Synthesis of functionalized maoecrystal V core structures†
K. C.
Nicolaou
*abc,
Lin
Dong
a,
Lujiang
Deng
a,
Adam C.
Talbot
a and
David Y.-K.
Chen
*a
aChemical Synthesis Laboratory@Biopolis, Institute of Chemical and Engineering Sciences (ICES), Agency for Science, Technology and Research (A*STAR), 11 Biopolis Way, The Helios Block #03-08, Singapore, 138667. E-mail: kcn@scripps.edu; david_chen@ices.a-star.edu.sg
bDepartment of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California, 92037, USA. E-mail: kcn@scripps.edu
cDepartment of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, California, 92093, USA
First published on 23rd October 2009
Abstract
Two strategies toward the total synthesis of maoecrystal V (1) culminating in the construction of core structures 2 and 3 are described.
Maoecrystal V (1, Fig. 1) is a recently reported terpenoid endowed with potent and selective cytotoxic properties and novel architectural motifs.1 Isolated from the medicinally used Chinese herb Isodon eriocalyx, collected from the Jiangchuan prefecture of Yunnan province, this natural product exhibits selective cytotoxicity against HeLa cells (IC50 = 0.02 μg mL−1) as compared to three other cell lines tested (K562, A549 and BGC-823). Its highly unusual structure was finally reported in 2004 after an X-ray crystallographic analysis, although it was suspected several years earlier on the basis of NMR spectroscopic and mass spectrometric experiments.1 By virtue of its appealing molecular structure and antitumor properties, maoecrystal V attracted the attention of synthetic organic chemists and, thus, studies toward its synthesis already began to emerge.2–4 In this communication, we report our early forays in this area that culminated in the construction of functionalized core structures 2 and 3 (Fig. 1) as well as preliminary biological evaluation of these compounds.
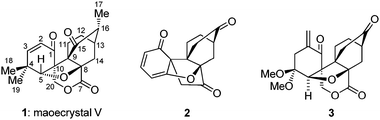 | ||
| Fig. 1 Structures of maoecrystal V (1) and functionalized core structures 2 and 3. | ||
The striking molecular structure of maoecrystal V (1) is characterized by its compact polycyclic nature, unprecedented ring framework, and five stereogenic centers (of which three are quaternary). Our first approach to the pentacyclic framework of this molecule was based on the premise that it could be formed from a bicyclic precursor through an intramolecular [4 + 2] cycloaddition5 that would introduce two additional rings, followed by an intramolecular cyclopropanation/dearomatization/ring opening cascade to forge the final ring (see Scheme 1). To this end, 2,6-dimethoxy benzoic acid (4) was reacted with cyclohexenone (5) in the presence of Pd(OCOCF3)2 catalyst and Ag2CO3 to afford arylated enone 6 through a decarboxylative Heck process,6 in 89% yield. Mono-demethylation of dimethoxy enone 6 (BBr3, 70% yield) followed by O-alkylation of the resultingphenol (7) with bromide 87 resulted in the formation of pyrrolidine derivate 9, whose conversion to the desired alkenyl methyl ester 10 required quaternization and extrusion of its nitrogen moiety (Na2CO3–MeI, 80% overall yield from 7). Subsequent reaction of enone10 with TBSOTf–Et3N furnished TBS enol ether11, which was subjected to the intended intramolecular [4 + 2] cycloaddition reaction directly and without purification in the presence of K2CO3 and hydroquinone (toluene, reflux). Upon exposure of the resulting cycloadducts to aq. HCl, the exo (major) Diels–Alder product 12 (64% yield from 10) and its endo isomer (C8-epi-12, not shown, 1.7% yield from 10) were obtained and chromatographically separated. The structural identity of 12 (m.p. 200–201 °C, EtOAc/hexanes) was unambiguously assigned through X-ray crystallographic analysis8 (see ORTEP, Fig. 2).9 The second of the two contiguous all carbon quaternary centers was fabricated through an intramolecular, carbene-mediated cyclopropanation/fragmentation process inspired by the work of Mander,10 as shown in Scheme 1. Thus, saponification of the methyl ester within 12 (1 N aq. NaOH/EtOH 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 60 °C), followed by acid chloride formation [(COCl)2] and exposure to TMSCHN2, led to α-diazo ketone 13 in 79% overall yield for the three steps. Treatment of diazoketone13 with Rh2(OAc)4 catalyst in CH2Cl2 at ambient temperature resulted, upon exposure of the resulting cyclopropane (14) to silica gel, in the formation of pentacyclic dienone maoecrystal V core 2 in 75% overall yield from 13. Assigned by NMR spectroscopic and mass spectrometric techniques, the structure of 2 was unambiguously confirmed through X-ray crystallographic analysis of its fully hydrogenated product [15, H2, Pd–C (cat), 87% yield] (m.p. 237–238 °C, EtOAc, see ORTEP,11Fig. 2). Compound 2 holds promise as a possible precursor to maoecrystal V (1).
:1, 60 °C), followed by acid chloride formation [(COCl)2] and exposure to TMSCHN2, led to α-diazo ketone 13 in 79% overall yield for the three steps. Treatment of diazoketone13 with Rh2(OAc)4 catalyst in CH2Cl2 at ambient temperature resulted, upon exposure of the resulting cyclopropane (14) to silica gel, in the formation of pentacyclic dienone maoecrystal V core 2 in 75% overall yield from 13. Assigned by NMR spectroscopic and mass spectrometric techniques, the structure of 2 was unambiguously confirmed through X-ray crystallographic analysis of its fully hydrogenated product [15, H2, Pd–C (cat), 87% yield] (m.p. 237–238 °C, EtOAc, see ORTEP,11Fig. 2). Compound 2 holds promise as a possible precursor to maoecrystal V (1).
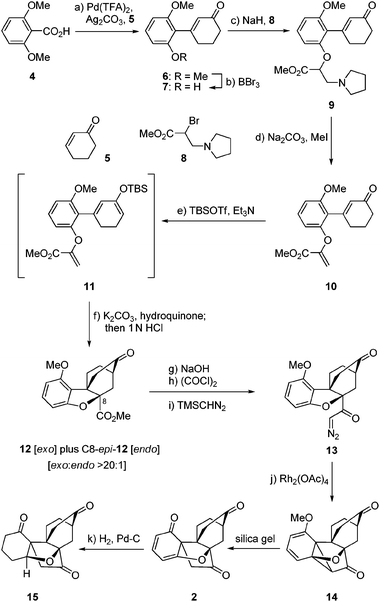 | ||
| Scheme 1 Construction of dienone 2. Reagents and conditions: (a) 5 (1.6 equiv.), Pd(TFA)2 (0.2 equiv.), Ag2CO3 (2.0 equiv.), DMF/DMSO (20:1), 80 °C, 3 h, 89%; (b) BBr3 (1.8 equiv.), CH2Cl2, −15 °C, 2.5 h, 70%; (c) NaH (2.1 equiv.), THF, 0 → 23 °C, 1 h; then 8 (5.0 equiv.) 0 → 23 °C, 16 h; (d) Na2CO3 (5.0 equiv.), MeI (10.0 equiv.), MeOH, reflux, 2 h, 80% over the two steps; (e) TBSOTf (1.5 equiv.), Et3N (3.0 equiv.), CH2Cl2, 0 °C, 1.5 h; (f) K2CO3 (4.8 equiv.), hydroquinone (1.2 equiv.), toluene, reflux, 16 h; then 1.0 N aq. HCl, 1 h, 23 °C, 64% over the two steps; (g) NaOH (1.0 N aq.)/EtOH (1:1), 60 °C, 5 h; (h) (COCl)2 (5.0 equiv.), DMF (1 drop), CH2Cl2, reflux, 1 h; (i) TMSCHN2 (5.0 equiv.), THF–CH3CN (1:1), 0 °C, 2 h, 79% over the three steps; (j) Rh2(OAc)4 (0.1 equiv.), CH2Cl2, 23 °C, 1 h, 75%; (k) 10% Pd/C (0.26 equiv.), H2, EtOAc, 23 °C, 24 h, 87%. | ||
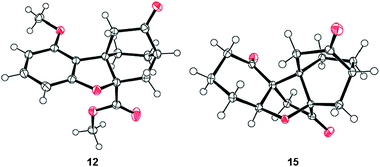 | ||
| Fig. 2 X-ray derived ORTEP of intramolecular Diels–Alder product 12 and triketone 15 with thermal ellipsoids shown at the 50% probability level. | ||
Our second foray toward maoecrystal V (1) involved the same type of intramolecular Diels–Alder reaction but a different dearomatization process to arrive at the even more advanced intermediate 3, as shown in Scheme 2. Thus, bis-demethylation of intermediate 6 (BBr3) followed by mono-protection of the resulting diphenol (16) as a MOM ether (NaH–MOMCl), afforded phenolic enone 17 in 55% overall yield. Attachment of the dienophile moiety onto 17 was carried out through the same sequence as described above for 7 → 10 (Scheme 1) (NaH, 8; Na2CO3, MeI),7 leading to enone alkenyl ester 18 in 57% overall yield. Silyl enol ether formation within the latter compound (TBSOTf–Et3N) then provided the desired Diels–Alder substrate 19, which was used without purification in the following steps as described above for the related substrate 11. In this instance, heating 19 in refluxing toluene in the presence of K2CO3 and hydroquinone provided, upon treatment with 1 N aq. HCl at ambient temperature, exo Diels–Alder product 20 as a single diastereoisomer in 50% overall yield from 18. The desired dearomatization of the benzenoid ring of 20 was performed, this time, through a sequence involving deprotection of its phenolic group (6 N aq. HCl/EtOH–CHCl3, 1:1:1, reflux) and subsequent treatment of the resulting phenol (21) with PIFA–KHCO3 in MeOH, furnishing dimethoxy dienone derivative 22 in 69% overall yield for the two steps. Hydrogenation of the latter compound [H2, 10% Pd–C (cat)] afforded a mixture of the desired diketone23 (epimeric to 1 at C5) and aromatized product 24 (24:23ca. 1:3 ratio, 99% combined yield). Chromatographic separation of 24 and 23 allowed recycling of the former to dienone 22 (PIFA-KHCO3, MeOH, 70% yield), and saponification of the latter to carboxylic acid 25 (aq. NaOH, EtOH, reflux). On exposure to KOtBu and ClCH2I in the presence of 18-crown-6 at ambient temperature, diketone carboxylic acid 25 underwent triple alkylation (two intermolecular and one intramolecular) to afford enone lactone 3 in 42% overall yield from methyl ester 23. Supported by NMR spectroscopic and mass spectrometric data, the structure of 3 (m.p. 173–175 °C, EtOAc/hexanes) was unambiguously proven by X-ray crystallographic analysis (see ORTEP,12Fig. 3). The precise sequence of events in the cascade leading from 25 to 3 has not been established as yet. Worthy of note, however, is the failure of chloroester 2613 to afford 3 (or any other γ-lactone product) under the conditions employed to convert 25 to 3, suggesting that C-alkylation may precede O-alkylation in this process.14 Containing the entire ring framework of maoecrystal V (1), core structure 3 (epimeric to 1 at C-5) may prove a viable precursor to this natural product.
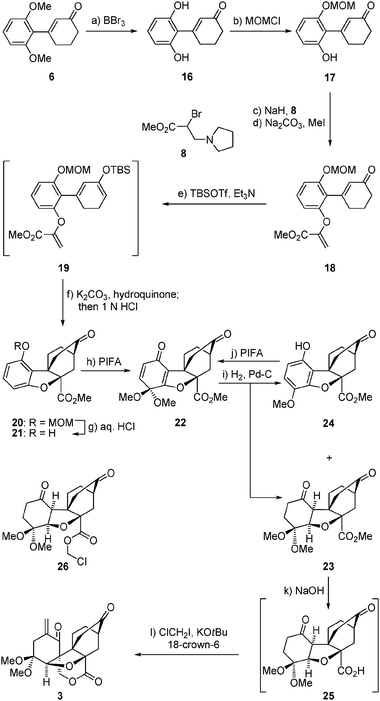 | ||
| Scheme 2 Construction of pentacyclic lactone3. Reagents and conditions: (a) BBr3 (3.5 equiv.), CH2Cl2, −78 → 0 °C, 2 h, 98%; (b) NaH (1.1 equiv.), MOMCl (1.2 equiv.), THF, 0 °C, 2 h, 56%; (c) NaH (2.1 equiv.), THF, 0 → 23 °C, 1 h; then 8 (4.0 equiv.), THF, 0 → 23 °C, 8 h; (d) Na2CO3 (2.6 equiv.), MeI (11 equiv.), MeOH, reflux, 2 h, 57% over the two steps; (e) TBSOTf (1.5 equiv.), Et3N (3.0 equiv.), CH2Cl2, 0 °C, 2 h; (f) K2CO3 (4.8 equiv.), hydroquinone (1.2 equiv.), toluene, reflux , 18 h; then 1.0 N aq. HCl, 23 °C, 1 h, 50% over the two steps; (g) HCl (6.0 N aq.)/EtOH–CHCl3 (1:1:1), reflux, 3 h, 83%; (h) PIFA (2.0 equiv.), KHCO3 (2.2 equiv.), MeOH, 0 → 23 °C, 0.5 h, 83%; (i) 10% Pd/C (0.33 equiv.), H2, EtOH, 23 °C, 10 h, 23: 66%, 24: 33%; (j) PIFA (2.0 equiv.), KHCO3 (2.2 equiv.), MeOH, 0 → 23 °C, 0.5 h, 70%; (k) 1.0 N aq. NaOH (7.0 equiv.), EtOH, reflux, 5 h; then 1.0 N aq. HCl; (l) ClCH2I (11 equiv.), KOtBu (5.0 equiv.), 18-crown-6 (5.5 equiv.), THF, 23 °C, 3 h, 42% over the two steps. | ||
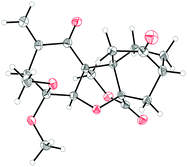 | ||
| Fig. 3 X-ray derived ORTEP of enone lactone 3 with thermal ellipsoids shown at the 50% probability level. | ||
Compounds 2, 3, and 15 were tested against a panel of cancer cells, including breast (MCF-7), CNS (SF268), lung (NCI-H460), and cervical (HeLa), using doxorubicin and Taxol® as standards, and the results are summarized in Table 1. While compounds 2 and 15 exhibited no significant cytotoxicity below 10 μM, the more advanced intermediate 3 showed moderate activity (3.6–6.7 μM) against these tumor cell lines (see Table 1).
| Entry | Compound | Cell line | |||
|---|---|---|---|---|---|
| MCF-7b | SF268b | NCI-H460b | HeLab | ||
| a Antiproliferative effects of tested compounds against human tumor cell lines and drug-resistant cell lines in a 48 h growth inhibition assay using the sulforhodamine B staining methods. Human cancer cell lines : breast (MCF-7), lung (NCI-H460), CNS (SF268), and cervical (HeLa). Growth inhibition of 50% (GI50) is calculated as the drug concentration which caused a 50% reduction in the net protein increase in control cells during drug incubation.15 GI50 values for each compound are given in μM and represent the mean of two independent experiments ± standard error of the mean. b These cell lines were provided by the National Cancer Institute (NCI), Division of Cancer Treatment and Diagnosis (DCTD). | |||||
| 1 | 2 | >10 | >10 | >10 | >10 |
| 2 | 3 | 3.6 ± 0.3 | 5.3 ± 0.1 | 5.6 ± 0.1 | 6.7 ± 0.2 |
| 3 | 15 | >10 | >10 | >10 | >10 |
| 4 | Doxorubicin | 0.089 ± 0.011 | 0.468 ± 0.003 | 0.054 ± 0.003 | 0.965 ± 0.002 |
| 5 | Taxol ® | 0.007 ± 0.000 | 0.033 ± 0.000 | 0.0065 ± 0.001 | 0.008 ± 0.000 |
The described chemistry is expected to facilitate the total synthesis of maoecrystal V (1) and related compounds for chemical and biological studies.
We thank Ms Doris Tan (ICES) for high resolution mass spectrometric (HRMS) assistance, Dr Srinivasulu Aitipamula (ICES) and Ms Chia Sze Chen (ICES) for X-ray crystallographic analysis, and Mr Rong-Ji Sum for his assistance in the biological studies. Financial support for this work was provided by A*STAR, Singapore.
Notes and references
- S.-H. Li, J. Wang, X.-M. Niu, Y.-H. Shen, H.-J. Zhang, H.-D. Sun, M.-L. Li, Q.-E. Tian, Y. Lu, P. Cao and Q.-T. Zheng, Org. Lett., 2004, 6, 4327–4330 CrossRef CAS.
- J. Gong, G. Lin, C.-C. Li and Z. Yang, Org. Lett., 2009 DOI:10.1021/ol9014392.
- P. J. Krawczuk, N. Schöne and P. S. Baran, Org. Lett. DOI:10.1021/o1901963v.
- F. Peng, M. Yu and S. J. Danishefsky, Tetrahedron Lett., 2009, 50, 6586–6587 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- D. Tanaka and A. G. Myers, Org. Lett., 2004, 6, 433–436 CrossRef CAS.
- K. F. W. Hekking, F. L. Van Delft and F. P. J. T. Rutjes, Tetrahedron, 2003, 59, 6751–6758 CrossRef CAS.
- CCDC 748550 contains the supplementary crystallographic data for compound 12. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif or in the ESI†.
- The minor isomer (C8-epi-12) (m.p. 182–183 °C, EtOAc/hexanes) was also subjected to X-ray crystallographic analysis that confirmed its structure. CCDC 742701 contains the supplementary crystallographic data for compound C8-epi-12†.
- J. C. Morris, L. N. Mander and D. C. R. Hockless, Synthesis, 1998, 455–467 CrossRef CAS.
- CCDC 748551 contains the supplementary crystallographic data for compound 15†.
- CCDC 742693 contains the supplementary crystallographic data for compound 3†.
- Chloroester 26 was prepared from 25 by reaction with K2CO3 (1.0 equiv.) and ClCH2I (11 equiv., DMF, 23 °C, 8 h) in 82% yield.
- Iodide catalysis in the presence of NaI, or the use of silver salts were also attempted to effect the γ-lactone formation.
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley, P. Cronise, A. Vaigro-Wolff, M. Gray-Goodrich, H. Campbell, J. Mayo and M. Boyd, J. Natl. Cancer Inst., 1991, 83, 757–766 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray crystallographic analysis and 1H and 13C NMR spectra of compounds. CCDC 742693, 748550 and 748551. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b917045f |
| This journal is © The Royal Society of Chemistry 2010 |