Nitroxide polymer networks formed by Michael addition: on site-cured electrode-active organic coating†
Takeshi
Ibe
a,
Rainer B.
Frings
b,
Artur
Lachowicz
b,
Soichi
Kyo
a and
Hiroyuki
Nishide
*a
aDepartment of Applied Chemistry, Waseda University, 169-8555 Tokyo, Japan. E-mail: nishide@waseda.jp; Fax: +81 (0)3 3209 5522; Tel: +81 (0)3 3200 2669
bDIC Berlin GmbH R&D Laboratory, 13403 Berlin, Germany. E-mail: lachowicz-a@dic-berlin.de; Fax: +49 (0)30 435 79 0 10; Tel: +49 (0)30 435 79 9 10
First published on 23rd April 2010
Abstract
Highly and homogeneously crosslinked poly(β-ketoester) networks densely bearing robust nitroxide radicals were prepared via a click-type and stepwise Michael polyaddition. A half-battery cell composed of the thermally-cured radical network coatings displayed a rapid, reversible, and almost stoichiometric redox-activity even with a thickness of ca. 10 μm, which may be applicable as the electrode of organic-based rechargeable devices.
The past few years have witnessed remarkable developments in click chemistry utilized to offer well-defined and tailor-made macromolecules.1–3 The Michael polyaddition of multifunctional monomers has been one of the most versatile click-type synthetic procedures for novel polymeric substances.4–10 The Michael addition of multifunctional acryloyl and protic compounds proceeds via a significantly rapid nucleophilic addition, which is utilized as a conventional tool particularly for the formation of dendritic,5 hyperbranched,6 and networked polymers.7 The bulk polymerization of multifunctional monomers or the three-dimensional polymer network formation through a curing processes has been widely studied for high performance coatings, adhesives, and laminates.10 In the study, the Michael addition was examined because of its characteristics, such as mild polymerization conditions, the wide variety of monomers, and the absence of undesirable byproducts. However, due to insulating aliphatic Michael adducts, applications of the Michael addition to produce electronic organic materials have not been reported.11
Recent interest has been stimulated by the potential use of organic radical molecules as an electronic and magnetic material.12–14 We have synthesized a series of aliphatic polymers bearing pendant nitroxide radical groups and successfully utilized them as an electrode-active material for use in rechargeable devices.15–18 The nitroxide radical moieties in the polymers displayed the rapid and stoichiometric redox process of the unpaired electron (>N–O˙ ⇌ >N+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and chemical robustness. For the fabrication process of the radical polymers to be used in devices, it is crucial to form an insoluble, but homogeneously swellable redox-active polymer layer on a current collector. For example, our previous work demonstrated the photocrosslinking of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)-substituted polynorbornenes using a bisazide additive15,16 or coumarine residues17 that resulted in the redox-active layer in which a charge very rapidly propagates and is reversibly stored. However, the thickness of the radical polymer layer (the charge-storage capacity) was limited to less than sub-micron levels.18 Side-reactions of the nitroxide radical moieties during the polymerization and/or the crosslinking process were also a problematic issue that decreased the content of the redox-active site.
O) and chemical robustness. For the fabrication process of the radical polymers to be used in devices, it is crucial to form an insoluble, but homogeneously swellable redox-active polymer layer on a current collector. For example, our previous work demonstrated the photocrosslinking of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)-substituted polynorbornenes using a bisazide additive15,16 or coumarine residues17 that resulted in the redox-active layer in which a charge very rapidly propagates and is reversibly stored. However, the thickness of the radical polymer layer (the charge-storage capacity) was limited to less than sub-micron levels.18 Side-reactions of the nitroxide radical moieties during the polymerization and/or the crosslinking process were also a problematic issue that decreased the content of the redox-active site.
In this study, the successfully designed Michael polyaddition of the acetoacetate derivatives with acryloyl derivatives and the following moderate curing process directly yielded nitroxide polymer network coatings, being almost stoichiometrically redox-active beyond a 10 μm layer thickness on any shaped current collectors without undergoing any tedious purification processes (Scheme 1). Besides the remarkable redox performances of the radical polymer coatings during rechargeable battery testing and the simple polymer coating formation process without quenching of the radical moieties, we describe, in this communication, the features of the step-growth mechanism of the Michael polyaddition of multifunctional monomers which forms the geometrically homogeneous network structure, that leads to an appropriate crosslink density, a decayed gel-point conversion, a lower shrinkage stress, and a high-adhesion on any type of current collector.
 | ||
| Scheme 1 Michael polyaddition of the TEMPO-substituted acetoacetate with polyacrylates to form nitroxide polymer networks. | ||
The TEMPO-substituted acetoacetate monomer, 2,2,6,6-tetramethylpiperidinyl-N-oxyl-4-yl acetoacetate (TEMPO-AcAc), was designed and prepared via the transesterification of tert-butyl acetoacetate with 4-hydroxy-2,2,6,6-tetramethylpiperidinyl-N-oxyl.† The two acidic protons on the acetoacetate group correspond to a difunctionality.6 The trimethylolpropane triacrylate (TMPTA) and, for comparison, pentaerythritol tetraacrylate (PETA) were selected as multifunctional Michael acceptors. A mixture of TEMPO-AcAc and TMPTA was cured by the addition of 1 wt% 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) at room temperature.‡ The pot-life of ca.10 min allowed a viscous liquid film to form. Consumption of the acrylate group was monitored by the decrease in the IR band area attributed to the acryloyl CC stretch at 1631 cm−1 (e.g., to one-fifth after 2 h), which followed pseudo-first-order kinetics (kobs = 5.1 × 10−4 s−1 at room temperature) during the early stages of the reaction (Fig. S2, ESI†). The post-curing at 80 °C for 2 h was carried out in order to enhance the crosslinking,§ yielding a film with the gel fraction¶ of 0.99 (Table 1); the network polymer soaked in organic solvents was not dissolved off except the elution out of DBU. Shrinkage (negative volume change) after the curing was estimated from the density change to be 1.0% for TEMPO-AcAc/TMPTA, which was surprisingly low among the shrinkages after polymerization of the multifunctional monomers and may be explained by the step-growth mechanism.19 The swelling degree of the films after soaking in organic solvents was 1.5–2.0 (e.g., in acetonitrile, Table 1) supporting the effective crosslinking structure. The resulting film was homogeneously red colored, which was ascribed to the homogeneous loading of the TEMPO residue (λmax = 430 nm, ε = 3.0 × 101 M−1 cm−1) in the film. The unpaired electron's concentration per acetoacetate monomer unit in the polymer film was estimated using SQUID measurements to be 0.92 and 0.93 for the TEMPO-AcAc/TMPTA and TEMPO-AcAc/PETA, respectively. The nitroxide radical moiety was hardly quenched during the thermal-curing process. The half-life of the nitroxide radical in the film state was more than one year at room temperature, and three months at 100 °C; the three-dimensional network remarkably prolonged the half-life. The self-standing films were bendable and mechanically tough both in the dry state and solvent-holding gel states (see the pictures in the ESI)†.
| Acrylate | Concentration of unpaired electrona (%) | Gel fraction (wt%) | Densityb (g cm−3) | Shrinkage (vol%) | Swelling degree in CH3CNc (v/v) | E′ at 120 °C (MPa) | T g (°C) | tan δ fwhm (°C) | Crosslink density (M) |
|---|---|---|---|---|---|---|---|---|---|
| a The concentration per the acetoacetate monomer unit, determined by the slope of Curie plots with SQUID measurements. b Measured by a sink/float method. c Defined as the ratio of volumes of the film soaked in solvent and in the dry state. d Estimated by E′′ maximum in the dynamic mechanical measurement. | |||||||||
| TMPTA | 92 | 99 | 1.1 | 1.0 | 1.8 | 3.0 | 58 | 10 | 0.31 |
| PETA | 93 | 99 | 1.2 | 6.2 | 1.5 | 5.0 | 71 | 12 | 0.50 |
The network structure and crosslink density of the films, TEMPO-AcAc/TMPTA and TEMPO-AcAc/PETA, were analyzed by dynamic mechanical measurement (Fig. 1). The elastic storage and loss modulus (E′ and E′′, respectively) of the films were comparable to the previously reported poly(β-ketoester) networks,7 indicating a network polymer formation based on a flexible main chain. The E′ value and Tg (determined by the maximum E′′) for PETA was greater than those for TMPTA, due to the functionality of the acryloyl monomer. The rubbery plateau modulus determined the crosslink density|| of the film (Table 1). The breadth of tan δ, determined as the full width at half maximum (fwhm), for the networks was very narrow compared to the chain growth networks (e.g., fwhm for fully-cured vinyl ester networks were reported to be in the range of 20 to 60 °C),20 suggesting a homogeneous and uniform network formation through the polymerization and curing process.
 | ||
| Fig. 1 Elastic storage modulus E′ (solid curve), loss modulus E′′ (dashed curve), and tan δ (red and blue curves for TEMPO-AcAc/TMPTA and TEMPO-AcAc/PETA, respectively) vs. temperature at 2 °C min−1 scan rate, 1.0 Hz frequency, and 10 mN preload force. | ||
Toluene solutions of TEMPO-AcAc and TMPTA (or PETA) containing 1 wt% DBU were coated onto a glassy carbon (GC) substrate using a bar coater with a 5–10 μm thickness. The mixtures were initially cured at room temperature for 24 h, then post-cured at 80 °C for 2 h in air. Using the resulting polymer film on the GC substrate as the working electrode, a half-cell was fabricated with a Pt mesh and Ag/AgCl as the counter and reference electrodes, respectively. The cyclic voltammogram of each sample displayed a chemically reversible redox wave at 0.81 V vs. Ag/AgCl in a (C4H9)4NClO4 acetonitrile solution (Fig. 2(a) and (b)).** Coulometric analysis by integrating the current obtained through a scan rate of 1 mV s−1 almost agreed with the nitroxide group content in the coating calculated from the film weight, even for the very thick films (0.19 C cm−2 for the TEMPO-AcAc/TMPTA film coating with a 10 μm thickness, and 0.20 C cm−2 for the TEMPO-AcAc/PETA film coating with a 10 μm thickness). The redox capacity under the slow scan rate conditions could be ascribed to the stoichiometric redox reaction, and the anodic peak current (ip) was proportional to the square root of the scan rate, which supported the diffusion-dominated electrochemical behavior of the coating (Fig. 2(c)). For TEMPO-AcAc/TMPTA, deviation from the linear fitting was small even at the higher scan rate in contrast to that of TEMPO-AcAc/PETA. The cell using TEMPO-AcAc/TMPTA allowed a smaller peak-to-peak separation in the cyclic voltammograms, indicating a more rapid charge diffusion in comparison to that of TEMPO-AcAc/PETA. The diffusion coefficient (D = 5.2 × 10−9 for TEMPO-AcAc/TMPTA and 2.6 × 10−9 cm2 s−1 for TEMPO-AcAc/PETA) was determined from the Cottrell plots for the semi-infinite diffusion prevailed at the early stages of the electrolysis (Fig. S3, ESI†). These results suggested that the charge transfer rate in such relatively thick films was dominated by the crosslink density of the network.
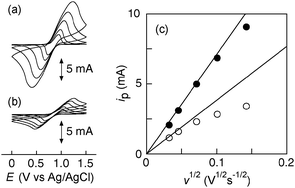 | ||
| Fig. 2 Cyclic voltammograms of (a) TEMPO-AcAc/TMPTA and (b) TEMPO-AcAc/PETA film with 10 μm thickness on the GC plate at the scan rates of 1, 2, 5, 10 and 20 mV s−1 in a 0.1 M (C4H9)4NClO4 acetonitrile solution. (c) The anodic peak current vs. square root of the scan rate for TEMPO-AcAc/TMPTA (●) and TEMPO-AcAc/PETA (○). | ||
The chrono-potentiometric analysis determined the charging and discharging performance of the half-cells (Fig. 3). The charging and discharging curves of the cell of TEMPO-AcAc/TMPTA and TEMPO-AcAc/PETA at the 1 C-rate†† displayed a plateau voltage at 0.81 V vs. Ag/AgCl. The discharging capacity was 54 and 55 mAh g−1, which almost coincided with the calculated capacity of the polymer loaded on the GC. The discharging capacity of the cell at 20 C (corresponding to the full discharge for 3 min) was maintained at more than 90% (49 mAh g−1) of the 1 C-discharging capacity.
 | ||
| Fig. 3 Charging–discharging of the half-cell composed of the (a) TEMPO-AcAc/TMPTA and (b) TEMPO-AcAc/PETA cathode and the 0.1 M (C4H9)4NClO4 acetonitrile electrolyte at the discharge rates of 1 and 20 C. (c) Discharge capacity (CD) for the half-cell of the TEMPO-AcAc/TMPTA (●) and TEMPO-AcAc/PETA (○) film in 0.1 M (C4H9)4NClO4 acetonitrile solution. | ||
The nitroxide polymer-coated GC felt electrode (a fiber diameter of ca. 20 μm) was also applied to the cathode of the half-cell.‡‡ The cyclic voltammogram at 1 mV s−1 for the half-cell using the felt also displayed a quantitative redox capacity (Fig. S4†, e.g., 4.1 C for the felt using 23 mg TEMPO-AcAc/TMPTA loaded on a 25 mg GC felt cathode).
A nitroxide polymer network coating was formed by the Michael polyaddition, which effectively offered three-dimensional networks with the appropriate crosslink density to fabricate a polymer-electrode, which exhibited a rapid, reversible and almost stoichiometric redox capability as a cathode-active material. A redox-active polymer with a well-defined network structure could provide an electronic material with tailor-made properties.
This work was partially supported by Grants-in-Aid for Scientific Research (No. 19105003 and 19655043) from MEXT, Japan, the NEDO Project on “Radical Battery for Ubiquitous Power”. The stay of T. I. in Berlin was supported by Waseda University Doctoral Student Career Center in the Program for Developing Innovative Researchers (No. 885100011) from MEXT, Japan. The authors thank Prof. Kenichi Oyaizu (Waseda University) for discussion on the electrochemistry of radical polymer coatings, and Dr Hisatomo Yonehara (Central Research Laboratories, DIC, Inc.) and Dr Kai-Uwe Gaudl (DIC Berlin GmbH R&D Laboratory) for advice on the thermal-curing reaction.
Notes and references
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- C. Remzi Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- Y. Hirayama, Y. Sakamoto, K. Yamaguchi, S. Sakamoto and M. Iwamura, Tetrahedron Lett., 2005, 46, 1027–1030 CrossRef CAS.
- Y. B. Kim, H. K. Kim, H. Nishida and T. Endo, Macromol. Mater. Eng., 2004, 289, 923–926 CrossRef CAS.
- S. R. Williams, B. D. Mather, K. M. Miller and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4118–4128 CrossRef CAS.
- Y. Lin, Z. M. Doing, X. H. Liu and Y. S. Li, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4309–4321 CrossRef CAS.
- D. A. Rothenfluh, H. Bermudez, C. P. O'Neil and J. A. Hubbell, Nat. Mater., 2008, 7, 248–254 CrossRef CAS.
- A. Noomen, Prog. Org. Coat., 1997, 32, 137–142 CrossRef CAS.
- K. Yamamoto, M. Higuchi, H. Takai and T. Nishiumi, Org. Lett., 2001, 3, 131–134 CrossRef CAS.
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737–738 CrossRef CAS.
- K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS.
- E. Fukuzaki and H. Nishide, J. Am. Chem. Soc., 2006, 128, 996–1001 CrossRef CAS.
- T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS.
- T. Suga, H. Konishi and H. Nishide, Chem. Commun., 2007, 1730–1732 RSC , highlighted in Chem. Technol., 2007, 4. T27.
- K. Oyaizu, Y. Ando, H. Konishi and H. Nishide, J. Am. Chem. Soc., 2008, 130, 14459–14461 CrossRef CAS.
- K. Koshika, N. Sano, K. Oyaizu and H. Nishide, Chem. Commun., 2009, 836–838 RSC.
- H. Lu, J. A. Carioscia, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2005, 21, 1129–1136 CrossRef CAS.
- T. F. Scott, W. D. Cook and J. S. Forsythe, Eur. Polym. J., 2002, 38, 705–716 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, pictures of the cured film samples, IR kinetic experiments, and electrochemical measurements. See DOI: 10.1039/c002797a |
| ‡ Mixing experiments of TEMPO-AcAc and TMPTA did not yield any decrease in the radical content at room temperature even after 1 day. |
| § Total consumption of the acrylates after the post-curing was estimated to be >90% by IR measurement. |
| ¶ The gel fraction of the polymer was determined as the weight ratio of gels after drying and before the 3 day-swelling in acetone. |
| || The crosslink density (ρ) of the bulk material is estimated from the rubbery modulus using the theory of rubber elasticity: ρ = E′/[2(1 + ν)RT], where ν is Poisson's ratio, R is the gas constant, T is the temperature, and E′ is the elastic modulus. |
| ** TMPTA did not display any redox-activity in the cyclic voltammetry in solution at room temperature. |
| †† The 1 C rate is defined as the current density at which the charging or discharging of the cell takes 1 h. |
| ‡‡ A piece of the felt (1.0 × 1.0 × 0.2 cm3) was dipped in the toluene solution of TEMPO-AcAc and TMPTA containing 1 wt% DBU for a few seconds, dried and heated to yield the polymer-coated felt, in which the weight of the loaded polymer increased with the concentration of the monomer solution. |
| This journal is © The Royal Society of Chemistry 2010 |