De Munno, M. Andruh, S. Uriel, R. Llusar, F. Lloret and M. Julve, Inorg. Chim. Acta, 2002, 336, 46–54 CrossRef CAS.
-
(a) F. D. Rochon, R. Melanson and M. Andruh, Inorg. Chem., 1996, 35, 6086–6092 CrossRef CAS;
(b) M. Andruh, R. Melanson, C. V. Stager and F. D. Rochon, Inorg. Chim. Acta, 1996, 251, 309–317 CrossRef CAS;
(c) S. Bruda, M. Andruh, H. W. Roesky, Y. Journaux, M. Noltemeyer and E. Rivière, Inorg. Chem. Commun., 2001, 4, 111–114 CrossRef CAS;
(d) E. Coronado, M. C. Giménez, C. J. Gómez-García and F. M. Romero, Polyhedron, 2003, 22, 3115–3122 CrossRef CAS;
(e) G. Marinescu, D. Visinescu, A. Cucos, M. Andruh, Y. Journaux, V. Kravtsov, Y. A. Simonov and J. Lipkowski, Eur. J. Inorg. Chem., 2004, 2914–2922 CrossRef CAS;
(f) S. Nastase, C. Maxim, F. Tuna, C. Duhayon, J. P. Sutter and M. Andruh, Polyhedron, 2009, 28, 1688–1693 CrossRef CAS.
- N. Sakagami, E. Kita, P. Kita, J. Wisniewska and S. Kaizaki, Polyhedron, 1999, 18, 2001–2007 CrossRef CAS.
- F. Bérézovsky, A. A. Hajem, S. Triki, J. S. Pala and P. Molinié, Inorg. Chim. Acta, 1999, 284, 8–13 CrossRef CAS.
- C. Yuste-Vivas, F. S. Delgado, C. Ruiz-Pérez and M. Julve, CrystEngComm, 2004, 6, 11–18 RSC.
-
(a) G. Marinescu, M. Andruh, M. Julve, F. Lloret, R. Llusar, S. Uriel and J. Vaissermann, Cryst. Growth Des., 2005, 5, 261–267 CrossRef CAS;
(b) D. Visinescu, J. P. Sutter, C. Ruiz-Pérez and M. Andruh, Inorg. Chim. Acta, 2006, 359, 433–440 CrossRef CAS.
-
A. Earnshaw, Introduction to Magnetochemistry, Academic Press, London, 1968 Search PubMed.
-
(a)
Bruker Analytical X-ray Systems, SAINT 5.0, Madison, WI, 1996 Search PubMed;
(b)
Bruker Analytical X-ray Systems, SAINT 6.45, Madison, WI, 2003 Search PubMed.
-
SADABS, Version 2.03, Bruker AXS Inc., Madison, WI, USA, 2000 Search PubMed.
-
G. M. Sheldrick, SHELXTL 5.10, Bruker Analytical X-ray Inc., Madison, WI, USA 1998 Search PubMed.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
-
DIAMOND 2.1d, Crystal Impact GbR, CRYSTAL IMPACT K; Brandeburg & H. Putz GBR, Bonn, Germany, 2000 Search PubMed.
-
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th edn, John Wiley & Sons Inc., 1986, p 228 Search PubMed.
- P. Pearson, C. M. Kepert, G. B. Deacon, L. Spiccia, A. C. Warden, B. W. Skelton and A. H. White, Inorg. Chem., 2004, 43, 683–691 CrossRef CAS.
- I. Dance and M. Scudder, Chem.–Eur. J., 1996, 2, 481–486 CrossRef CAS.
- R. Boca, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef CAS.
- C. J. O'Connor, Prog. Inorg. Chem., 1982, 29, 203–283 CAS.
- M. Wesolek, D. Meyer, J. A. Osborn, A. De Cian, J. Fischer, A. Derory, P. Legoll and M. Drillon, Angew. Chem., Int. Ed. Engl., 1994, 33, 1592–1594 CrossRef.
- A. J. Amoroso, J. C. Jeffery, P. L. Jones, J. A. McCleverty, P. Thornton and M. ward, Angew. Chem., Int. Ed. Engl., 1995, 34, 1443–1446 CrossRef CAS.
- H. M. McConnell, J. Chem. Phys., 1963, 39, 1910 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table of hydrogen bonding interactions for compounds 1, 3 and 4. Figures of the [Cr(dmbipy)(ox)2]− anion of 1 and supramolecular interactions between the PPh4+ cations in 2. CCDC reference numbers 740221–740224 (1–4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b914419f |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.