Copper(I) complexes with N-thiophosphorylated thioureas and phosphines with versatile structures and luminescence†
Maria G.
Babashkina
a,
Damir A.
Safin
*a,
Michael
Bolte
b and
Axel
Klein
*a
aInstitut für Anorganische Chemie, Universität zu Köln, Greinstrasse 6, D-50939, Köln, Germany. E-mail: damir.safin@ksu.ru; axel.klein@uni-koeln.de; Fax: +49 221 4705196; Tel: +49 221 4702913
bInstitut für Anorganische Chemie J.-W.-Goethe-Universität, Frankfurt/Main, Germany
First published on 30th September 2009
Abstract
Reaction of the potassium salts of RC(S)NHP(S)(OiPr)2 (R = MeNH, HLI; iPrNH, HLII; tBuNH, HLIII; Me2N, HLIV; PhNH, HLV; 2,6-Me2C6H3NH, HLVI; 2,4,6-Me3C6H2NH, HLVII) with [Cu(PPh3)3I] or a mixture of CuI and Ph2P(CH2)nPPh2 (n = 1, 2, 3) in aqueous EtOH/CH2Cl2 leads to mononuclear 1–6, 8–10, 12–27 or binuclear 7, 11 complexes. The structures of these compounds were investigated by 1H, 31P{1H} NMR spectroscopy and elemental anayses. The crystal structures of 1, 2, 4, 5, 11, 18, 20 and 21 were determined by single crystal X-ray diffraction. The luminescence properties of mononuclear complexes 1, 21, 22 and 27, and binuclear complexes 7 and 11·C3H6O are reported.
Introduction
In preceding papers, we have described the complexation properties of the alkaline salts of N-thiophosphorylated thioureas and thioamide RC(S)NHP(S)(OiPr)2 (HQ) [R = morpholin-N-yl (HQI), piperidin-N-yl (HQII);1 PhNH (HQIII);2α-naphthylNH (HQIV);3 NH2 (HQV);1,4 pyridin-2-ylNH (HQVI), pyridin-3-ylNH (HQVII), 6-amino-pyridin-2-ylNH (HQVIII);5 Ph (HQIX), Et2N (HQX);6–8 (EtO)2P(O)CH2C6H4-4-NH (HQXI)9] towards CuI. Starting from CuI as the CuI-source we obtained polynuclear complexes [CuQ]n. The structures of the complexes [CuQI,III,V,IX]n were established by single crystal X-ray diffraction (Chart 1). The polynuclear structures are formed through the C = S sulfur atoms, which are coordinated towards two or three CuI atoms. | ||
| Chart 1 | ||
According to the crystal structure data, the complex [Cu3QI3] forms a trinuclear cyclic compound, while the complex [Cu3QIII3]2 is a hexamer, containing two cyclic trimeric units, connected by a pair of Cu–S(P)–Cu bridges. This complex is the first example of polynuclear CuI complexes with RC(S)NHP(S)(OiPr)2 ligands, where the P = S sulfur atoms take part in the aggregate formation.1
In the complex [(Cu6QV6)(Cu3QV3)·4Me2CO] the [CuQV] moieties associate to form independent trimer [Cu3QV3] and hexamer [Cu6QV6] units.1,4 The space group is P-1 with a single six-membered ring alternating with a double symmetry sandwich six-membered ring which is connected by Cu–S bonding in one unit cell. The six-membered rings are linked by thiocarbonyl sulfurs resulting in a central six-membered Cu3S3 ring that holds the trimer together. Contrary to our expectations, the hexameric unit is also formed by Cu–S interactions through the thiocarbonyl sulfur atoms, while the thiophosphoryl sulfur atoms in each ligand moiety still form only one Cu–S bond. A central hexagonal-prismatic C6S12 cluster links the six chelate units together. Trimeric and hexameric units are interlinked by hydrogen bonds. As a result the 3D structure consists of two parallel layers of trimer molecules which are linked by means of hydrogen bonds to the hexamers. Some cavities within the formed “honeycomb” structure are occupied by acetone molecules and the other solvate molecules are located in the space between the two dimensional layers.
The complex [Cu4QIX4] is similar to [Cu3QI3], but shows a cyclic tetramer structure.8 Moreover, the second product in the reaction of KQIX with CuI is a [KCuQIX2]n polynuclear complex. The [CuQIX2]− anions are combined by the potassium cations, through the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S and PS sulfur atom, and the OiPr oxygen atoms, thus forming polymeric chains. Using the lithium or sodium salts the compound [Cu4QIX4] is the only product.
S and PS sulfur atom, and the OiPr oxygen atoms, thus forming polymeric chains. Using the lithium or sodium salts the compound [Cu4QIX4] is the only product.
When using [Cu(PPh3)2NO3] or [Cu(PPh3)3I] instead of CuI mononuclear complexes [Cu(PPh3)2QI–VII,IX,X] or [Cu(PPh3)QVIII,XI] were formed (Chart 2). It was supposed that the coordination of only one molecule of triphenylphosphine to CuI atom in the latter complexes is due to the formation of a hydrogen bonded dimer through the hydrogen atom of the substituent at the C=S group of one molecule and the oxygen atom, corresponding to the second molecule, while the hydrogen bonded dimers in the former complexes are of ”classical type” and formed through the hydrogen atom of the NH group and the sulfur atom of the C=S fragment. Reaction of alkaline salts of the tetrakis-thiourea, containing a cyclam fragment, with [Cu(PPh3)3I] leads to the tetranuclear CuI complex [{Cu(PPh3)2}4(cyclam)].10
 | ||
| Chart 2 | ||
Furthermore, there are numerous papers, describing complexes of CuX (X = Cl, Br, I, CN, SCN, ClO4, BH4, O3SCF3, PF6, CCPh) salts with various pnictide containing ligands EPh3, Ph2E–Z–EPh2 [E = P, As, Sb; Z = (CH2)1–6, (C5H4)Fe(C5H4)], MeC(PPh2CH2)3.11–23 A multitude of structures is reported. The structure of the obtained complexes depends on the reagents stoichiometry, solvents and anion X.
A number of heteroligand CuI complexes, containing both aryl-substituted pnictines and cyclic thioureas or pyridine thiolates, were also described.21–23 Combination of these two types of ligands might lead to interesting and unusual photophysical and electrochemical properties.20
In this contribution we will report on a systematic study on heteroligand CuI complexes with HQ and PPh3 or Ph2P–Z–PPh2 ligands with the focus on the structural characterisation and luminescence properties investigation.
Results and discussion
The N-thiophosphorylated thioureas HLI–VI were prepared by addition of the corresponding amine to O,O'-diisopropylthiophosphoric acid isothiocyanate (iPrO)2P(S)NCS (Scheme 1).24 Reaction of the potassium salts of HLI–VI with [Cu(PPh3)3I] or a mixture of CuI and Ph2P(CH2)nPPh2 (n = 1–3) in aqueous EtOH/CH2Cl2 leads to mononuclear 1–6, 8–10, 12–27 or binuclear 7, 11·C3H6O complexes (Schemes 2 and 3).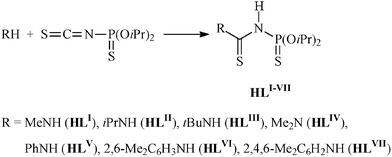 | ||
| Scheme 1 Preparation of HLI–VI. | ||
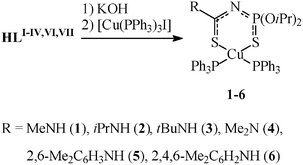 | ||
| Scheme 2 Preparation of 1–6. | ||
 | ||
| Scheme 3 Preparation of 7–27. | ||
The obtained complexes are colorless crystalline powders, soluble in acetone, benzene, dichloromethane, DMSO, DMF and insoluble in n-hexane. 1H, 31P{1H} NMR data indicated that the deprotonated thioureas LI–VI are 1,5-S,S'-ligands in all the present cases studied.
In the 31P{1H} NMR spectra of the complexes, the resonances in the range 51.7–59.2 ppm correspond to the phosphorus atoms of the thiophosphoryl group. The signals of triphenylphosphine groups in the spectra of the complexes 1–6 are shown at –4.2–2.9 ppm, while the signals for the phosphine phosphorus atoms in the 31P{1H} NMR spectra of the complexes 7–27 exhibit chemical shifts from −25.2 to −7.0 ppm.
Fast exchange between free and bound phosphine in solutions of studied complexes results in phosphine signal broadening in the 31P{1H} NMR spectra, as was observed for the CuI complexes with thioether ligands25 and N-(thio)phosphorylated thioamides and thioureas.1–3,5–7,9,10
The 1H NMR spectra of the complexes contain only signals which correspond to the proposed structures. Signals of the Me2N group in 4, 10, 17 and 24 are doubled due to hindered rotation. The same was observed for the complex [Cu(PPh3)2QX].6,7 Methyl protons in isopropyl groups are diastereotopic and show two signals in spectra. A rather high 4JP,H coupling constant (7.4–9.6 Hz) for the NH proton in 2, 3, 5, 6 and 15 is explained by the so-called W-criterion for the PNCNH chain.24 The contribution of the observed splitting to the presence of 4JP,H spin–spin coupling was unambiguously confirmed by recording 1H NMR spectra with 31P decoupling. The signals for the phenyl protons are observed as a multiplet at 6.34–8.20 ppm. The 1H NMR spectra of 7–27 contain the signals for the CH2 protons of the phosphines Ph2P(CH2)nPPh2 at 1.50–3.06 ppm.
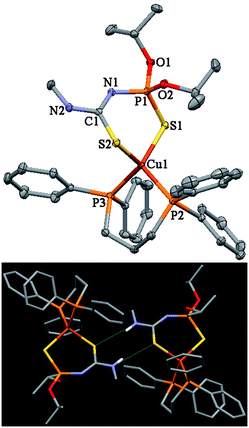
Crystals of 1, 2, 4, 5, 11·C3H6O, 18, 20 and 21 were obtained by slow evaporation of the solvent from dichloromethane or acetone–n-hexane mixtures, v/v 1:3 (Table 1). The molecular structures of 1, 18 and 21 are shown in Figs. 1–3, respectively. Selected bond lengths and bond angles are given in Table 2.
| 1 | 2 | 4 | 5 | 11·C3H6O | 18 | 20 | 21 | |
|---|---|---|---|---|---|---|---|---|
| Empirical formula | C44H48CuN2O2P3S2 | C46H52CuN2O2P3S2 | C45H50CuN2O2P3S2 | C51H54CuN2O2P3S2 | C63H64Cu2IN2O2P5S2·C3H6O | C39H44CuN2O2P3S2 | C42H50CuN2O2P3S2 | C35H44CuN2O2P3S2 |
| Formula weight | 857.41 | 885.47 | 871.47 | 947.53 | 1412.19 | 793.36 | 835.41 | 745.29 |
| Crystal system | triclinic | monoclinic | monoclinic | triclinic | monoclinic | triclinic | monoclinic | monoclinic |
| Space group | P-1 | P21/n | Cc | P-1 | P21/c | P-1 | P21/n | P21/n |
| a/Å | 10.7497(7) | 12.8828(15) | 12.7328(8) | 11.4650(5) | 13.5587(6) | 10.7751(5) | 12.7625(5) | 10.3331(3) |
| b/Å | 12.5836(8) | 21.032(2) | 21.3708(12) | 14.3083(7) | 17.5724(4) | 11.7916(5) | 28.9176(12) | 30.3668(6) |
| c/Å | 16.4356(11) | 17.227(2) | 16.9009(10) | 17.0891(8) | 27.298(1) | 16.6198(8) | 22.7401(8) | 11.9019(3) |
| α/° | 83.842(5) | 90 | 90 | 109.818(4) | 90 | 76.507(4) | 90 | 90 |
| β/° | 83.922(5) | 109.286(3) | 105.617(5) | 90.221(4) | 98.651(3) | 83.057(4) | 96.515(3) | 96.807(2) |
| γ/° | 78.782(5) | 90 | 90 | 112.994(4) | 90 | 71.394(4) | 90 | 90 |
| V/Å3 | 2159.9(2) | 4405.7(8) | 4429.1(5) | 2397.5(2) | 6430.0(4) | 1943.57(16) | 8338.3(6) | 3708.29(16) |
| Z | 2 | 4 | 4 | 2 | 4 | 2 | 8 | 4 |
| D calc/g cm−3 | 1.318 | 1.335 | 1.307 | 1.313 | 1.459 | 1.356 | 1.331 | 1.335 |
| T/K | 173(2) | 153(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) |
| F(000) | 896 | 1856 | 1824 | 992 | 2888 | 828 | 3504 | 1560 |
| µ/mm−1 | 0.751 | 0.739 | 0.734 | 0.684 | 1.378 | 0.829 | 0.776 | 0.864 |
| Reflections collected | 21993 | 35270 | 16080 | 32277 | 36380 | 19046 | 49783 | 58933 |
| Unique reflections | 8049 | 7763 | 7765 | 8952 | 12457 | 7229 | 14692 | 7396 |
| Observed reflections | 6350 (Rint = 0.067) | 5860 (Rint = 0.072) | 7329 (Rint = 0.037) | 7644 (Rint = 0.045) | 9953 (Rint = 0.049) | 6131 (Rint = 0.057) | 8713 (Rint = 0.113) | 7093 (Rint = 0.038) |
| R indices, (all data) | R 1 = 0.0392 | R 1 = 0.0558 | R 1 = 0.0305 | R 1 = 0.0305 | R 1 = 0.0314 | R 1 = 0.0374 | R 1 = 0.0654 | R 1 = 0.0257 |
| wR 2 = 0.1003 | wR 2 = 0.1460 | wR 2 = 0.0795 | wR 2 = 0.0773 | wR 2 = 0.0776 | wR 2 = 0.0917 | wR 2 = 0.1179 | wR 2 = 0.0642 |
 | ||
| Fig. 1 Thermal ellipsoid representation and hydrogen bonded dimer of 1. Ellipsoids are drawn at the 30% probability level. | ||
 | ||
| Fig. 2 Thermal ellipsoid representation and hydrogen bonded dimer of 18. Ellipsoids are drawn at the 30% probability level. | ||
 | ||
| Fig. 3 Thermal ellipsoid representation and hydrogen bonded dimer of 21. Ellipsoids are drawn at the 30% probability level. | ||
| 1 | 2 | 4 | 5 | 11·C3H6O | 18 | 20, Aa | 20, Ba | 21 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| a The data for two independent molecules (A) and (B). b The data for the Cu2 atom. | ||||||||||
| CS |
1.738(2) | 1.726(4) | 1.743(3) | 1.727(2) | 1.758(3) | 1.742(2) | 1.752(4) | 1.755(4) | 1.748(2) | |
| PS |
1.9694(9) | 1.974(2) | 1.9841(10) | 1.9750(7) | 1.9746(9) | 1.9824(8) | 1.974(2) | 1.977(2) | 1.9764(5) | |
| P–N | 1.606(2) | 1.618(4) | 1.603(3) | 1.592(2) | 1.628(2) | 1.607(2) | 1.608(5) | 1.604(5) | 1.6080(13) | |
| P–O | 1.574(2) | 1.580(3) | 1.573(3) | 1.583(2) | 1.568(2) | 1.577(2) | 1.584(4) | 1.582(3) | 1.5843(12) | |
| 1.585(2) | 1.592(4) | 1.593(2) | 1.594(2) | 1.584(2) | 1.589(2) | 1.586(3) | 1.588(4) | 1.5895(11) | ||
| C–N(P) | 1.324(3) | 1.211(6) | 1.327(4) | 1.315(2) | 1.303(3) | 1.319(3) | 1.313(6) | 1.320(6) | 1.319(2) | |
| C–N(C) | 1.336(3) | 1.377(6) | 1.351(4) | 1.355(2) | 1.359(3) | 1.357(3) | 1.342(7) | 1.329(7) | 1.339(2) | |
| Cu–S(C) | 2.3125(7) | 2.3032(13) | 2.3025(7) | 2.2926(6) | 2.3042(6) | 2.3377(7)b | 2.2937(6) | 2.329(2) | 2.326(2) | 2.3199(4) |
| Cu–S(P) | 2.3457(7) | 2.3563(13) | 2.3581(7) | 2.3728(5) | 2.3595(7) | 2.3144(7) | 2.3462(14) | 2.3459(14) | 2.3664(4) | |
| Cu–P | 2.2684(7) | 2.2803(12) | 2.2709(7) | 2.2822(6) | 2.2763(7) | 2.2718(6)b | 2.2826(7) | 2.2829(14) | 2.2804(14) | 2.2615(4) |
| 2.2942(7) | 2.3049(13) | 2.3353(7) | 2.3056(6) | 2.3076(6) | 2.2895(7)b | 2.3047(7) | 2.324(2) | 2.320(2) | 2.2965(4) | |
| Cu–I | 2.7096(4) | |||||||||
| Cu⋯Cu | 3.2457(4) | |||||||||
| S–C–N(C) | 115.98(18) | 112.3(3) | 118.1(2) | 114.85(14) | 113.1(2) | 114.7(2) | 117.1(3) | 117.3(3) | 116.33(11) | |
| S–C–N(P) | 127.53(19) | 129.1(4) | 125.6(2) | 129.13(13) | 126.5(2) | 127.0(2) | 125.7(4) | 125.4(4) | 126.80(12) | |
| N–C–N | 116.5(2) | 118.6(4) | 116.2(3) | 116.0(2) | 120.4(2) | 118.3(2) | 117.2(4) | 117.2(4) | 116.87(14) | |
| C–N–P | 129.7(2) | 129.4(3) | 129.9(2) | 133.11(14) | 123.5(2) | 125.3(2) | 127.4(3) | 127.5(3) | 124.96(11) | |
| N–P–S | 120.25(8) | 120.6(2) | 118.74(9) | 121.44(7) | 119.36(8) | 117.71(8) | 114.6(2) | 115.1(2) | 119.60(5) | |
| S–Cu–S | 106.98(2) | 106.35(5) | 107.40(3) | 109.69(2) | 102.05(2) | 108.53(2) | 109.86(5) | 109.89(5) | 106.63(1) | |
| P–Cu–P | 117.02(3) | 110.17(4) | 118.99(3) | 118.19(2) | 109.77(3) | 108.81(3)b | 89.17(2) | 90.30(5) | 90.04(5) | 98.91(2) |
| P–Cu–S(C) | 106.01(3) | 107.61(5) | 104.09(3) | 109.41(2) | 104.17(3) | 103.27(2)b | 109.14(2) | 116.08(5) | 116.46(5) | 108.66(2) |
| 116.79(3) | 109.84(5) | 112.84(3) | 113.91(2) | 119.27(2) | 122.65(3)b | 113.96(3) | 118.08(5) | 117.74(5) | 112.67(1) | |
| P–Cu–S(P) | 99.26(3) | 103.82(5) | 106.30(3) | 96.77(2) | 104.77(2) | 116.32(3) | 109.40(6) | 108.91(6) | 114.74(2) | |
| 110.17(3) | 118.81(5) | 106.58(3) | 107.79(2) | 117.26(2) | 118.50(3) | 111.75(6) | 112.38(6) | 115.18(2) | ||
| Cu–S–C | 104.38(8) | 109.0(2) | 104.67(9) | 109.03(6) | 107.29(8) | 114.81(8)b | 105.64(8) | 98.82(18) | 99.22(18) | 101.61(5) |
| Cu–S–P | 101.07(3) | 98.50(6) | 99.32(4) | 100.19(2) | 101.44(3) | 96.44(3) | 94.56(7) | 94.65(7) | 96.99(2) | |
| I–Cu–P | 106.09(2) | 113.81(2) | ||||||||
| I–Cu–S | 102.40(2) | |||||||||
| Cu–S–Cu | 88.72(2) |
In the structure of 1, 2, 4, 5, 18, 20 and 21, the CuI cation is in a P2S2 tetrahedral environment formed by two sulfurs of the corresponding deprotonated N-thiophosphoryl thiourea ligand and two PPh3 molecules (1, 2, 4, 5,) or two phosphorus atoms of one Ph2P(CH2)nPPh2 molecule (18, 20 and 21) (Figs. 1–3). The asymmetric unit of 20 contains two independent molecules.
In the complex 11·C3H6O, the deprotonated thiourea LV is coordinated towards two CuI atoms, showing a tridentate coordination mode, in which the Cu–S–Cu bridging bond is formed by the C=S sulfur atom (Fig. 4). The same type of the anionic thiourea ligand was observed in the structure of polynuclear CuI complexes (Chart 1).1,2,4,8 Both CuI atoms in 11·C3H6O are in a tetrahedral environment. Besides two sulfur atoms the Cu1 cation is coordinated with two phosphorus atoms of the two phosphine molecules, while further two phosphorus are bound to Cu2. The coordination sphere of Cu2 is completed by the iodine atom. The distance Cu1⋯Cu2 3.2457(4) Å (Table 2) is longer than the sum of the van der Waals radius of CuI, 2.80 Å.26 This indicates lack of any distinct Cu⋯Cu interactions.
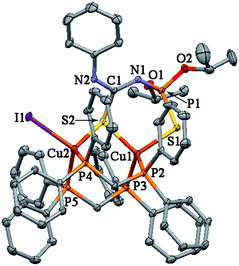 | ||
| Fig. 4 Thermal ellipsoid representation of 11·C3H6O. Ellipsoids are drawn at the 30% probability level. | ||
The small distortions relative to a perfect tetrahedron in the structure of 1, 2, 4, 5, reveal the absence of major steric hindrance as it was observed for the analogous complexes [Cu(PPh3)2QII–IV,VII,IX,X].1–3,5–7 The values of bond angles around Cu are in the range from 99.26(3) to 118.99(3)°. Cu–P bonds are longer than they are in the trigonal complexes of [Cu(PPh3)QVIII,XI] (2.2161(7) and 2.2142(11) Å),5,9 [Cu(PPh3)N{Ph2P(X)}2] (X = S, 2.222; Se, 2.190 Å),27,28 and [Cu(PPh3)PhC(O)NP(S)Ph2] (2.216 Å),29 and tetrahedral [Cu(PPh3)2N{Ph2P(O)}2] (2.255 and 2.245 Å).30 The difference in P–Cu bond lengths in comparison with trigonal complexes testifies for some sterical hindering of triphenylphosphine molecules. An example of stronger hindrance is found for the [Cu(PPh3)3I] molecule. The Cu–P bond length here is 2.362 Å.31
Complexes 18, 20 and 21 have similar bond parameters and angle parameters as for 1, 2, 4, 5, except for the P–Cu–P angle, which is in the range of 89.17(2)–98.91(2)°. As it could be expected, the increase of the bite angle value is observed from the complex 18 to 20 and 21 due to the increase in length of the alkylen bridging chain. Thus, the formation of 7 and 11·C3H6O could be explained by the rather small bite angle excerted by Ph2PCH2PPh2. We also suppose that the other complexes 8–10, 12 and 13, containing the same phosphine, do not form a structure similar to 7 and 11·C3H6O because of the steric influence of the bulky substituents at the C=S group. In this case the chelate-type coordination of Ph2PCH2PPh2 to one CuI atom, even with small bite angle value, seems to be more efficient.
The six-membered CuSPNCS metallocycles have the conformation of a distorted boat. The fragment NC(S)NP is almost planar, the sulfur of thiophosphoryl group is significantly deviated from the average planes of these fragments. This deviation of the phosphoryl sulfur is common for complexes of N-thioacylamidophosphates.24
In the crystal two molecules of complexes 1, 5, 18, 20 and 21 form centrosymmetric dimers caused by the formation of intermolecular hydrogen bonds (Figs. 1–3, Table 3). These intermolecular H-bonds are formed by the hydrogen atom of the NH group and the sulfur atom of the C=S group of a further molecule. In the crystal of 11·C3H6O there is an intramolecular H-bond formed by the NH hydrogen atom and the iodine atom (Table 3).
| D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A | |
|---|---|---|---|---|---|
| a Symmetry codes: #1 −x + 1, −y + 1, −z + 1. b Symmetry codes: #1 −x + 1/2, y + 1/2, −z + 3/2; #2 −x + 1/2, y −1/2, −z +3/2. | |||||
| 1 | N2–H2⋯S2#1 | 0.85(3) | 2.59(3) | 3.415(2) | 163(3) |
| 5 | N2–H2⋯S2#1 | 0.85(3) | 2.55(3) | 3.383(2) | 165(2) |
| 11·C3H6O | N2–H2⋯I1 | 0.84(3) | 2.85(3) | 3.681(2) | 175(3) |
| 18 | N2–H2⋯S2#1 | 0.902(10) | 2.526(14) | 3.400(2) | 163(3) |
| 20 | N2–H2⋯ S2A#1 | 0.879(10) | 2.681(14) | 3.552(4) | 171(5) |
| N2A–H2A⋯S2#2 | 0.881(10) | 2.691(16) | 3.558(4) | 168(5) | |
| 21 | N2–H2⋯S2#1 | 0.84(2) | 2.55(2) | 3.3469(14) | 160(2) |
The luminescence properties of 1–27 in the solid state at room temperature were investigated. It was found that complexes 1, 21, 22 and 27 show blue emission bands with maxima ranging from 423 to 430 nm upon excitation at 346 nm, while for complexes 7 and 11 this band seems to be red-shifted to 455 and 467 nm, respectively, and further, much more intense emission bands occur at 607 and 613 nm, respectively, under the same conditions (Fig. 5, Table 4).
| Complex | Emission max/nm |
|---|---|
| 1 | 429 |
| 7 | 455, 607 |
| 11 | 467, 613 |
| 21 | 423 |
| 22 | 430 |
| 27 | 426 |
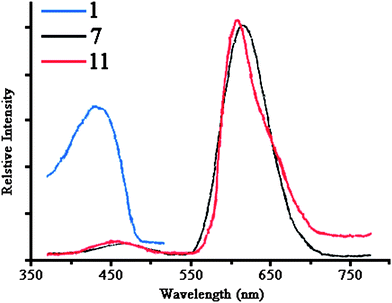 | ||
| Fig. 5 Emission spectra of complexes 1 (spectra of complexes 21, 22 and 27 are similar), 7 and 11 (the excitation wavelength was 346 nm). | ||
All other complexes reported here did no show emission under the same experimental conditions, which is puzzling in view of the highly similar structures e.g.1 (luminescent) and 5 (non-luminescent) or 26 and 27. On the basis of these experiments we can only draw the vague conclusion that the presence of the iodine ligands in 7 and 11 are connected to the observation of the long-wavelength emissions around 610 nm. Presumably they originate from ligand (I−) to metal (CuI) charge transfer transition.
The emissions around 425 or 460 nm are typically observed for CuI complexes containing phosphines.32 On the other hand, quite similar emissions have been observed for ZnII complexes of N-phosphorylated thioamides and thioureas RC(S)NHP(O)(OiPr)2. Based on the present (poor) data we cannot discriminate between these two possiblities.
This demands for a thorough investigation of the luminescence properties, including low-temperature experiments (we might expect luminescence for most of the other species at low temperatures), determination of quantum yields and time-resolved spectroscopy (to assign the character of the excited states), combined with quantum chemical calculations. Such a study is underway at present.
Conclusions
Reaction of [Cu(PPh3)3I] or a mixture of CuI and Ph2P(CH2)nPPh2 (n = 1–3) with the potassium salts of the N-thiophosphorylated thioureas HLI–VI has allowed us to obtain mononuclear 1–6, 8–10, 12–27 or binuclear complexes 7 and 11·C3H6O, containing tetracoordinated CuI.The crystal structures of 1–5, 11·C3H6O, 18, 20 and 21 were determined by single crystal X-ray diffraction. According to the X-ray data, the CuI atom in 1–5, 18, 20 and 21 is coordinate with one phosphorus containing anion through the C=S and P = S sulfur atoms and two molecules of PPh3 (1–5) or one molecule of the bisphosphine ligand in a chelate manner (18, 20 and 21). In the complex 11·C3H6O, the deprotonated thiourea is coordinated towards two CuI atoms, showing a tridentate coordination mode, in which the Cu–S–Cu bridging bond is formed by the C=S sulfur atom. Besides two sulfur atoms the Cu1 cation is coordinated with two phosphorus atoms of the two phosphine molecules, while further two phosphorus atoms are bound to Cu2. The coordination sphere of Cu2 is completed by an iodine atom.
The formation of two binuclear complexes 7 and 11 is favored by the small bite angle of Ph2PCH2PPh2 combined with a small steric demand of the MeNH– or PhNH–group.
Furthermore, the two binuclear complexes 7 and 11 exhibit marked long-wavelength emission in the solid state at ambient temperature, of which the origin is not yet clear.
Experimental
General procedures
NMR spectra were obtained on a Bruker Avance 300 MHz spectrometer at 25 °C. 1H and 31P{1H} NMR spectra (CDCl3) were recorded at 299.948 and 121.420 MHz, respectively. Chemical shifts are reported with reference to SiMe4 (1H) and H3PO4 (31P{1H}). Fluorescence measurements on solid samples were carried out on a Spex FluoroMax-3 spectrofluorometer at room temperature. Elemental analyses were performed on a CHNS HEKAtech EuroEA 3000 analyzer.Syntheses
1 . Yield: 0.369 g (86%). Mp 81–82 °. 1H NMR (acetone-d6): δ 1.26 (d, 3JH,H = 6.2 Hz, 12 H, CH3, iPr), 2.80 (d, 3JH,H = 4.6 Hz, 3 H, CH3, Me), 4.71 (d. sept, 3JH,H = 6.2 Hz, 3JP,H = 10.7 Hz, 2 H, OCH), 7.31–7.45 (m, 30 H, Ph) ppm. 31P{1H} NMR (acetone-d6): δ –4.2 (s, 2 P, PPh3), 55.5 (s, 1 P, NPS) ppm. C44H48CuN2O2P3S2 (857.46): calcd. C 61.63, H 5.64, N 3.27; found C 61.46, H 5.75, N 3.22.
2 . C46H52CuN2O2P3S2 (885.52): calcd. C 62.39, H 5.92, N 3.16; found C 62.25, H 5.82, N 3.28.
3 . C47H64CuN2O2P3S2 (899.54): calcd. C, 62.75; H, 6.05, N, 3.11. Found: C, 62.59; H, 6.12; N, 3.06.
4 . Yield: 0.318 g (73%). Mp 116–117 °. 1H NMR (CDCl3): δ 1.26 (d, 3JH,H = 6.3 Hz, 6 H, CH3, iPr), 1.31 (d, 3JH,H = 6.2 Hz, 6 H, CH3, iPr), 3.13 (s, 3 H, CH3, Me), 3.33 (s, 3 H, CH3, Me), 4.72 (d. sept, 3JH,H = 6.2 Hz, 3JP,H = 10.8 Hz, 2 H, OCH, OiPr), 7.25–7.53 (m, 30 H, Ph) ppm. 31P{1H} NMR (CDCl3): δ 1.5 (s, 2 P, PPh3), 55.0 (s, 1 P, NPS) ppm. C45H50CuN2O2P3S2 (871.49): calcd. C 62.02, H 5.78, N 3.21; found C 62.19, H 5.66, N 3.16.
5 . Yield: 0.337 g (71%). Mp 168–169 °. 1H NMR (DMSO-d6): δ 0.95 (d, 3JH,H = 6.2 Hz, 6 H, CH3, iPr), 1.03 (d, 3JH,H = 6.2 Hz, 6 H, CH3, iPr), 2.15 (s, 6 H, CH3, Ar), 4.33 (d. sept, 3JH,H = 6.0 Hz, 3JP,H = 10.6 Hz, 2 H, OCH), 7.03 (s, 3 H, C6H3), 7.25–7.55 (m, 30 H, Ph), 9.37 (d, 4JP,H = 9.3 Hz, 1 H, NH) ppm. 31P{1H} NMR (DMSO-d6): δ –3.1 (s, 2 P, PPh3), 51.7 (s, 1 P, NPS) ppm. C51H54CuN2O2P3S2 (947.59): calcd. C, 64.64; H, 5.74, N, 2.96. Found: C, 64.80; H, 5.65; N, 3.00.
6 . Yield: 0.308 g (64%). Mp 153–154 °. 1H NMR (CDCl3): δ 1.06 (d, 3JH,H = 6.1 Hz, 6 H, CH3, iPr), 1.14 (d, 3JH,H = 6.0 Hz, 6 H, CH3, iPr), 2.25 (s, 9 H, CH3, Ar), 4.473 (d. sept, 3JH,H = 6.2 Hz, 3JP,H = 10.7 Hz, 2 H, OCH), 6.87 (s, 2 H, C6H2),7.01 (d, 4JP,H = 9.3 Hz, 1 H, NH), 7.29–7.52 (m, 30 H, Ph) ppm. 31P{1H} NMR (CDCl3): δ –2.7 (s, 2 P, PPh3), 52.3 (s, 1 P, NPS) ppm. C52H56CuN2O2P3S2 (961.62): calcd. C, 64.95; H, 5.87, N, 2.91. Found: C, 65.13; H, 5.80; N, 2.86.
7 . Yield: 0.203 g (63%). Mp 208–209 °. 1H NMR (DMSO-d6): δ 1.33 (d, 3JH,H = 5.9 Hz, 6 H, CH3, iPr), 1.38 (d, 3JH,H = 6.0 Hz, 6 H, CH3, iPr), 2.38–2.69 (m, 4 H, CH2), 3.24 (br. s, 3 H, CH3, Me), 4.76 (br. s, 2 H, OCH), 6.39–7.94 (m, 40 H, Ph), 10.41 (br. s, 1 H, NH) ppm. 31P{1H} NMR (DMSO-d6): δ –24.8 — –15.6 (m, 4 P, PPh2), 57.9 (s, 1 P, NPS) ppm. C58H62Cu2IN2O2P5S2 (1292.13): calcd. C 53.91, H 4.84, N 2.17; found C 54.14, H 4.76, N 2.14.
8 . Yield: 0.329 g (88%). Mp 127–128 °. 1H NMR (CDCl3): δ 1.14–1.32 (m, 18 H, CH3, iPrN + iPrO), 2.84–3.02 (m, 2 H, CH2), 3.85–4.18 (m, 1 H, NCH), 4.54–4.86 (m, 2 H, OCH), 6.93–7.89 (m, overlapped with the solvent signal, Ph + NH) ppm. 31P{1H} NMR (CDCl3): δ –25.2 — –8.3 (m, 2 P, PPh2), 56.7 (s, 1 P, NPS) ppm. C35H44CuN2O2P3S2 (745.33): calcd. C 56.40, H 5.95, N 3.76; found C 56.49, H 6.03, N 3.70.
9 . Yield: 0.281 g (74%). Mp 142–143 °. 1H NMR (CDCl3): δ 1.10–1.39 (m, 21 H, CH3, tBu + iPrO), 2.78–2.96 (m, 2 H, CH2), 4.78 (br. s, 2 H, OCH), 7.14–8.05 (m, overlapped with the solvent signal, Ph + NH) ppm. 31P{1H} NMR (CDCl3): δ −24.1— –7.0 (m, 2 P, PPh2), 57.2 (s, 1 P, NPS) ppm. C36H46CuN2O2P3S2 (759.36): calcd. C 56.94, H 6.11, N 3.69; found C 57.11, H 6.24, N 3.62.
10 . Yield: 0.307 g (84%). Mp 114–115 °. 1H NMR (CDCl3): δ 1.17 (d, 3JH,H = 6.1 Hz, 12 H, CH3, iPr), 2.63–2.80 (m, 2 H, CH2), 3.02 (s, 3 H, CH3, Me), 3.25 (s, 3 H, CH3, Me), 4.57–4.89 (m, 2 H, OCH), 7.15–7.98 (m, overlapped with the solvent signal, Ph) ppm. 31P{1H} NMR (CDCl3): δ –22.3 — –10.1 (m, 2 P, PPh2), 58.0 (s, 1 P, NPS) ppm. C34H42CuN2O2P3S2 (731.31): calcd. C 55.84, H 5.79, N 3.83; found C 56.01, H 5.70, N 3.88.
11 . Yield: 0.196 g (58%). Mp 229–2300 °. 1H NMR (DMSO-d6): δ 1.36 (d, 3JH,H = 6.0 Hz, 12 H, CH3, iPr), 2.51 (br. s, 4 H, CH2), 4.78 (br. s, 2 H, OCH), 6.34–7.99 (m, 45 H, Ph), 10.40 (br. s, 1 H, NH) ppm. 31P{1H} NMR (DMSO-d6): δ –25.7 — –12.2 (m, 4 P, PPh2), 58.0 (s, 1 P, NPS) ppm. C63H64Cu2IN2O2P5S2 (1354.20): calcd. C 55.88, H 4.76, N 2.07; found C 59.05, H 4.66, N 2.02.
12 . Yield: 0.278 g (69%). Mp 162–163 °. 1H NMR (CDCl3): δ 0.97 (d, 3JH,H = 5.9 Hz, 6 H, CH3, iPr), 1.06 (d, 3JH,H = 5.9 Hz, 6 H, CH3, iPr), 2.29 (s, 6 H, CH3, Ar), 3.06 (br. s, 2 H, CH2), 4.22–4.57 (m, 2 H, OCH), 6.87–7.89 (m, overlapped with the solvent signal, Ph + C6H3 + NH) ppm. 31P{1H} NMR (CDCl3): δ –17.8 (br. s, 2 P, PPh2), 53.3 (s, 1 P, NPS) ppm. C40H46CuN2O2P3S2 (807.40): calcd. C 59.50, H 5.74, N 3.47; found C 59.37, H 5.81, N 3.40.
13 . Yield: 0.386 g (94%). Mp 155–156 °. 1H NMR (CDCl3): δ 0.83–1.12 (m, 12 H, CH3, iPr), 2.26 (s, 9 H, CH3, Ar), 3.05 (br. s, 2 H, CH2), 4.20–4.63 (m, 2 H, OCH), 6.78–8.03 (m, overlapped with the solvent signal, Ph + C6H2 + NH) ppm. 31P{1H} NMR (CDCl3): δ –19.9 (br. s, 2 P, PPh2), 53.2 (s, 1 P, NPS) ppm. C41H48CuN2O2P3S2 (821.43): calcd. C 59.95, H 5.89, N 3.41; found C 59.81, H 5.93, N 3.35.
14 . Yield: 0.292 g (80%). Mp 83–84 °. 1H NMR (CDCl3): δ 1.29 (t, 3JH,H = 6.0 Hz, 12 H, CH3, iPr), 2.13–2.36 (m, 4 H, CH2), 2.86 (d, 3JH,H = 4.0 Hz, 3 H, CH3, Me), 4.73 (d. sept, 3JH,H = 6.0 Hz, 3JP,H = 10.5 Hz, 2 H, OCH), 6.88–8.20 (m, overlapped with the solvent signal, Ph + NH) ppm. 31P{1H} NMR (CDCl3): δ –9.8 (br. s, 2 P, PPh2), 56.2 (s, 1 P, NPS) ppm. C34H42CuN2O2P3S2 (731.31): calcd. C 55.84, H 5.79, N 3.83; found C 55.68, H 5.83, N 3.78.
15 . Yield: 0.292 g (80%). Mp 95–96 °. 1H NMR (CDCl3): δ 1.13 (d, 3JH,H = 6.6 Hz, 6 H, CH3, iPrN), 1.24 (t, 3JH,H = 6.2 Hz, 12 H, CH3, iPr), 2.15–2.32 (m, 4 H, CH2), 4.10 (d. sept, 3JH,H = 6.6 Hz, 3JP,H = 8.0 Hz, 1 H, NCH), 4.74 (d. sept, 3JH,H = 6.2 Hz, 3JP,H = 10.4 Hz, 2 H, OCH), 5.80 (t, 3JH,H ≈ 4JP,H = 7.5 Hz, 1 H, NH), 6.97–7.87 (m, overlapped with the solvent signal, Ph) ppm. 31P{1H} NMR (CDCl3): δ –12.9 (br. s, 2 P, PPh2), 57.1 (s, 1 P, NPS) ppm. C36H46CuN2O2P3S2 (759.36): calcd. C 56.94, H 6.11, N 3.69; found C 57.09, H 6.05, N 3.75.
16 . Yield: 0.290 g (75%). Mp 117–118 °. 1H NMR (CDCl3): δ 1.08–1.41 (m, 21 H, CH3, tBu + iPr), 2.18–2.39 (m, 4 H, CH2), 4.70 (d. sept, 3JH,H = 6.1 Hz, 3JP,H = 10.2 Hz, 2 H, OCH), 5.85 (br. s, 1 H, NH), 7.06–7.94 (m, overlapped with the solvent signal, Ph) ppm. 31P{1H} NMR (CDCl3): δ –13.5 (br. s, 2 P, PPh2), 57.4 (s, 1 P, NPS) ppm. C37H48CuN2O2P3S2 (773.39): calcd. C 57.46, H 6.26, N 3.62; found C 57.31, H 6.22, N 3.69.
17 . Yield: 0.231 g (62%). Mp 103–104 °. 1H NMR (CDCl3): δ 1.12 (d, 3JH,H = 6.0 Hz, 12 H, CH3, iPr), 2.07–2.29 (m, 4 H, CH2), 3.15 (br. s, 6 H, CH3, Me), 4.61 (d. sept, 3JH,H = 6.2 Hz, 3JP,H = 10.6 Hz, 2 H, OCH), 6.94–7.80 (m, overlapped with the solvent signal, Ph) ppm. 31P{1H} NMR (CDCl3): δ –14.8 (br. s, 2 P, PPh2), 56.4 (s, 1 P, NPS) ppm. C35H44CuN2O2P3S2 (745.33): calcd. C 56.40, H 5.95, N 3.76; found C 56.23, H 6.02, N 3.71.
18 . Yield: 0.365 g (92%). Mp 132–133 °. 1H NMR (CDCl3): δ 1.23 (d, 3JH,H = 6.0 Hz, 12 H, CH3, iPr), 2.28 (br. s, 4 H, CH2), 3.06 (br. s, 2 H, CH2), 4.74 (br. s, 2 H, OCH), 6.93–7.79 (m, overlapped with the solvent signal, Ph + NH) ppm. 31P{1H} NMR (CDCl3): δ –13.2 (br. s, 2 P, PPh2), 57.3 (s, 1 P, NPS) ppm. C39H44CuN2O2P3S2 (793.38): calcd. C 59.04, H 5.59, N 3.53; found C 59.12, H 5.41, N 3.50.
19 . Yield: 0.222 g (54%). Mp 140–141 °. 1H NMR (CDCl3): δ 1.03 (d, 3JH,H = 5.9 Hz, 6 H, CH3, iPr), 1.12 (d, 3JH,H = 5.9 Hz, 6 H, CH3, iPr), 2.26 (s, 6 H, CH3, Ar), 2.40 (br. s, 4 H, CH2), 4.29–4.63 (m, 2 H, OCH), 6.89–7.83 (m, overlapped with the solvent signal, Ph + C6H3 + NH) ppm. 31P{1H} NMR (CDCl3): δ –12.9 (br. s, 2 P, PPh2), 52.6 (s, 1 P, NPS) ppm. C41H48CuN2O2P3S2 (821.43): calcd. C 59.95, H 5.89, N 3.41; found C 60.13, H 5.82, N 3.34.
20 . Yield: 0.330 g (79%). Mp 152–153 °. 1H NMR (CDCl3): δ 0.99 (d, 3JH,H = 6.1 Hz, 6 H, CH3, iPr), 1.06 (d, 3JH,H = 6.1 Hz, 6 H, CH3, iPr), 2.13–2.33 (m, 13 H, CH3 (Ar) + CH2), 4.48 (d. sept, 3JH,H = 6.0 Hz, 3JP,H = 10.7 Hz, 2 H, OCH), 6.70–7.89 (m, overlapped with the solvent signal, Ph + C6H2 + NH) ppm. 31P{1H} NMR (CDCl3): δ –13.1 (br. s, 2 P, PPh2), 55.9 (s, 1 P, NPS) ppm. C42H50CuN2O2P3S2 (835.46): calcd. C 60.38, H 6.03, N 3.35; found C 60.19, H 5.97, N 3.40.
21 . Yield: 0.227 g (61%). Mp 70–71 °. 1H NMR (CDCl3): δ 1.05–1.42 (m, 12 H, CH3, iPr), 1.50–2.89 (m, 9 H, CH3 (Me) + CH2), 4.52–4.86 (m, 2 H, OCH), 7.04–7.28 (m, overlapped with the solvent signal, Ph + NH) ppm. 31P{1H} NMR (CDCl3): δ –21.0 (br. s, 2 P, PPh2), 59.2 (s, 1 P, NPS) ppm. C35H44CuN2O2P3S2 (745.33): calcd. C 56.40, H 5.95, N 3.76; found C 56.23, H 5.89, N 3.81.
22 . Yield: 0.321 g (83%). Mp 78–79 °. 1H NMR (CDCl3): δ 1.02–1.32 (m, 18 H, CH3, iPrN + iPrO), 1.56–1.91, 2.24–2.55 (m, 6 H, CH2), 4.03 (br. s, 1 H, iPrN), 4.68 (d. sept, 3JH,H = 6.1 Hz, 3JP,H = 10.6 Hz, 2 H, OCH), 6.95–7.97 (m, overlapped with the solvent signal, Ph + NH) ppm. 31P{1H} NMR (CDCl3): δ –21.3 (br. s, 2 P, PPh2), 54.5 (s, 1 P, NPS) ppm. C37H48CuN2O2P3S2 (773.39): calcd. C 57.46, H 6.26, N 3.62; found C 57.34, H 6.37, N 3.58.
23 . Yield: 0.260 g (66%). Mp 65–66 °. 1H NMR (CDCl3): δ 1.12–1.39 (m, 21 H, CH3, tBu + iPr), 1.52–1.84, 2.20–2.53 (m, 6 H, CH2), 4.78 (d. sept, 3JH,H = 6.0 Hz, 3JP,H = 10.2 Hz, 2 H, OCH), 6.73–7.84 (m, overlapped with the solvent signal, Ph + NH) ppm. 31P{1H} NMR (CDCl3): δ –18.4 (br. s, 2 P, PPh2), 56.0 (s, 1 P, NPS) ppm. C38H50CuN2O2P3S2 (787.41): calcd. C 57.96, H 6.40, N 3.56; found C 58.10, H 6.46, N 3.49.
24 . Yield: 0.361 g (95%). Mp 93–94 °. 1H NMR (CDCl3): δ 1.09–1.36 (m, 12 H, CH3, iPr), 1.83–2.18, 2.31–2.57 (m, 6 H, CH2), 3.32 (br. s, 6 H, CH3, Me), 4.47 (d. sept, 3JH,H = 6.0 Hz, 3JP,H = 10.1 Hz, 2 H, OCH), 6.81–7.72 (m, overlapped with the solvent signal, Ph) ppm. 31P{1H} NMR (CDCl3): δ –20.5 (br. s, 2 P, PPh2), 55.3 (s, 1 P, NPS) ppm. C36H46CuN2O2P3S2 (759.36): calcd. C 56.94, H 6.11, N 3.69; found C 57.15, H 6.07, N 3.74.
25 . Yield: 0.327 g (81%). Mp 126–127 °. 1H NMR (CDCl3): δ 1.20 (d, 3JH,H = 6.1 Hz, 6 H, CH3, iPr), 1.26 (d, 3JH,H = 6.1 Hz, 6 H, CH3, iPr), 2.43 (br. s, 4 H, CH2), 2.93 (br. s, 2 H, CH2), 4.70 (br. s, 2 H, OCH), 6.90–7.86 (m, overlapped with the solvent signal, Ph), 8.03 (s, 1 H, NH) ppm. 31P{1H} NMR (CDCl3): δ –17.7 (br. s, 2 P, PPh2), 56.7 (s, 1 P, NPS) ppm. C40H46CuN2O2P3S2 (807.40): calcd. C 59.50, H 5.74, N 3.47; found C 59.68, H 5.67, N 3.52.
26 . Yield: 0.234 g (56%). Mp 139–140 °. 1H NMR (CDCl3): δ 0.87 (d, 3JH,H = 6.0 Hz, 6 H, CH3, iPr), 0.97 (d, 3JH,H = 5.9 Hz, 6 H, CH3, iPr), 2.12 (s, 6 H, CH3, Ar), 2.16–2.48 (m, 6 H, CH2), 4.17–4.50 (m, 2 H, OCH), 6.91–7.69 (m, overlapped with the solvent signal, Ph + C6H3 + NH) ppm. 31P{1H} NMR (CDCl3): δ –21.2 (br. s, 2 P, PPh2), 55.6 (s, 1 P, NPS) ppm. C42H50CuN2O2P3S2 (835.46): calcd. C 60.38, H 6.03, N 3.35; found C 60.22, H 5.97, N 3.38.
27 . Yield: 0.297 g (70%). Mp 181–182 °. 1H NMR (CDCl3): δ 0.90 (d, 3JH,H = 6.0 Hz, 6 H, CH3, iPr), 0.99 (d, 3JH,H = 6.0 Hz, 6 H, CH3, iPr), 2.09 (s, 6 H, CH3, Ar), 2.15–2.54 (m, 6 H, CH2), 4.37 (d. sept, 3JH,H = 6.0 Hz, 3JP,H = 10.6 Hz, 2 H, OCH), 6.71–7.76 (m, overlapped with the solvent signal, Ph + C6H2 + NH) ppm. 31P{1H} NMR (CDCl3): δ –21.3 (br. s, 2 P, PPh2), 55.4 (s, 1 P, NPS) ppm. C43H52CuN2O2P3S2 (849.49): calcd. C 60.80, H 6.17, N 3.30; found C 60.62, H 6.24, N 3.27.
X-Ray crystallography
The X-ray diffraction data for the crystals of 1, 4, 5, 11·C3H6O, 18, 20 and 21 were collected on a STOE IPDS-II diffractometer. The images were indexed, integrated and scaled using the X-Area package.34 Data were corrected for absorption using the PLATON35 program. The structures were solved by direct methods using SHELXS36 program and refined first isotropically and then anisotropically using SHELXL97.36 Hydrogen atoms were revealed from Δρ maps and refined using a riding model. All figures were generated using the program XP.37 The N–H distances in 18 and 20 were restrained to 0.90(1) Å.The X-ray diffraction data for 2 were collected on a Bruker AXS APEX CCD diffractometer equipped with a rotation anode. Diffraction data were collected over the full sphere and were corrected for absorption. The data reduction was performed with the Bruker SMART program package. Structure solutions were found with the SHELXS9736 package using the heavy-atom method and were refined with SHELXL9736 against F2 using first isotropic and later anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms were added to the structure models on calculated positions.
Acknowledgements
This work was supported by the Russian Science Support Foundation. DAS and MGB thank DAAD for the scholarships (Forschungsstipendien 2008/2009).Notes and references
- F. D. Sokolov, M. G. Babashkina, D. A. Safin, A. I. Rakhmatullin, F. Fayon, N. G. Zabirov, M. Bolte, V. V. Brusko, J. Galezowska and H. Kozlowski, Dalton Trans., 2007, 4693 RSC.
- F. D. Sokolov, M. G. Babashkina, F. Fayon, A. I. Rakhmatullin, D. A. Safin, T. Pape and F. E. Hahn, J. Organomet. Chem., 2009, 694, 167 CrossRef CAS.
- M. G. Babashkina, D. A. Safin, Ł. Szyrwiel, M. Kubiak, F. D. Sokolov, Y. V. Starikov and H. Kozlowski, Z. Anorg. Allg. Chem., 2009, 635, 554 CrossRef CAS.
- R. C. Luckay, X. Sheng, C. E. Strasser, H. G. Raubenheimer, D. A. Safin, M. G. Babashkina and A. Klein, Dalton Trans., 2009, 4646 RSC.
- D. A. Safin, M. G. Babashkina, M. Bolte, F. D. Sokolov and V. V. Brusko, Inorg. Chim. Acta, 2009, 362, 1895 CrossRef CAS.
- N. G. Zabirov, A. Yu. Verat, F. D. Sokolov, M. G. Babashkina, D. B. Krivolapov and V. V. Brusko, Mendeleev Commun., 2003, 13, 163 CrossRef.
- A. Y. Verat, F. D. Sokolov, N. G. Zabirov, M. G. Babashkina, D. B. Krivolapov, V. V. Brusko and I. A. Litvinov, Inorg. Chim. Acta, 2006, 359, 475 CAS.
- D. A. Safin, M. G. Babashkina, F. D. Sokolov, A. Klein, D. B. Krivolapov, I. A. Litvinov, in preparation.
- D. A. Safin, M. G. Babashkina, T. R. Gimadiev, M. Bolte, M. V. Pinus, D. B. Krivolapov and I. A. Litvinov, Polyhedron, 2008, 27, 2978 CrossRef CAS.
- D. A. Safin, M. G. Babashkina, F. D. Sokolov, N. G. Zabirov, J. Galezowska and H. Kozlowski, Polyhedron, 2007, 26, 1113 CrossRef CAS.
- C. Di Nicola, Effendy, F. Fazaroh, C. Pettinari, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2005, 358, 720 CrossRef CAS.
- A. Cingolani, C. Di Nicola, Effendy, C. Pettinari, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2005, 358, 748 CrossRef CAS.
- Effendy, C. Di Nicola, M. Fianchini, C. Pettinari, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2005, 358, 763 CrossRef.
- C. Di Nicola, C. Pettinari, M. Ricciutelli, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2005, 358, 4003 CrossRef CAS.
- C. Di Nicola, Effendy, C. Pettinari, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2006, 359, 53 CrossRef CAS.
- Effendy, C. Di Nicola, C. Pettinari, A. Pizzabiocca, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2006, 359, 64 CrossRef.
- C. Di Nicola, G. A. Koutsantonis, C. Pettinari, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2006, 359, 2159 CrossRef CAS.
- R. D. Hart, J. D. Kildea, C. Pettinari, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2006, 359, 2178 CrossRef CAS.
- M. I. Bruce, N. A. Zaitseva, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2007, 360, 681 CrossRef CAS.
- V. W.-W. Yam, K. K.-W. Lo, C.-R. Wang and K.-K. Cheung, J. Phys. Chem. A, 1997, 101, 4666 CrossRef CAS.
- A. Kaltzoglou, P. J. Cox and P. Aslanidis, Inorg. Chim. Acta, 2005, 358, 3048 CrossRef CAS.
- P. Aslanidis, P. J. Cox, S. Divanidis and A. C. Tsipis, Inorg. Chem., 2002, 41, 6875 CrossRef CAS.
- T. S. Lobana, R. Sharma, R. Sharma, S. Mehra, A. Castineiras and P. Turner, Inorg. Chem., 2005, 44, 1914 CrossRef CAS.
- F. D. Sokolov, V. V. Brusko, N. G. Zabirov and R. A. Cherkasov, Curr. Org. Chem., 2006, 10, 27 CrossRef CAS.
- C. Ohrenberg, L. M. Liable-Sands, A. L. Rheingold and C. G. Riordan, Inorg. Chem., 2001, 40, 4276 CrossRef CAS.
- J. E. Huheey, E. A. Keiter, R. L. Keiter (Eds.), Inorganic Chemistry: Principles of Structure and Reactivity, fourth ed., Harper Collins College Publishers, New York, 1993 Search PubMed.
- I. Haiduc, R. Cea-Olivares, R. A. Toscano and C. Silvestru, Polyhedron, 1995, 14, 1067 CrossRef CAS.
- J. Novosad, M. Necas, J. Marek, P. Veltsistas, C. Papadimitriou, I. Haiduc, M. Watanabe and J. D. Woollins, Inorg. Chim. Acta, 1999, 290, 256 CrossRef CAS.
- D. J. Birdsal, J. Green, T. Q. Ly, J. Novosad, M. Necas, A. M. Z. Slawin, J. D. Woollins and Z. Zak, Eur. J. Inorg. Chem., 1999, 1445 CrossRef CAS.
- A. Silvestru, A. Rotar, J. E. Drake, M. B. Hursthouse, M. E. Light, S. I. Farcas, R. Roesler and C. Silvestru, Can. J. Chem., 2001, 79, 983 CrossRef CAS.
- P. F. Barron, J. C. Dyason, P. C. Healy, L. M. Engelhardt, C. Pakawatchai, V. A. Patrick and A. H. White, J. Chem. Soc., Dalton Trans., 1987, 1099 RSC.
- A. Vogler and H. Kunkely, Coord. Chem. Rev., 2002, 230, 243 CrossRef CAS.
- M. G. Babashkina, PhD thesis, Kazan State University, Kazan, Russian Federation, 2006.
- Stoe & Cie. X-Area. Area-Detector Control and Integration Software. Stoe & Cie, Darmstadt, Germany, 2001 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 64, 112 CrossRef.
- G. M. Sheldrick, SHELXTL-Plus. Release 4.1. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA, 1991 Search PubMed.
Footnote |
| † CCDC reference numbers 730348 (1), 717086 (2), 712924 (4), 730349 (5), 730350 (11·C3H6O), 730351 (18), 730352 (20) and 730353 (21), respectively. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b914163d |
| This journal is © The Royal Society of Chemistry 2010 |