A predicted dimer-based polymorph of 10,11-dihydrocarbamazepine (Form IV)†
Jean-Baptiste
Arlin
a,
Andrea
Johnston
a,
Gary J.
Miller
a,
Alan R.
Kennedy
b,
Sarah L.
Price
c and
Alastair J.
Florence
*a
aSolid-State Research Group, Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, 27 Taylor Street, Glasgow, UK G4 0NR. E-mail: alastair.florence@strath.ac.uk; Fax: +0141-552-2565; Tel: +0141-548-4877
bWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, UK G1 1XL
cDepartment of Chemistry, University College London, 20 Gordon St, London, UK WC1H 0AJ
First published on 5th October 2009
Abstract
A novel polymorph of 10,11-dihydrocarbamazepine (form IV), which had been predicted to be thermodynamically feasible, was obtained from the vapour phase and displays an R22(8) hydrogen bonded dimer motif in contrast to the catemeric motifs in forms I–III.
10,11-Dihydrocarbamazepine (DHC) is a recognised impurity in carbamazepine (CBZ), a dibenzazepine drug used in the treatment of epilepsy.1 Here, DHC is of particular interest as part of a wider investigation that combines experimental searches with crystal structure prediction (CSP) to explore the basic science underpinning structural diversity in CBZ and the closely related analogues, DHC, cyheptamide (CYH) and cytenamide (CYT) (Fig. 1). In this communication, we present the single-crystal structure of a new dimer-based polymorph of DHC, form IV,† that was successfully predicted by CSP methods. We also report on the structural relationships between DHC polymorphs as indicated by the XPac program.2
 | ||
| Fig. 1 CBZ and related structural analogues DHC, CYH and CYT. | ||
DHC forms I (monoclinic),3 II (orthorhombic)4 and III (triclinic)5 all display the catemeric C(4) hydrogen-bonded motif shown in Fig. 2, as does a 1:1 CBZ:DHC6 solid solution. Notably, the dominance of the catemer motif in non-solvated DHC structures is in marked contrast to the known polymorphs of CBZ7 and CYT8 that contain only the dimer motif. CSP studies on DHC (this work) as well as CBZ,9 CYH and CYT† have identified thermodynamically competitive populations of hypothetical crystal structures that are based on either the catemer motif or an R22(8)10 dimer. However, prior to this report only CYH had been crystallised in both dimer and catemer motifs.11
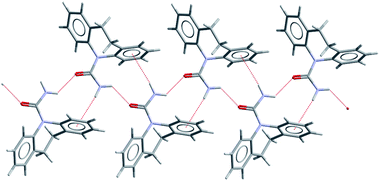 | ||
| Fig. 2 The C(4) catemer motif observed in DHC forms I–III (form I shown) and also in CYH form I and in a 1:1 solid solution of DHC:CBZ (N–H⋯O and N–H⋯π contacts shown as dashed lines). | ||
The CSP results for DHC were challenged by employing a diverse range of experimental crystallisation conditions in a search for predicted polymorphs and, in particular, potential dimeric structures. The crystallisation methods used included an automated parallel solution crystallisation study12,13 supplemented by manual solution crystallisations, solvate desolvation plus recrystallisation from both the melt and vapour phase. In total, 170 automated and manual solution recrystallisations were implemented using a library of 71 solvents under 4 different conditions (details in the ESI).† These produced DHC I, II and III in addition to six crystalline solvates (formic acid,14 formamide,15 acetic acid,16 butyric acid, triflouroacetic acid and DMSO17). A dimeric DHC motif is observed in the crystalline DMSO solvate, but in the other four known solvate structures (see ESI)† DHC molecules form an R22(8) motif with the carboxylic acid or amide group of the solvent molecule in preference to a solvated DHC dimer or catemer. Desolvation of polycrystalline samples of these solvates, including DHC:DMSO, produced DHC I and/or II (see ESI). Crystallisation of DHC from the melt failed due to rapid and complete decomposition upon melting to produce 10,11-dihydro-5H-dibenzo[b,f]azepine.18
Single-crystals of form IV were obtained, together with those of form I and II, by crystallisation from the vapour phase via sublimation.‡ Single-crystal analysis was carried out on a small needle shaped crystal at 123(2) K. The single-crystal structure is shown in Fig. 3 overlaid with the matching predicted structure (ak2, Table S4 in ESI†) found in the CSP search ∼10 kJ mol−1 above the global minimum.
 | ||
| Fig. 3 Crystal structure of DHC IV viewed down the b-axis showing the centrosymmetric R22(8) dimers (N–H⋯O contacts shown as dashed lines). The predicted structure ak2 is overlaid (red sticks). A comparison of form IV and ak2 using the packing similarity tool in Mercury 2.219 yields an RMS value of 0.33 Å for a 15 molecule coordination group. | ||
DHC can adopt one of two molecular conformations depending on the orientation of the carboxamide group relative to the C10–C11 bond (Fig. 4). The lower energy anti-conformer is observed in DHC I, II and III and the known solvate structures except that of the disordered DHC:DMSO, in which both the anti- and syn-conformers are both present with fractional occupancies of 0.81 and 0.19 respectively. DHC form IV however contains the syn-conformer that is estimated to be <2 kJ mol−1 less stable than the anti,§ a value consistent with the values expected for conformational polymorphism. Using the approximate crystal energy landscape (as in ref. 9, details in the ESI),† generated assuming the molecule adopted one of these gas-phase optimised conformations, DHC II was found to be the most stable structure. It also showed catemer-based structures generated from the syn conformation that were almost as stable as forms I and III (e.g. fc13 in the ESI).† Dimer-based structures were significantly less stable (by about 5 kJ mol−1) with DHC form IV further destabilised by the conformational energy penalty associated with the syn-conformer. A previously published CSP study on DHC20 also predicted form II as the global minimum, and found the lowest energy syn-conformer (catemeric) structure and lowest energy dimeric structure to be ca. 7 and 6 kJ mol−1 above the global minimum, respectively. At the time, these authors estimated that structures greater than 5 kJ mol−1 above the global minimum were unlikely to be observed.20
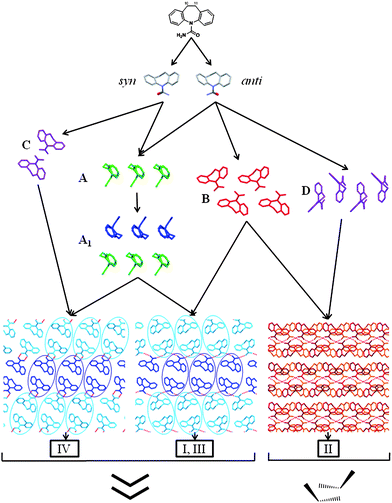 | ||
| Fig. 4 Tree diagram illustrating the similarity relationships between DHC I, II, III and IV. From top to bottom: DHC and the syn- and anti-conformers; SC A, a 1D stack; 1D SC A1 composed of two inversion related A stacks (A1 comprises anti-conformers in I and III and syn-conformers in IV); SC B, a 1D catemeric arrangement. A, A1 and B are each viewed perpendicular to the SC translation vector. Motifs C, a 0D dimer, and D, an offset 1D stack are observed only in forms IV and II, respectively. The packing diagrams for I, III and IV are viewed parallel to the A1 translation vector and the blue ovals highlight the different relative orientations of the A1 stacks in the catemeric (I, III) and dimeric (IV) structures. The diagrams at the base of the figure illustrate which of the two principle 1D stacking arrangements are observed in the structures (i.e. A1 in I, III and IV and D in II). | ||
The packing arrangement of DHC molecules in form IV was compared to the other three polymorphs using the XPac method and the same procedures applied previously to 25 CBZ related structures,21 CYH I and II11 and CYT I and II.8 Three supramolecular constructs2 (SCs) are observed more than once across the four DHC polymorph structures and their structural relationships are illustrated in Fig. 4. The 1D SCs A and A1 are observed in DHC I, III and IV, whilst the catemer, B, is observed in I, II and III. Fig. 4 also highlights two motifs that occur only once across the four DHC structures considered in the analysis, namely the 0D hydrogen-bonded dimer in IV (C) and an offset 1D stack of DHC molecules (D) in form II. The combination of SCs A1 and B in DHC I (monoclinic) and III (triclinic) results in structures with a high degree of 3D correspondence.
A previous XPac analysis of forms I and II of the related molecule CYH,11 identified that the 1D stack equivalent to A in Fig 4. is preserved in both structures, despite the change in hydrogen bonding (catemer → dimer) that accompanies the CYH I → II thermal phase transition. Similarly for DHC, the importance of space-filling effects is evident, with non-hydrogen bonded 1D stacks of DHC molecules (SC A1) observed in forms I, III and IV. The catemer and dimer hydrogen bonding motifs in these structures therefore arise from altered packing of the common molecular stack, A1, as highlighted in the packing diagrams in Fig. 4.
In general terms, the discovery of this new dimer-based polymorph of DHC provides another example of the successful application of CSP technologies to identify favourable packing motifs and crystal structures. An important caveat in the context of informing experimental polymorph discovery however, is the need to consider the predicted structures within a reasonably large energy range as being possible, given the likely error in calculating relative crystal energies. It has recently been shown that the relative energies of predicted CBZ structures are sensitive to the modelling of molecular flexibility22,23 and intermolecular forces, although the explicit modelling of the distortion of the charge distribution within the crystal24 may be less important for DHC.20 Whilst reliable calculations of the relative ambient free energies of organic crystals are a major challenge to computational chemistry, the discovery of DHC IV confirms that the approximate lattice energy landscape provides a meaningful indication of potential solid-state diversity, establishing a rationale for experimental investigation of the true crystal energy landscape. In the case of DHC, there are other predicted structures that may reasonably be expected to crystallise in preference to the syn-dimer based DHC IV, such as ak61 (see ESI) that shows identical packing of the more stable anti-conformer. Clearly, nucleation is important in determining which energetically feasible structures are actually observed as polymorphs, however, the concomitant formation of DHC I, II and IV from the vapour phase illustrates the considerable challenge of predicting the impact of kinetic factors involved in crystallisation.
In conclusion, DHC IV represents an important advance in defining the true extent of solid-state structural diversity of the molecules in Fig. 1, as summarised in Table 1. With two of the four compounds (DHC and CYH) having now been shown to adopt both dimer and catemer based crystal structures in accordance with CSP, the continued challenge to experimental and computational technologies is to provide confirmation of whether CBZ and CYT can or cannot form polymorphs based on the catemer motif.
| Polymorphs | R22(8) | C(4) | |
|---|---|---|---|
| CBZ | 4 | 4 | — |
| DHC | 4 | 1 | 3 |
| CYH | 2 | 1 | 1 |
| CYT | 2 | 2 | — |
Acknowledgements
We thank the Basic Technology programme of the UK Research Councils for funding this work under the project Control and Prediction of the Organic Solid State (www.cposs.org.uk), EPSRC funded Glasgow Centre for Physical Organic Chemistry for access to single-crystal diffraction facilities, AstraZenenca for funding GM and Prof M. B. Hursthouse and Dr G. Tizzard, University of Southampton, and Dr T. Gelbrich, University of Innsbruck, for advice in the use of the XPac program.Notes and references
- T. D. Cyr, F. Matsui, R. W. Sears, C.N.M and E. G. Lovering, J. Assoc. Off. Anal. Chem., 1987, 70, 836–840 CAS.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2005, 7, 324–336 RSC.
- G. Bandoli, M. Nicolini, A. Ongaro, G. Volpe and A. Rubello, J. Crystallogr. Spectrosc. Res., 1992, 22, 177–183 CrossRef CAS.
- W. T. A. Harrison, H. S. Yathirajan and H. G. Anilkumar, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o240–O242 CrossRef.
- C. K. Leech, A. J. Florence, K. Shankland, N. Shankland and A. Johnston, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o675–o677 CrossRef.
- A. J. Florence, C. K. Leech, N. Shankland, K. Shankland and A. Johnston, CrystEngComm, 2006, 8, 746–747 RSC.
- A. L. Grzesiak, M. D. Lang, K. Kim and A. J. Matzger, J. Pharm. Sci., 2003, 92, 2260–2271 CrossRef CAS.
- A. J. Florence, C. T. Bedford, F. P. A. Fabbiani, K. Shankland, T. Gelbrich, M. B. Hursthouse, N. Shankland, A. Johnston and P. Fernandes, CrystEngComm, 2008, 10, 811–813 RSC.
- A. J. Florence, A. Johnston, S. L. Price, H. Nowell, A. R. Kennedy and N. Shankland, J. Pharm. Sci., 2006, 95, 1918–1930 CrossRef.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- A. J. Florence, K. Shankland, T. Gelbrich, M. B. Hursthouse, N. Shankland, A. Johnston, P. Fernandes and C. K. Leech, CrystEngComm, 2008, 10, 26–28 RSC.
- A. J. Florence, B. Baumgartner, C. Weston, N. Shankland, A. R. Kennedy, K. Shankland and W. I. F. David, J. Pharm. Sci., 2003, 92, 1930–1938 CrossRef CAS.
- A. J. Florence, A. Johnston, P. Fernandes, N. Shankland and K. Shankland, J. Appl. Crystallogr., 2006, 39, 922–924 CrossRef CAS.
- A. Johnston, A. J. Florence, P. Fernandes, N. Shankland and A. R. Kennedy, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o1469–o1470 CrossRef.
- A. Johnston, A. J. Florence, P. Fernandes, N. Shankland and A. R. Kennedy, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3888–o3889 CrossRef.
- A. Johnston, A. J. Florence, P. Fernandes, N. Shankland and A. R. Kennedy, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o5361–o5362 CrossRef.
- A. Johnston, A. J. Florence, K. Shankland, C. K. Leech, N. Shankland and P. Fernandes, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3918–o3919 CrossRef.
- J. P. Reboul, B. Cristau, J. Estienne and A.J.P, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2108–2112 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. J. C. Cabeza, G. M. Day, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2007, 7, 100–107 CrossRef.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2006, 8, 449–460 RSC.
- A. J. C. Cabeza, G. M. Day, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2006, 6, 1858–1866 CrossRef CAS.
- P. G. Karamertzanis and S. L. Price, J. Chem. Theory Comput., 2006, 2, 1184–1199 CrossRef CAS.
- G. W. A. Welch, P. G. Karamertzanis, A. J. Misquitta, A. J. Stone and S. L. Price, J. Chem. Theory Comput., 2008, 4, 522–532 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Summary of results for solution crystallisations and CSP on DHC, also further details on results of XPac comparisons of DHC I–IV. [Note, detailed CSP results for CYH and CYT are outwith the scope of this report and will be published in detail in a subsequent report]. CCDC reference number 739821. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b914365c |
| ‡ 20 mg of DHC, obtained from Aldrich, was placed in a 10 mL glass volumetric flask in which a glass fibre was suspended. A small crystal of CYT I was attached to the fibre. The container was sealed and placed on a hot plate at 100 °C and left for 24 h after which fine crystals were observed on the glass fibre. Individual crystals were identified using single-crystal diffraction, confirming the presence of forms I, II and IV in the recrystallised sample. Although there is no evidence to suggest DHC IV grew on the surface of the CYT I crystal, further studies are ongoing to assess the reproducibility of DHC IV in the absence of heterochemical seeds and to obtain bulk samples of form IV for characterisation of the physical properties. DHC IV. C15H14N2O, Mr = 238.28, Bruker-AXS Kappa Apex II instrument, Mo radiation (λ = 0.71073 Å), Experimental structure [values for predicted ak2]: monoclinic, P21/c, a = 13.207(6) [13.897], b = 5.347(2) [5.215], c = 18.891(7) [19.430] Å, β = 116.37(2) [117.13]°, V = 1195.23 Å3, T = 123(2) K, µ = 0.085 mm−1, 16837 reflections measured, 2353 unique (Rint = 0.0824), θmax = 26.0°, 171 parameters, R = 0.043 [based on F and 1566 data with F2 > 2σ (F2), Rw = 0.098 (based on F2 and all 2353 unique reflections), S = 1.03. |
| § The syn-conformer is estimated to be less stable than the anti by 1.7 kJ mol−1 at the MP2 level (ESI) and 1.43 at the SCF level with a 6–31G(d,p) basis set, and 1.67 kJ mol−1 (DFT calculations with PW91 functional and DNP basis set) ref 20, by full optimisation of the two conformers. |
| This journal is © The Royal Society of Chemistry 2010 |