New synthesis of meso-free-[14]triphyrin(2.1.1) by McMurry coupling and its derivatization to Mn(I) and Re(I) complexes†
Daiki
Kuzuhara
a,
Hiroko
Yamada
*a,
ZhaoLi
Xue
a,
Tetsuo
Okujima
a,
Shigeki
Mori
b,
Zhen
Shen
c and
Hidemitsu
Uno
a
aGraduate School of Science and Engineering, Ehime University, 2–5, Bunkyo-cho, Matsuyama, 790-8577, Japan. E-mail: yamada@chem.sci.ehime-u.ac.jp; Fax: +81 89-927-9613; Tel: +81 89-927-9613
bDepartment of Molecular Science, Integrated Center for Sciences, Ehime University, Matsuyama 790-8577, Japan
cState Key Laboratory of Coordination Chemistry, Nanjing National Laboratory of Microstructures, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China
First published on 16th November 2010
Abstract
The metal-free and meso-free [14]triphyrin(2.1.1) compound was successfully prepared based on the intramolecular McMurry coupling reaction of diformyl-tripyrrane in 16% yield, and was converted to the bowl-shaped Mn(I)(TriP)(CO)3 and Re(I)(TriP)(CO)3.
Triphyrins are porphyrin analogues which contain only three pyrrole or pyrrole related rings linked through meso-sp2carbon atoms.1–4 There has been a considerable interest because of their potential applications as the functional materials. In 2006, Osuka and coworkers reported the synthesis of [14]triphyrin(1.1.1), so-called tribenzosubporphyrins, as 14 π-electron porphyrin analogues.2a Subsequently, Kobayashi and Osuka et al. independently developed synthetic protocols for meso-aryl substituted subporphyrins as their boron-complexes.2,3 The triphyrins reported to date have all been boron complexes with highly nonplanar dome-shaped conformations, with the exception of subpyriporphyrin5a and triphyrins with more than four meso carbons.5b–g In 2008, we have succeeded in preparing nearly planar metal-free meso-aryl-substituted [14]triphyrin(2.1.1) compounds by acid-catalyzed condensation of bicyclo[2.2.2]octadiene(BCOD)-fused pyrrole and aromatic aldehydes.6 These compounds had never been reported until they were found serendipitously during the synthesis of tetraarylporphyrins by the well-known Lindsey method. The [14]triphyrin(2.1.1) compounds represent the first examples of near planar metal-free contracted porphyrinoids with a 14 π-electron aromatic system containing only the pyrrole moieties.
Including the [14]triphyrin(2.1.1), however, the previously reported triphyrins and their analogues have been mainly meso-substituted compounds. One exception is meso-free benzotriphyrin prepared by Osuka et al.; heating of the mixture of 2-(3-oxo-2,3-dihydro-1H-isoindole-1-yl) acetic acid and boronic acid at 350 °C.2a For the further generalization of the [14]triphyrin chemistry, their meso-free and/or metal complexes are desired. We will report here the new synthetic method of metal-free and meso-free [14]triphyrin(2.1.1) 1 based on the intramolecular McMurry coupling reaction of diformyl-tripyrrane. The Mn(I) and Re(I) triphyrins were also successfully obtained by the reaction of triphyrin 1 with Mn(CO)5Br or Re(CO)5Cl, respectively.
A synthetic scheme is shown in Scheme 1. Intramolecular McMurry coupling of diformyltripyrrane 37 was performed in THF and the reaction was quenched with 10% aqueous K2CO3. After a filtration by celite, the solvent of the organic layer was removed under a reduced pressure. The residue was purified by alumina column chromatography with CHCl3 and the obtained green residue was oxidized by DDQ. The purification by silica gel column chromatography (20% EtOAc in n-hexane) gave triphyrin 1 as red solid in 16% yield. The triphyrin 1 is soluble in common organic solvents, such as CHCl3, CH2Cl2, THF, n-hexane, and MeOH.
 | ||
| Scheme 1 Synthetic schemes of triphyrins 1, 1–Mn, and 1–Re. | ||
The NMR spectrum of triphyrin 1 measured in CDCl3 is shown in Fig. 1a. It is symmetrical and shows peaks of two kinds of meso-H (8.88 and 8.87 ppm). Inner NH proton is observed at 7.25 ppm as a broad signal. The FAB-mass spectrum shows the peak at 442, which corresponds to [M + H]+ of triphyrin 1 (Fig. S1, ESI†). The absorption spectrum of the triphyrin 1 was measured in CH2Cl2 (Fig. 2). The intense peak (similar to Soret peak of porphyrins) is shown at 336 nm and the broad weaker bands (similar to Q bands) in the 440–560 nm with broad peaks at 470 and 550 nm. They are 20 nm blue-shifted from those of meso-aryl-substituted [14]triphyrin(2.1.1).6
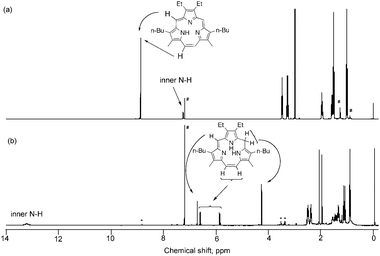 | ||
| Fig. 1 1H-NMR spectra of (a) 1 and (b) intermediate 2 in CDCl3. # represents the solvent peaks. (b) * represents the peaks of triphyrin 1 generated during the measurement of 2. | ||

To clarify the reaction mechanism, the green intermediate after the rough purification by alumina chromatography was further purified by silica-gel column chromatography with CH2Cl2. The NMR spectrum of the intermediate 2 is shown in Fig. 1b. It is not symmetrical and the meso-peaks of 1 at 8.87 and 8.88 ppm disappeared. Meso-methine proton is observed at 6.77 ppm as singlet and ethynyl protons at 5.90 and 6.66 ppm as doublets (J = 12 Hz). The singlet peak for 2H is observed at 4.31 ppm, which is characterized as methylene protons between pyrrole rings. The intermediate was not stable under air and the color changed gradually in solution. Small peaks of triphyrin 1 are observed in Fig. 1b, which appeared during the measurement. These results suggested the structure of the intermediate 2 as shown in Scheme 1.
The crystals of triphyrin 1 were obtained in MeOH–ethylene glycol. The X-ray crystal structure of 1 is shown in Fig. 3.‡ The diethylpyrrole unit is slightly tilted with respect to macrocycle and the dihedral angle between the mean planes of diethylpyrrole and the other part of triphyrin plane is 10.68°. N–N distances are 2.504(2), 2.502(2), and 2.552(2) Å for N1–N2, N2–N3, and N3–N1, respectively. The four carbons, C2, C1, C14, and C13, are almost in line but the angle made by C7–C3–C4 was 20.01(9)°. This means that the hydrogen atom on N1 was directed to N3 rather than to N2, and therefore NH hydrogen atom was mainly delocalized on the N1 and N3.
 | ||
| Fig. 3 X-Ray crystallographic structures of 1. (a) Top view and (b) side view. Hydrogen atoms and alkyl groups for side views are omitted for clarity. The nitrogen–nitrogen distances of N1–N2, N2–N3, and N3–N1 are 2.504(2), 2.502(2), and 2.552(2) Å, respectively. | ||
To confirm the structure of 1, NMR spectra in CD2Cl2 were measured from room temperature to −90 °C (Fig. S2, ESI†). At room temperature the inner proton is broad at 6.68 ppm and disappeared by lowering the temperature. It re-observed at 7.27 ppm at −60 °C and shifted to 7.30 ppm at −90 °C. The peak at 6.68 ppm disappeared by addition of CD3OD, to confirm the peaks to inner NH proton. At room temperature two singlet peaks due to meso-protons are observed at 8.90 and 8.82 ppm. The peak at 8.82 ppm was broadened at −40 °C with methylene quartet peaks at 3.2 ppm, but the other meso-peak remained sharp. The broadened meso-proton was re-observed at 8.89 ppm at −90 °C. These results indicated that the diethylpyrrole flip-flopped at room temperature but methylbutylpyrrole did not because of the hydrogen bonding between inner hydrogen and nitrogen atoms.
Electron distributions of LUMO and HOMO of tautomers of triphyrin 1 with inner proton on N1 or N2 were estimated by density functional theory (DFT) calculations where the geometry optimization was carried out at the B3LYP/6-31G(*) level (Fig. S3, ESI†).8 When the inner proton is located on N2, the triphyrin is flat. However when the proton is located on N1, diethylpyrrole is twisted and N2 is raised from the triphyrin plane. The latter structure is well-coincident with the crystal structure of 1 in Fig. 1a.
We have also succeeded in preparing Mn(I)-coordinated triphyrin (MnTriP(CO)3: 1–Mn) and Re(I)-coordinated triphyrin (ReTriP(CO)3: 1–Re). Triphyrin 1 was treated with MnBr(CO)5 in toluene in the presence of NaOAc. After 1 h reflux of the solution, the solvent was removed and the residue was purified by silica-gel column chromatography (CH2Cl2) to give 1–Mn in 80% yield. For 1H-NMR, the NH proton of 1 disappeared. IR spectra showed clear absorptions at 2007 and 1889 cm−1, due to typical symmetric and asymmetric vibration modes for three CO ligands which were not observed for free-base 1. UV-vis spectrum of 1–Mn is shown in Fig. 1. The Soret-like peak is broadened and 5 nm blue-shifted. The Q-like band appears around 470 nm and the edge reaches around 750 nm.
In a similar procedure with the synthesis of 1–Mn, 1–Re was also prepared in 71% yield. The Soret peak is observed at 347 nm and 10 and 6 nm red-shifted from triphryin 1 and 1–Mn, respectively. The Q-like band is very similar to that of 1–Mn and it appears around 470 nm and the edge reaches around 750 nm.
The crystals of triphyrin 1–Mn and 1–Re were obtained in MeOH–ethylene glycol. The structures are shown in Fig. 4.‡ Three inner nitrogen atoms and three CO moieties are coordinated to the MnI and ReI ions, respectively. The triphyrins are bowl-shaped and Mn and Re ions are above the plane made by three inner nitrogens by 1.300 Å and 1.455 Å, respectively. The distances between C2–C13 and C7–C16 are 5.986(3) and 6.496(3) Å for 1–Mn and 5.981(3) and 6.383(3) Å for 1–Re, respectively. The nitrogen–nitrogen distances of N1–N2, N2–N3, and N3–N1 were 2.586(2), 2.594(2), and 2.696(2) Å for 1–Mn and 2.650(2), 2.654(2), and 2.779(2) for 1–Re, respectively. Each N–N length of 1–Mn is 0.06 Å shorter than that of 1–Re, because of the larger size of ReI ion than MnI ion. This also influences the shape of the bowl: the curve of 1–Re is deeper than that of 1–Mn.
 | ||
| Fig. 4 X-Ray crystallographic structures of (a) 1–Mn and (b) 1–Re. Upper: top views; lower: side views. Hydrogen atoms and alkyl groups for side views are omitted for clarity. The nitrogen–nitrogen distances of N1–N2, N2–N3, and N3–N1 are 2.586(2), 2.594(2), and 2.696(2) Å for 1–Mn and 2.650(2), 2.654(2), and 2.779(2) Å for 1–Re, respectively. | ||
The cyclic voltammogram of 1 in CH2Cl2 containing 0.1 M Bu4NPF6 at room temperature consists of a single electron reduction process (E0red = −1.77 V vs. Fc/Fc+) and a single one-electron oxidation process (E0ox = 0.63 V vs. Fc/Fc+), as shown in Fig. S4a and Table S1 (ESI†). The reduction was reversible but the oxidation was irreversible. The cyclic voltammogram of 1–Mn consists of a single electron reduction process (E0red = −1.79 V vs. Fc/Fc+) and two one-electron oxidation processes (E0ox = 0.25 V and Epox = 0.97 V vs. Fc/Fc+). The reduction and the first oxidation reactions were reversible but the second oxidation was irreversible. The reversible first oxidation occurred at Mn(I) center. The cyclic voltammogram of 1–Re is very similar to that of 1–Mn: E0red = −1.63 V, E0ox = 0.48 and 1.10 V vs. Fc/Fc+, and the reversible first oxidation corresponded to the oxidation of Re(I) center.
In conclusion we have successfully prepared meso-free hexaalkyl[14]triphyrin(2.1.1) from diformyltripyrrane by McMurry coupling. The reaction mechanism of the reductive cyclization mechanism was clarified by the NMR measurement. The Mn(I) and Re(I) complexes were also prepared and the bowl-shaped structures were confirmed by the X-ray analysis. With this method we can freely design the substituents at β- and/or meso-positions of triphyrins. These triphyrins will be promising as new ligands in the coordination chemistry.
Authors thank the Venture Business Laboratory of Ehime University, for its help on TOF-MS spectroscopy. D. K. thanks JSPS for a Research Fellowship for Young Scientists and Z. X. thanks JSPS for a postdoctoral fellowship for foreign researchers. This work was partially supported by Grants-in-Aid for the Scientific Researches on Innovative Areas (No 22350083 to H. Y., No 20550047 and π-Space to H. U.) from the Japanese Ministry of Education, Culture, Sports, Science and Technology. H. Y. also thanks JST, CREST for the partial support.
Notes and references
- Y. Inokuma and A. Osuka, Dalton Trans., 2008, 2517 RSC.
- (a) Y. Inokuma, J. H. Kwon, T. K. Ahn, M.-C. Yoo, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 961 CrossRef CAS; (b) Y. Inokuma, Z. S. Yoon, D. Kim and A. Osuka, J. Am. Chem. Soc., 2007, 129, 4747 CrossRef CAS; (c) Y. Inokuma and A. Osuka, Chem. Commun., 2007, 2938 RSC; (d) S. Saito, K. S. Kim, Z. S. Yoon, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2007, 46, 5591 CrossRef CAS; (e) Y. Inokuma, S. Easwaramoorthi, S. Y. Jang, K. S. Kim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 4848; (f) Y. Inokuma, S. Easwaramoorthi, Z. S. Yoon, D. Kim and A. Osuka, J. Am. Chem. Soc., 2008, 130, 12234 CrossRef CAS; (g) E. Tsurumaki, Y. Inokuma, S. Easwaramoorthi, J. M. Lim, D. Kim and A. Osuka, Chem.–Eur. J., 2009, 15, 237 CrossRef CAS; (h) S. Easwaramoorthi, J.-Y. Shin, S. Cho, P. Kim, Y. Inokuma, E. Tsurumaki, A. Osuka and D. Kim, Chem.–Eur. J., 2009, 15, 12005 CrossRef CAS; (i) Y. Inokuma and A. Osuka, Chem.–Eur. J., 2009, 15, 6863 CrossRef CAS; (j) S. Hayashi, Y. Inokuma, S. Easwaramoorthi, K. S. Kim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 321 CAS; (k) S. Hayashi, Y. Inokuma and A Osuka, Org. Lett., 2010, 12, 4148 CrossRef CAS.
- (a) N. Kobayashi, Y. Takeuchi and A. Matsuda, Angew. Chem., Int. Ed., 2007, 46, 758 CrossRef CAS; (b) Y. Takeuchi, A. Matsuda and N. Kobayashi, J. Am. Chem. Soc., 2007, 129, 8271 CrossRef CAS; (c) E. A. Makarova, S. Shimizu, A. Matsuda, E. A. Luk'yanets and N. Kobayashi, Chem. Commun., 2008, 2109 RSC; (d) S. Shimizu, A. Matsuda and N. Kobayashi, Inorg. Chem., 2009, 48, 7885 CrossRef CAS.
- (a) T. Xu, R. Lu, X. Liu, P. Chen, X. Qiu and Y. Zhao, Eur. J. Org. Chem., 2008, 1065 CrossRef; (b) X. Liu, R. Lu, T. Xu, D. Xu, Y. Zhan, P. Chen, X. Qiu and Y. Zhao, Eur. J. Org. Chem., 2009, 53.
- (a) R. Myśliborski, L. Latos-Grażyński, L. Szterenberg and T. Lis, Angew. Chem., Int. Ed., 2006, 45, 3670 CrossRef CAS; (b) E. Pacholska, L. Latos-Grażyński and Z. Ciunik, Chem.–Eur. J., 2002, 8, 5403 CrossRef CAS; (c) A. Berlicka, L. Latos-Grażyński and T. Lis, Angew. Chem., Int. Ed., 2005, 44, 5288 CrossRef CAS; (d) A. Krivokapic, A. R. Cowley and H. L. Anderson, J. Org. Chem., 2003, 68, 1089 CrossRef CAS; (e) G. M. Badger, J. A. Elix and G. E. Lewis, Proc. Chem. Soc., London, 1964, 82 Search PubMed; (f) G. M. Badger, J. A. Elix and G. E. Lewis, Aust. J. Chem., 1965, 18, 70 CAS; (g) Z. Hu, J. L. Atwood and M. P. Cava, J. Org. Chem., 1994, 59, 8071 CrossRef CAS.
- Z.-L. Xue, Z. Shen, J. Mack, D. Kuzuhara, H. Yamada, T. Okujima, N. Ono, X.-Z. You and N. Kobayashi, J. Am. Chem. Soc., 2008, 130, 16478 CrossRef CAS.
- N. Ono, K. Kuroki, E. Watanabe, N. Ochi and H. Uno, Heterocycles, 2004, 62, 365 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 03, R. C., Gaussian, Inc., Wallingford CT, 2004. The full list of authors is given in ESI† Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis and Fig. S1–S4 and Table S1. CCDC 790488 (1), 790489 (1-Mn) and 794381 (1-Re). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc04286b |
‡ Crystal data for 1 (C30H39N3): MW = 441.66, monoclinic P21/c, a = 14.5650(3), b = 6.28328(13), c = 28.6812(6) Å, β = 105.3790(10)°, V = 2530.81(9) Å3, T = 100(2) K, Z = 4. 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 028 reflections were measured, and Rmerge = 0.0752, R1 = 0.0558 (3297, I > 2σ(I)), wR2 (all) = 0.1487 (4635), GOF = 1.057. Crystal data for 1–Mn (C33H38MnN3O3): MW = 579.62, monoclinic P21/c, a = 10.844(2), b = 15.867(3), c = 17.615(4) Å, β = 99.781(3)°, V = 2986.8(11) Å3, T = 100(2) K, Z = 4. 48856 reflections were measured, and Rmerge = 0.0657, R1 = 0.0485 (6371, I > 2σ(I)), wR2 (all) = 0.1069 (6835), GOF = 1.111. Crystal data for 1–Re (C33H38ReN3O3): MW = 710.87, triclinic P 028 reflections were measured, and Rmerge = 0.0752, R1 = 0.0558 (3297, I > 2σ(I)), wR2 (all) = 0.1487 (4635), GOF = 1.057. Crystal data for 1–Mn (C33H38MnN3O3): MW = 579.62, monoclinic P21/c, a = 10.844(2), b = 15.867(3), c = 17.615(4) Å, β = 99.781(3)°, V = 2986.8(11) Å3, T = 100(2) K, Z = 4. 48856 reflections were measured, and Rmerge = 0.0657, R1 = 0.0485 (6371, I > 2σ(I)), wR2 (all) = 0.1069 (6835), GOF = 1.111. Crystal data for 1–Re (C33H38ReN3O3): MW = 710.87, triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.1361(17), b = 10.5681(18), c = 13.789(2) Å, α = 87.855(4)°, β = 87.178(5)°, γ = 84.036(6)°, V = 1466.5(4) Å3, T = 100(2) K, Z = 2. 24082 reflections were measured, and Rmerge = 0.0257, R1 = 0.0156 (6578, I > 2σ(I)), wR2 (all) = 0.0387 (6703), GOF = 1.108. CCDC 790488 (1), CCDC 790489 (1–Mn) and CCDC 794381 (1–Re). , a = 10.1361(17), b = 10.5681(18), c = 13.789(2) Å, α = 87.855(4)°, β = 87.178(5)°, γ = 84.036(6)°, V = 1466.5(4) Å3, T = 100(2) K, Z = 2. 24082 reflections were measured, and Rmerge = 0.0257, R1 = 0.0156 (6578, I > 2σ(I)), wR2 (all) = 0.0387 (6703), GOF = 1.108. CCDC 790488 (1), CCDC 790489 (1–Mn) and CCDC 794381 (1–Re). |
| This journal is © The Royal Society of Chemistry 2011 |