Rh(I)-catalyzed intramolecular [3 + 2] cycloaddition reactions of 1-ene-, 1-yne- and 1-allene-vinylcyclopropanes†
Lei
Jiao
,
Mu
Lin
and
Zhi-Xiang
Yu
*
Beijing National Laboratory of Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering, College of Chemistry, Peking University, Beijing, 100871, China. E-mail: yuzx@pku.edu.cn; Fax: +86-10-6275-1708; Tel: +86-10-6276-7735
First published on 14th December 2009
Abstract
New Rh(I)-catalyzed intramolecular [3 + 2] cycloaddition reactions of 1-ene-, 1-yne and 1-allene-vinylcyclopropanes have been developed, affording an efficient and versatile synthesis of cyclopentane- and cyclopentene-embedded bicyclic structures.
A five-membered carbocycle is an ubiquitous skeleton in organic molecules. Due to this, developing methods to synthesize five-membered carbocycles has been intensively pursued by the synthetic community.1 To this end, many powerful synthetic methodologies have been developed (e.g., the Pauson–Khand reaction,2 the Nazarov cyclization,3 and various [3 + 2] cycloadditions4). Nevertheless, even with these marvellous reactions in hand, discovering new reactions for the construction of five-membered carbocycles, especially five-membered-ring-embedded polycyclic structures, is still highly in demand.
One straightforward but challenging way was to develop new intramolecular [3 + 2] cycloadditions between unique three-carbon and two-carbon components. Vinylcyclopropane (VCP) was reported as a good intramolecular cycloaddition participant.1c VCP substrates bearing an olefin or alkyne functionality at the C(β)-position (named as β-ene/yne-VCP) were employed in various transition-metal catalyzed [5 + x] cycloadditions, where VCP moiety acts as a five-carbon component (Scheme 1, pathways a and b).5 Recently, we discovered that trans-2-ene-VCP could participate in the intramolecular [3 + 2] cycloaddition, rendering the first example that VCP serves as an unconventional three-carbon component in Rh(I)-catalyzed cycloadditions (Scheme 1, pathway c).6 However, this [3 + 2] cycloaddition suffers from the limitations that only 5,5-bicyclic skeleton could be generated, carbon-tethered substrates were not tolerated, the alkyne as the 2π-component was not compatible and the quaternary carbon center could not be established at the bridgehead.6 Based on the knowledge that these VCP-participating cycloadditions proceeded via a π-allyl rhodacyclohexene intermediate (Scheme 1, intermediates in the boxes),7 we hypothesized to derive a new type of substrate, 1-ene/yne-VCP, by tethering the 2π component to the C(1)-position of VCP to achieve the rhodacyclohexene intermediate with a new substitution pattern. We envisioned that this new intermediate could lead to a novel [3 + 2] cycloaddition mode (Scheme 1, pathway d) and may overcome the limitations associated with the previous [3 + 2] reaction. More attractive was that the designed [3 + 2] cycloaddition could install an all-carbon quaternary stereocenter at the bridgehead carbon of the cycloadduct. Herein, we wish to report our endeavours towards the development of this new Rh(I)-catalyzed intramolecular [3 + 2] cycloaddition reaction.
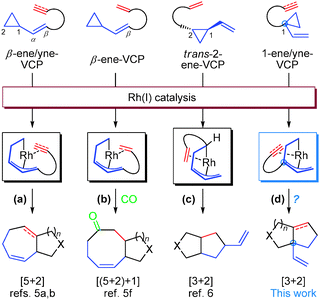 | ||
| Scheme 1 Reaction modes of ene/yne-VCPs under Rh(I)-catalysis and a new reaction design. | ||
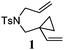
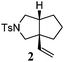
Our test of the new design started with the tosylamide-tethered 1-ene-VCP substrate 1. The first run was conducted under the Wender [5 + 2] cycloaddition conditions ([Rh(CO)2Cl]2 as the catalyst, toluene as the solvent, a reaction temperature of 110 °C).5b To our delight, the proposed bicyclic [3 + 2] cycloadduct 2 was generated as a single diastereomer, albeit accompanied by a minor amount of β-hydride elimination byproduct 3 (Table 1, entry 1). This result indicates that our designed process is operative and thus opens up a new reaction type of VCP derivatives.
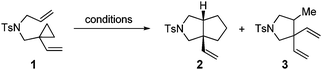
| Entry | Catalyst | T/°C | Time | Ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3b :3b |
Yield (%)c |
|---|---|---|---|---|---|
| a Reaction conditions: 5 mol% Rh(I) catalyst, anhydrous dichloroethane (DCE) as solvent (substrate concentration 0.05 M), argon atmosphere. b Determined by 1H NMR. c Combined isolated yield of inseparable product mixture of 2 and 3. d Toluene as solvent. e See ESI† for experimental details. f No [3 + 2] cycloadduct 2 was generated. | |||||
| 1 | [Rh(CO)2Cl]2 | 110d | 4.5 h | 4:1 |
57 |
| 2 | [Rh(PPh3)3]OTfe | 110d | 3 h | —f | —f |
| 3 | [Rh(CO)2]SbF6e | 80 | 1 h | 1:1 |
15 |
| 4 | [Rh(NBD)]SbF6e | 80 | 2 h | 5:1 |
38 |
| 5 | [Rh(dppe)]SbF6e | 80 | 2 h | 15:1 |
93 |
| 6 | [Rh(dppp)]SbF6e | 80 | 2 h | 40:1 |
93 |
| 7 | [Rh(dppb)]SbF6e | 80 | 2.5 h | 8:1 |
88 |
With the above encouraging results, we then concentrated our efforts on finding the optimal reaction conditions that can selectively produce the [3 + 2] cycloadduct 2. Several cationic Rh(I) catalysts proved ineffective for promoting this [3 + 2] process (Table 1, entries 2 to 4). We observed that cationic Rh(I)-bidentate phosphine complexes8 could suppress the undesired β-hydride elimination pathway (Table 1, entries 5–7). Among them, [Rh(dppp)]SbF6 gave the best selectivity (40:1) and the highest reaction yield (93%). Therefore, we chose this optimal reaction condition (Table 1, entry 6) to further study the scope of the [3 + 2] cycloaddition.
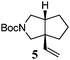
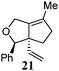
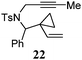
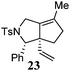
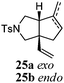
Various 1-ene/yne-VCP substrates were submitted to the optimized reaction conditions (Table 2). It was found that the yields were generally moderate to excellent and no β-hydride elimination byproduct was observed. Nitrogen-, oxygen- and gem-diester-tethered substrates could be employed to construct hetero- and carbo-bicyclic skeletons. In addition to a tosyl group, this [3 + 2] reaction also tolerates a N-Boc protecting group (entry 2). For 1-ene-VCPs, the formation of a 5,5-bicyclic skeleton favors a cis ring-fusion (entries 1–3); while the formation of a 6,5-ring system prefers a trans ring-fusion (entry 4). Except for 1-ene-VCPs, both terminal and internal 1-yne-VCPs serve as good substrates, giving rise to the corresponding [3 + 2] cycloadducts in good yields (entries 5–11). Two 1-yne-VCPs with a stereocenter neighbouring C(1), 20 and 22, were tested to examine the stereoinduction in this new [3 + 2] cycloaddition process (entries 10 and 11). A good level of stereoinduction was achieved, albeit the stereochemical outcomes of oxygen-tethered 1-yne-VCP 20 and tosylamide-tethered substrate 22 are opposite (cycloadduct 21versus23). This [3 + 2] reaction can also be extended to 1-allene-substituted VCP (entry 12). Under the optimized conditions, 1-allene-VCP 24 produced a mixture of cycloadducts 25a and 25b with exo and endo C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, respectively. An identical reaction using [Rh(CO)2Cl]2 as the catalyst afforded 25a as the major product.
C bond, respectively. An identical reaction using [Rh(CO)2Cl]2 as the catalyst afforded 25a as the major product.
| Entry | Substrate | Cycloadductb | Conditions | Yieldc |
|---|---|---|---|---|
| a Reaction conditions: 5 mol% [Rh(dppp)]SbF6 as catalyst, DCE as solvent, unless otherwise indicated. Ts = tosyl, Boc = tert-butoxycarbonyl, Ph = phenyl, E = COOMe. b The cycloadducts were obtained as racemic compounds. c Isolated yields after column chromatography. d Recovered 32% of 14. e Determined by 1H NMR. f Use 4 mol% [Rh(CO)2Cl]2 as catalyst, toluene as solvent. | ||||
| 1 |
|
|
80 °C, 2 h | 93% |
| 2 |

|
|
80 °C, 13 h | 66% |
| 3 |

|
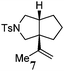
|
90 °C, 3.5 h | 53% |
| 4 |

|
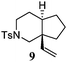
|
80 °C, 12 h | 98% |

|
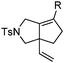
|
|||
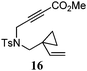
| 5 | 10 R = H | 11 | 80 °C, 5 h | 82% |
| 6 | 12 R = Me | 13 | 80 °C, 23 h | 78% |
| 7 | 14 R = i-Pr | 15 | 80 °C, 48 h | 35%d |
| 8 |
|
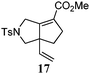
|
80 °C, 13 h | 66% |
| 9 |
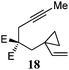
|

|
80 °C, 39 h | 59% |
| 10 |
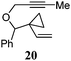
|
|
80 °C, 11.5 h | 74% dr 19:1e |
| 11 |
|
|
80 °C, 5 h | >99% dr 6:1e |
| 12 |

|
|
80 °C, 13 h | 48% a:b 3.6:1e |
| 110 °C, 2 hf | 41% a:b 10:1e |
The 1-ene-VCP substrates 26 and 28 bearing a disubstituted ene-moiety were also tested to further explore the substrate scope (Scheme 2). Under the standard conditions, the reaction of 26 gave no desired [3 + 2] cycloadduct but a monocyclic byproduct 27, which is assumed to be generated via β-hydride elimination on the methyl group. The reaction of 28 to construct a bicyclic structure with two quaternary bridgehead carbons was feasible, but the reaction was sluggish and only a minor amount of the desired cycloadduct 29 was obtained.
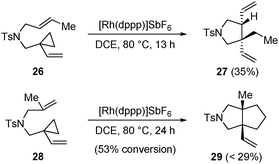 | ||
| Scheme 2 Further exploration of the substrate scope. | ||
Control experiments were conducted by submitting ene-cyclopropane 1a and yne-cyclopropane 1b to the cycloaddition reaction to probe the mechanism of this [3 + 2] cycloaddition. It was found that under identical reaction conditions, no corresponding [3 + 2] cycloadduct was observed and both substrates remained intact (Scheme 3). This result clearly identified the crucial function of the vinyl group in this transformation.
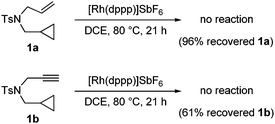 | ||
| Scheme 3 Control experiments. | ||
A proposed mechanism of this [3 + 2] process was shown in Fig. 1. The catalytic cycle commences on the binding of the cationic catalytic species Rh(dppp)+ to the alkene moiety of VCP to give intermediate A, followed by cyclopropane ring cleavage to generate the key π-allyl rhodacyclo-hexene intermediate B.7 Then insertion of a CC or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond to the C(1)–Rh bond occurs to form the intermediate C, which undergoes reductive elimination to furnish the bicyclic [3 + 2] cycloadduct, with the concomitant generation of the catalytic species for the next catalytic cycle. A minor amount of the observed byproduct 3 is probably generated through β-hydride elimination of intermediate C. In the reaction process, the vinyl group plays an important role as a “spectator” binding group to facilitate the ring-opening of a cyclopropane ring, well explaining the lack of activity for ene/yne-cyclopropanes.
C bond to the C(1)–Rh bond occurs to form the intermediate C, which undergoes reductive elimination to furnish the bicyclic [3 + 2] cycloadduct, with the concomitant generation of the catalytic species for the next catalytic cycle. A minor amount of the observed byproduct 3 is probably generated through β-hydride elimination of intermediate C. In the reaction process, the vinyl group plays an important role as a “spectator” binding group to facilitate the ring-opening of a cyclopropane ring, well explaining the lack of activity for ene/yne-cyclopropanes.
![Plausible catalytic cycle of the [3 + 2] reaction.](/image/article/2010/CC/b922417c/b922417c-f1.gif) | ||
| Fig. 1 Plausible catalytic cycle of the [3 + 2] reaction. | ||
In conclusion, we have developed a new type of Rh(I)-catalyzed intramolecular [3 + 2] cycloaddition of 1-ene-, 1-yne- and 1-allene-VCP substrates. The experimental findings represent the second example where VCP serves as a three-carbon component in Rh(I)-catalyzed cycloadditions. The present methodology provides an efficient, versatile and diastereoselective approach to carbo- and hetero-bicyclic compounds. The formation of a vinyl substituted quaternary stereocenter in this process enables further access to functionalized quaternary-stereocenter-containing cycloadducts. Further studies on the reaction mechanism, origins of the stereoinduction and the application of these cycloaddition reactions are ongoing.
We thank Peking University, the Natural Science Foundation of China (20825205-National Science Fund for Distinguished Young Scholars and 20672005) and the Ministry of Education of China (108001) for financial support.
Notes and references
- (a) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49 CrossRef CAS; (b) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS; (c) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS.
- J. Blanco-Urgoiti, L. Anorbe, L. Perez-Serrano, G. Dominguez and J. Perez-Castells, Chem. Soc. Rev., 2004, 33, 32 RSC.
- (a) C. Santelli-Rouvier and M. Santelli, Synthesis, 1983, 429 CrossRef CAS; (b) H. Pellissier, Tetrahedron, 2005, 61, 6479 CrossRef CAS.
- Selected recent reports on [3 + 2] cycloadditions: (a) J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1426 CrossRef CAS; (b) M. Gulías, R. García, A. Delgado, L. Castedo and J. L. Mascarenas, J. Am. Chem. Soc., 2006, 128, 384 CrossRef CAS; (c) L. Liu and J. Montgomery, J. Am. Chem. Soc., 2006, 128, 5348 CrossRef CAS; (d) S. Ogoshi, M. Nagata and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 5350 CrossRef CAS; (e) B. M. Trost, J. P. Stambuli, S. M. Silverman and U. Schwörer, J. Am. Chem. Soc., 2006, 128, 13328 CrossRef CAS; (f) P. A. Wender, T. J. Paxton and T. J. Williams, J. Am. Chem. Soc., 2006, 128, 14814 CrossRef CAS; (g) H.-T. Chang, T. T. Jayanth and C.-H. Cheng, J. Am. Chem. Soc., 2007, 129, 4166 CrossRef CAS; (h) X. Huang and L. Zhang, J. Am. Chem. Soc., 2007, 129, 6398 CrossRef CAS; (i) T. Nishimura, Y. Yasuhara and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 7506 CrossRef CAS; (j) B. M. Trost, N. Cramer and S. Silverman, J. Am. Chem. Soc., 2007, 129, 12396 CrossRef CAS; (k) S. S. Yudha, Y. Kuninobu and K. Takai, Angew. Chem., Int. Ed., 2008, 47, 9318 CrossRef CAS.
- For representative [5 + x] cycloadditions employing VCP as five-carbon unit, see: (a) P. A. Wender, H. Takahashi and B. Witulski, J. Am. Chem. Soc., 1995, 117, 4720 CrossRef CAS; (b) P. A. Wender and D. Sperandio, J. Org. Chem., 1998, 63, 4164 CrossRef CAS; (c) B. M. Trost, F. D. Toste and H. Shen, J. Am. Chem. Soc., 2000, 122, 2379 CrossRef CAS; (d) P. A. Wender, G. G. Gamber, R. D. Hubbard and L. Zhang, J. Am. Chem. Soc., 2002, 124, 2876 CrossRef CAS; (e) P. A. Wender, G. G. Gamber, R. D. Hubbard, S. M. Pham and L. Zhang, J. Am. Chem. Soc., 2005, 127, 2836 CrossRef CAS; (f) Y. Wang, J. Wang, J. Su, F. Huang, L. Jiao, Y. Liang, D. Yang, S. Zhang, P. A. Wender and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 10060 CrossRef CAS.
- L. Jiao, S. Ye and Z.-X. Yu, J. Am. Chem. Soc., 2008, 130, 7178 CrossRef CAS.
- Z.-X. Yu, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2004, 126, 9154 CrossRef CAS.
- Cationic Rh(I)-phosphine complexes were employed as efficient catalysts in intramolecular cycloaddition reactions, see: B. Wang, P. Cao and X. Zhang, Tetrahedron Lett., 2000, 41, 8041 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds. See DOI: 10.1039/b922417c |
| This journal is © The Royal Society of Chemistry 2010 |