Mechanically interlocked and switchable molecules at surfaces
Jason J.
Davis
*,
Grzegorz A.
Orlowski
,
Habibur
Rahman
and
Paul D.
Beer
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, UK OX1 3TA. E-mail: Jason.davis@chem.ox.ac.uk
First published on 22nd October 2009
Abstract
Mechanically interlocked molecules are of considerable interest from data storage, molecular scale or sensory device perspectives. Though the solution phase characterisation of these compounds has been extensively explored, progress towards real world application will, in many cases, necessitate a detailed understanding of their interfacing with supportive, optically transparent or electroactive surfaces. This feature article summarises the developments made in surface assembly and characterisation including recent progress in exploiting templating methods to interlock molecular systems on surfaces.
 Jason J. Davis | Jason Davis obtained a PhD in 1998 from the Inorganic Chemistry Laboratory, University of Oxford and was awarded a Royal Society University Research Fellowship in 1999. He was made a University Lecturer and Official Student and Tutor in Chemistry at Christ Church in 2003 and elected to a Readership in Chemistry in 2008. His work has focused on the molecular and nanometre-scale construction and analysis of bioinorganic, sensory, electronic and optical systems. |
 Grzegorz A. Orlowski | Grzegorz Orlowski obtained his PhD in 2007 from the University of Saskatchewan, Canada. From 2007 to 2008 he was a Postdoctoral Fellow at the University of Guelph, Canada. In 2008 he joined the group of Dr Davis at the University of Oxford. His research interests cover various electrochemical aspects of the solution/metal interface. |
 Habibur Rahman | Habibur Rahman gained an MEng degree in Materials Science and Engineering from Imperial College London in 2008. Habibur is currently a student at Christ Church, Oxford where he is reading for a DPhil in Inorganic Chemistry. He is working with Dr Jason Davis on surface assembled interlocked rotaxanes and catenanes. |
 Paul D. Beer | Paul Beer gained a PhD from King’s College, London in 1982. After a Royal Society European Postdoctoral Fellowship and a Demonstratorship at the University of Exeter, he took up a Lectureship at the University of Birmingham in 1984. In 1990 he moved to the Inorganic Chemistry Laboratory, University of Oxford where he was made a University Lecturer and Tutorial Fellow at Wadham College. He became a Professor of Chemistry in 1998. His research interests cover many areas of coordination and supramolecular chemistry. |
Introduction
Interlocked molecules such as catenanes and rotaxanes have captured the imagination of the scientific world for more than two decades. During this time, our understanding of the synthetic routes and solution phase characteristics of these interlocked molecules has advanced considerably.1–5 In surveying the range of molecular architectures now reported, we can broadly recognize three distinct classes, namely those where assembly has been achieved by π–π stacking interactions or hydrogen bonding, and those where component association is achieved by ion templation methodologies. In many cases the structures created possess a unique and demonstrable ability to rotate or translate about defined axes. In observing and defining such motion NMR spectroscopy6 and solution-phase voltammetry have been central.7Whether the proposed application of these mechanically interlocked structures, such as data storage or sensing, be molecular-scale or macroscale (the latter ideally achieved by a direct extrapolation of the former), triggered isolated or synchronous motion at a transducing surface is a requirement. This inherently requires both an ability to interface with solid surfaces (be these optically active or electronically addressable) and a detailed understanding of how this interfacing effects characteristics analyzed in solution. Single molecule thick two dimensional self-assembled monolayers (SAMs) represent an ideal means of bringing molecular scale function to the macro world (amplification).8 Their potentially robust, spectroscopically characterizable, and re-useable characteristics are particularly advantageous in numerous application settings where molecular switchability can be utilized. To date, however, only a handful of reports of switchable surface confined interlocked molecular systems have been published. This lies in part with both the synthetic demands of fabricating complicated interlocked scaffolds with suitable surface active moieties, and the complexity of analyzing surfaces which are likely to be non-crystalline, disordered and potentially fluxional. The aim of this feature article is to survey progress made in surface assembling rotaxanes and catenanes and, specifically, to highlight recent advances in both templated surface assembly and subsequent sensing. Progress in this area lies central not only to the utilization of interlocked assemblies in highly specific, multiplexable and environmentally flexible sensing, but also in the development of “molecular machines” where nanometre-scale motion is triggered by an externally applied field or recognition event.
Mechanically interlocked molecular architectures: solution phase characteristics
A rotaxane is a mechanically interlocked, kinetically trapped, assembly of a macrocycle and a dumbbell-shaped axle. Catenanes (“the Latin “catena” means “chain”) are composed of mutually interlocked rings. Solution phase studies of these species have highlighted, in many cases, a unique bistability (and therein, potential “molecular switch” characteristics) by virtue of different recognition sites, or stations, within their interlocked architecture.9,10 In the absence of sterically bulky end groups (stoppers) at either end of the thread/axle component, rotaxanes become “pseudorotaxanes”.The solution phase mechanical interlocking of molecular components can be driven by any or a mixture of π-electron donor–acceptor interactions, hydrogen bonding interactions,11–14 and cation15 or anion templation.16,17 Pioneering work by the group of Stoddart and others based on the π–π stacking interaction between a π-electron deficient cyclophane host, cyclobis(paraquat-p-phenylene) (CBPQT4+), and π-electron rich guests have, for example, been extensively utilised in rotaxane assembly.3,9,10,18–21
In cation guided assembly, the ion can be used not only in templating the interlocking in the first instance but also in presenting an interchangeable coordination environment that can feasibly be used to trigger motion. A number of solution phase studies have demonstrated both redox and chemically triggered rotary motion in derived rotaxanes and catenanes.15,22–24 The group of Sauvage et al. has, in particular, highlighted the use of copper(I) cations in both templating assembly and subsequently driving a shuttling process.15,22
It is only in the past few years that anion templated supramolecular assembly has attracted significant attention.25 We have previously described, for example, the highly specific anion templated synthesis of rotaxanes and catenanes, with particular focus on constructing novel anion sensing systems with highly specific binding characteristics.16,17,26–28 More recently we have described the first example of a catenane which undergoes anion induced circumrotation upon addition of base (a process which can, additionally, be inhibited by chloride anions).29
There are, then, a number of well-developed strategies for assembling the potentially functional components of an interlocked architecture. For a more detailed discussion of the principal methodologies of mechanical bond directed assembly, we direct readers to a number of review articles.9,15,22,26,30–33
Surface assembling of interlocked molecular systems
In order to utilise the switchable characteristics of bi- (or multi-) stable interlocked molecules in sensing, electronics, data storage or macroscale applications, controlled interfacing with electroactive or optically transparent solid surfaces is necessary. The first reported surface assembly of an interlocked supramolecular system was that of a catenane by Gokel, Kaifer et al.20 This involved the formation of a pseudorotaxane inclusion complex driven by stacking interactions between a π-electron acceptor (CBPQT4+ macrocycle) and a π-electron donor (bis-thiol derivative of a hydroquinol axle) (Fig. 1). The subsequent surface assembly of the bis-thiolated axle led to chemisorbed catenanes which could be probed by cyclic voltammetry.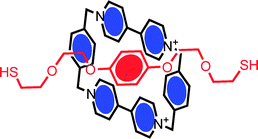 | ||
| Fig. 1 The solution phase assembly of a bis-thiolated hydroquinol thread and cyclophane macrocycle generates a pseudorotaxane which, on chemisorption, becomes a redox active catenane.20 | ||
In the decade or so which followed this work, only a handful of related surface assemblies have been reported. Among the more notable of these are by the group of Sauvage.34–36 Using standard thiolate chemistry, the group templated the assembly of catenanes and rotaxanes containing derivatives of 1,10-phenanthroline, and macrocycles containing phenanthroline or terpyridine moieties on gold electrodes using copper(I) ions (Fig. 2). Significant in the analogous solution phase work are the preferential coordination characteristics of the mono- and di-cation states of copper, something which facilitates a redox driven ring circumrotation (followed by NMR spectroscopy). Though the same molecules are redox switchable when immobilized on gold surfaces, no redox triggered rotary motion has, as yet, been demonstrated. In related work, Gavina, Romero et al. have reported the solution phase copper(I) templated assembly of axle and phenanthroline macrocycle components and subsequent chemisorption on gold electrodes.37
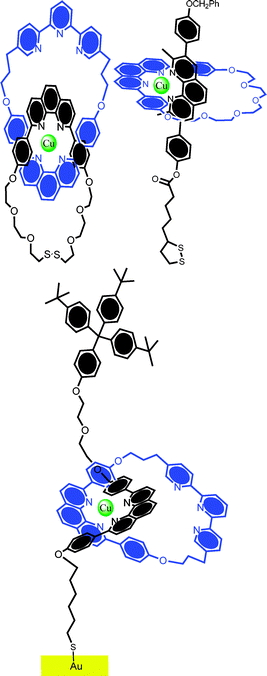 | ||
| Fig. 2 Examples of “Sauvage-type” catenanes and rotaxanes which have been immobilized on gold surfaces with good coverage and stability. (Top left) catenane; (top right) pseudorotaxane; and (bottom) surface bound rotaxane. Though the redox activity of the metal core is readily detectable, no evidence for triggered rotary or shuttling motion has been reported in these systems thus far. | ||
Surface confined interlocked molecular assemblies are most effectively characterized by voltammetry (molecular density, chemisorbed character, redox activity), FTIR (functional group assignment and molecular orientation), AFM (surface organization, layer homogeneity) and XPS (chemisorbed state, coverage). High-resolution electron energy-loss spectroscopy (HREELS) and X-ray absorption spectroscopy (XAS) have, additionally, been applied in the analysis of surface bonding and molecular orientation in both rotaxanes and catenanes.38,39 In work by Leigh and co-workers,38 for example, HREELS experiments have been used to characterize coherent domain sizes within surface confined Gly–Gly containing rotaxane and benzylic amide [2]catenane monolayers (Fig. 3). Though little long range film order was apparent, this was observed to increase with both immersion time and thermal annealing.
![The Gly–Gly rotaxane and dithiolated benzylic amide [2]catenane (composed of two interlocked thiolated macrocycles) used in the XPS and HREELS studies performed by Leigh et al.38 It was proposed in this work that the combined effects of molecular steric bulk and a preferential horizontal surface orientation serve to reduce order in chemisorbed films.](/image/article/2010/CC/b915122b/b915122b-f3.gif) | ||
| Fig. 3 The Gly–Gly rotaxane and dithiolated benzylic amide [2]catenane (composed of two interlocked thiolated macrocycles) used in the XPS and HREELS studies performed by Leigh et al.38 It was proposed in this work that the combined effects of molecular steric bulk and a preferential horizontal surface orientation serve to reduce order in chemisorbed films. | ||
In an angle dependent XAS study of the anthracene stoppered [3]rotaxane shown in Fig. 4, Vance and co-workers were able to demonstrate the importance of preassembly of the pseudorotaxane elements in solution.39 Specifically, the authors were able to highlight the impact of relative solution ratios of dialkylammonium axle and macrocycle (dibenzo-24-crown-8) on the degree of surface confined pseudorotaxane subsequently generated. Interestingly, monolayers could be created from the disulfide-containing [3]rotaxane (the sulfur–sulfur bond presumably cleaving on chemisorption) despite the expected low solution exposure of this functionality. In fact, XPS analyses support an improved surface assembly of this rotaxane in comparison to the pseudorotaxane, an observation loosely ascribed to the increased propensity of the anthracene moiety to π-stack at the surface (an interaction which will potentially prohibit chemisorption in the latter). In both cases, the absence of X-ray absorption angle dependency in subsequent XAS analyses of the rotaxane monolayer, is (again) indicative of surface disorder.
![The pseudorotaxane and [3]rotaxane studied by Vance et al.39 The disulfide bond present in the latter is effectively cleaved on exposure to a gold electrode such that two independent (surface capped) chemisorbed rotaxanes are generated.](/image/article/2010/CC/b915122b/b915122b-f4.gif) | ||
| Fig. 4 The pseudorotaxane and [3]rotaxane studied by Vance et al.39 The disulfide bond present in the latter is effectively cleaved on exposure to a gold electrode such that two independent (surface capped) chemisorbed rotaxanes are generated. | ||
In an attempt to generate spatially isolated, independent, surface confined rotaxanes Azehara et al. studied the effects of dilution within an n-hexanethiol coadsorbate (Fig. 5).40 Notable in this work was the preferential adsorption of the rotaxane for short surface immersion times and the apparent molecularly resolved AFM imaging of the rotaxane.
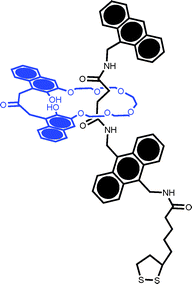 | ||
| Fig. 5 The lipoic acid derived pseudorotaxane studied by Azehara et al.40 Coadsorption with n-hexanethiol enabled coverage to be controlled to the point where individual molecules could be resolved by ambient AFM. | ||
The potential value of computational methods in predicting the adlayer characteristics associated with rotaxane films has been considered in a number of publications. In one example, DFT calculations have predicted a hexagonal surface packing arrangement in a chemisorbed bistable [2]rotaxane (Fig. 6).41 DFT methods have also been applied to the prediction of stable rotaxane surface conformation; within monolayers where a CBPQT4+ macrocycle ring is either encircling tetrathiafulvalene (TTF) or 1,5-dioxynaphthalene (DNP) unit, for example, calculations have suggested energy minimisation where the macrocycle ring is oriented almost in parallel to the underlying surface. Tilt angles for both conformers (i.e. macrocycle encircling either the TTF or DNP units of the axle) were very similar and found to be strongly dependent on the surface concentration.
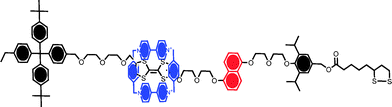 | ||
| Fig. 6 A surface assembling “Stoddart” rotaxane containing a cyclobis(paraquat-p-phenylene) CPBQT4+ ring and π-electron rich tetrathiafulvalene (TTF) and 1,5-dioxynaphthalene (DNP) sites on the axle. The stable resting conformation is that with the macrocycle encircling the TTF moiety. Shuttling between the two stations can be induced by electrochemical or chemical means, specifically by oxidising the TTF moiety.18 | ||
In most published examples of interlocked surface assembly to date, the molecular system has been preassembled in solution before being subsequently chemisorbed through standard silane or noble metal–sulfur chemistry. Work in our own laboratories has demonstrated the viability of templating pseudorotaxanes and rotaxanes from surface confined non-stoppered axles with suitable anions.17,26,27,42–47 Supported by concurrent solution phase NMR analyses, this surface assembly is, in some cases, demonstrably highly template specific.24,44–46,48,49 One example of such surface association has been the assembly of a chemisorbed redox-active bisferrocenyl functionalised rotaxane.42 This templation is chloride specific and could be followed by both surface plasmon resonance (SPR) and electrochemical methods. Interestingly, the three dimensional receptive cavity generated by interlocking the one dimensional (axle) and two dimensional (macrocycle) components can subsequently be utilised in analyte specific electrochemical detection (see later). Assembly such as this involves the coupling of anion recognition with ion-pairing; in non-competitive solvent media, a coordinatively unsaturated chloride anion of a tight ion-pair axle component facilitates, for example, the threading of pyridinium, imidazolium or guanidinium cationic axles through the annulus of an isopthalamide macrocycle.16,28,44
To further extend this work, we have explored the potential of anions to direct the assembly of mechanically interlocked structures composed of charge neutral axle and macrocycle components. Disulfide or thiolate appended indolocarbazole axles are initially chemisorbed. These adlayers can be studied by grazing FTIR, X-ray reflectometry and reductive stripping voltammetry, the latter resolving a molecular footprint of ∼1 nm2, consistent with the relative axle steric bulk. The subsequent threading of the hydrogen bond donating rigid indolocarbazole through isophthalamide macrocycles can, for example, be templated by either fluoride or sulfate (Fig. 7).45–47 This process can be followed by NMR in solution and by SPR at metallic surfaces (Fig. 8). In cases such as this where anions promote the delivery and threading of macrocycles from solution (onto surface confined non-stoppered axles), the process of threading and dethreading can be reversed by changing the solvent (and thereby influencing the templating interactions of the anion) (Fig. 8). Physically adsorbed, non-threaded macrocycles can be removed by gently flushing the surface with solvent to give a stable low pseudorotaxane coverage, corresponding to 30–40% of axle threading.
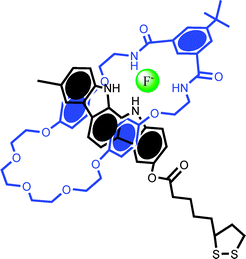 | ||
| Fig. 7 The pseudorotaxane used by Beer and Davis et al. in studying anion recognition and template interlocking on gold surfaces.46 A surface confined non-stoppered indolocarbazole axle and isophthalamide macrocycle are found to reversibly thread in the presence of fluoride or sulfate. | ||
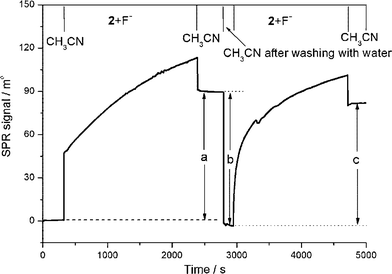 | ||
| Fig. 8 SPR sensorgram showing the reversible threading, dethreading, and rethreading of an isophthalamide macrocycle over a surface confined SAM of non-stoppered indolocarbazole axle in the presence of fluoride (tetra-n-butylammonium fluoride, TBAF) in acetonitrile: (a) the fluoride induced threading of the macrocycle over a SAM of axle; (b) macrocycle dethreading after washing the surface with water; and (c) the fluoride solvent mediated template induced macrocycle rethreading. The upper axis represents regions of flow in the indicated solvent. (© The American Chemical Society, reprinted with permission).46 | ||
In making rotaxanes of increasing potential functionality, one is likely to both increase molecular bulk and decrease adlayer crystallinity. Since our ability to analyse such surfaces will, in general, depend on both surface coverage and molecular order, this is not an insignificant problem. In recent work46 we have shown, for example, that the surface assembly of long axles (long to amplify potentially induced molecular motion, pegylated for solubility/synthetic reasons) generates axle layers which are both disordered and fluxional. The delivery of solution phase macrocycles and subsequent templated threading at the surface becomes, under such circumstances, non-viable and molecular interlocking must then be carried out in solution prior to any surface being presented.
Macrocycles threaded over axles which are stoppered at one end in solution are subsequently trapped at a gold electrode surface by chemisorption (surface stoppering of the other axle terminus). In demonstrating this with thioctic acid derived indolocarbazole axles and macrocycles that do not exhibit electrochemical or optical signatures, we have recently shown that this threading process can be usefully probed through the controlled electrochemical stripping of the axle and subsequent macrocycle release into solution (Fig. 9).
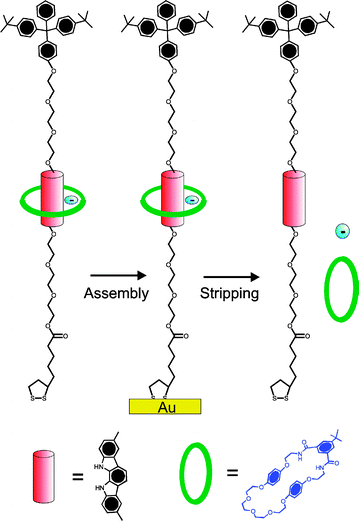 | ||
| Fig. 9 A neutral isophthalamide macrocycle can be threaded over a stoppered indolocarbazole axle in the presence of specific anions (F− or SO42−) and trapped at an electrode surface. Reductive cleavage of the gold–sulfur bonds releases the macrocycle which can be detected by high resolution electrospray mass spectroscopy.46 | ||
Surface assembly on nanoparticles
Nanoparticles (NP) offer highly tuneable optical and electrical characteristics, are potentially diffusive in solution, and present an interface on which host–guest receptors can be assembled.50 From a surface chemistry/surface assembly perspective, much of what is accessible on a planar surface is equally possible on the highly curved surface of a nanoparticle. Despite this, only a comparatively few relevant studies have been reported to date. Work by Fitzmaurice et al. demonstrated the predictable feasibility of rotaxane self assembly on a titanium oxide nanoparticle (Fig. 10).51–53![Tripodal [2]rotaxanes have been successfully immobilized on titanium dioxide nanoparticles. The electron-poor viologen moieties of the axle thread through the electron-rich crown ether macrocycle. 1H NMR spectroscopy and cyclic voltammetry have been employed to study the shuttling/switching characteristics on the nanoparticle.52,53](/image/article/2010/CC/b915122b/b915122b-f10.gif) | ||
| Fig. 10 Tripodal [2]rotaxanes have been successfully immobilized on titanium dioxide nanoparticles. The electron-poor viologen moieties of the axle thread through the electron-rich crown ether macrocycle. 1H NMR spectroscopy and cyclic voltammetry have been employed to study the shuttling/switching characteristics on the nanoparticle.52,53 | ||
More recently, work by Stoddart, Grzybowski and co-workers has shown that Pt, Pd and Au nanoparticles are all feasible supports for bistable donor–acceptor rotaxane and catenane surface self assembly.54Fig. 11 shows the structure of a catenane that has been used by Stoddart et al. to trigger switching/circumrotation, as followed by solution phase cyclic voltammetry and ζ potential measurements, on AuNPs.
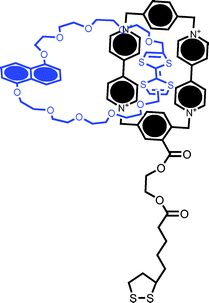 | ||
| Fig. 11 Structural formula of the catenane used to functionalize metal nanoparticles containing a CBPQT4+ group and TTF and DNP subunits in the macrocycle. Chemically triggered switching of the NP-immobilized catenane has been followed by voltammetric and ζ potential measurements.54 | ||
The group of Stoddart has also reported the immobilisation of a number of bistable pseudorotaxanes on mesoporous silica nanoparticles.55 We have recently reported pseudorotaxane formation via the surface covalent attachment (through silane coupling chemistry) of fluorescent indolocarbazole-based axles on silica nanoparticles.47 The axle footprint in this work was estimated to be 20–30 nm2, a value indicative of a low surface packing density and consistent with an emission spectrum unchanged from that resolved for the free axle in solution. The projection of these axles into solution enables subsequent anion templated threading of neutral isophthalamide macrocycles from solution (Fig. 12).47 The sensitivity of the axle fluorescence emission can be utilised as a sensory tool for this threading process and, thus, of the anions which template it.47
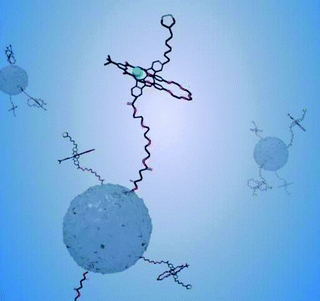 | ||
| Fig. 12 A schematic representation of the anion templated threading of a solution phase macrocycle over a nanoparticle confined indolocarbazole thread. The delivery of macrocycle from solution to the axle is mediated only by specific anions and can be followed by threading induced changes in the axle fluorescence emission.47 | ||
To summarise thus far, there now exist a number of reports of the controlled (chemisorbed and physisorbed56,57) surface assembly of interlocked rotaxanes and catenanes. In some cases these are redox active and responsive to analytes in solution. Though spectroscopic analyses are, in many cases, hampered both by low molecular coverage (arising in part from the steric bulk of these molecules) and low crystallinity, it is demonstrably possible to surface assemble mechanically interlocked structures on gold or oxide surfaces (planar or curved) with good levels of reproducibility. This may be achieved by exposure of a pristine surface to a template solution phase assembly or by carrying out the assembly on the surface itself.
Triggered molecular motion on surfaces
Key in developing potential applications for the increasingly sophisticated synthetic armoury available in this field is an understanding of how resolved solution phase mobility can be translated to a solid surface. One may expect mobility to be impeded by the energetics associated with (potentially dense) surface assembly, for example. The development of methods (spectroscopic or electrochemical) by which mobility/switching can be followed on a surface is, in itself, demanding. Perhaps the principal tool of current choice is that of electrochemistry and pioneering applications of its applicability to rotaxane monolayers have been described by the group of Willner.58 Conceivably, the most powerful means of scrutinizing mobility of surface confined supramolecular architectures is to utilise the redox activity either engendered through their synthesis or subsequently appended. Variable scan rate cyclic voltammetry or pulsed voltammetry methods then become directly analogous to variable temperature solution phase NMR spectroscopy in their ability to probe the environmental switching of a switchable (redox as opposed to nuclear resonant) centre. Electrochemical methods enable a direct resolution of the rate of electron transfer between a redox active component and its supporting electrode surface. If the redox moiety is mobile then its position relative to this surface should be sensitively reflected in the resolved electron transfer kinetics. The position of this redox group may depend on its interactions with an analyte and/or its redox state.Stoddart and co-workers have, for example, used such methods to test the bistability of a surface assembled redox switchable [2]rotaxane,60 where variable temperature and scan rate analyses provided insight into the dynamic behaviour of an immobilised bistable [2]rotaxane. The redox-controlled electromechanical switching mechanism exhibited by the surface immobilized [2]rotaxane is described (Fig. 13). The first redox induced translocation of the macrocycle i.e. movement to the TTF acceptor site was too fast to be detected by cyclic voltammetry (CV). However, the kinetics of the second redox induced translocation i.e. movement from TTF to DNP were analyzable from variable temperature and scan rate dependent CV. From such experiments, the lifetime (period of time the macrocycle rests at each acceptor site) and activation energy associated with this motion can be determined.
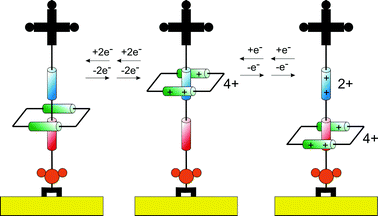 | ||
| Fig. 13 Schematic illustration of the reversible shuttling mechanism of surface bound rotaxane presented by Stoddart et al.18,19,59 Upon chemical oxidation the macrocycle shuttles to the upper position of the thread. In the second step, the acceptor unit on the thread is oxidized, generating an electrostatic repulsion that drives the macrocycle to the lower position. | ||
In double potential step chronoamperometry, it is possible to trigger motion of the redox group by switching the redox state and then probing its position through a second voltage pulse.
This method has been applied to analyse viologen macrocycle motion within rotaxanes, for example.62 Ring translocation has also been characterized by impedance spectroscopy which resolves switchable changes in surface capacitance; this was higher when the shuttling ring was in close proximity to the surface and notably lower when the ring remained at the π-electron binding site (Fig. 14). The translocation rate, and the effect of temperature on this, was resolved in this study.62
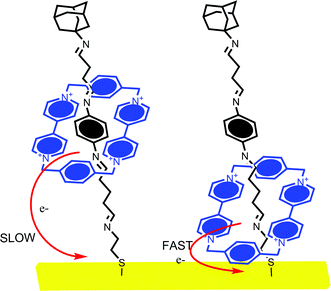 | ||
| Fig. 14 Willner and Katz et al. have investigated the effect of electrochemically induced cyclophane translocation on the electron transfer rate. When the cyclophane ring is close to the electrode surface, the resolved electron transfer rate is more than 10 times faster than that observed when it is at the opposite extreme of the axle.58,61 | ||
Willner and Katz et al. provided an in depth study of temperature dependency on electrochemically induced shuttling of the redox active cyclophane (CBPQT4+) ring along a molecular axle.62 It was demonstrated that at lower temperature the population of oxidized cyclophane was smaller and showed slower kinetics upon re-reduction where at high temperature much faster electron transfer rates were noted. Interestingly, the electron transfer rate of the redox active cyclophane was not affected by temperature change when in close proximity to the gold surface.
In cases where shuttling components are not themselves electroactive, voltammetry can still provide a powerful, indirect means of following molecular motion. In work presented by Kim and co-workers,63 the cucurbituril-macrocyclic cage components of a rotaxane were demonstrated to act as effective “ion gates” in reversibly blocking or facilitating access of solution phase redox probes to the underlying surface (Fig. 15).
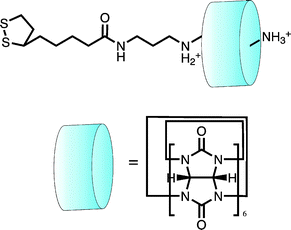 | ||
| Fig. 15 A surface confined pseudorotaxane can be used to modulate access of electroactive analyte to an electrode surface. Here it is shown in the “ON” position where access is restricted. In the “OFF” position (bare thread without the cucurbituril cap) Fe(CN)63− had unobstructed access to the electrode surface indicated by a significant increase in the Faradaic current.63 | ||
In an extension of the enzyme reconstitution methodologies pioneered by this group, the shuttling motion of a redox active macrocycle along a FAD stoppered thread has been applied to facilitate electrical contact between the enzyme glucose oxidase (GOx) and a gold electrode (Fig. 16).61 In effectively shuttling electrons between the electrode and the enzyme active site, the overpotential requirements of turnover are dramatically reduced.
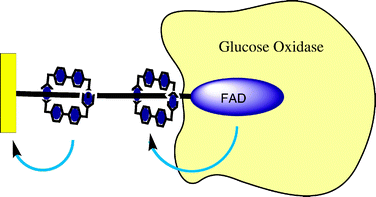 | ||
| Fig. 16 The shuttling motion of a CBPQT4+ ring can be applied as an electron transfer relay for a surface immobilized enzyme. In acting as an electron transfer mediator, the CBPQT4+ unit effectively lowers the overpotential required for electrocatalytic glucose oxidation.61 | ||
The induced mobility of interlocked molecular systems has also been analysed on a number of nanoparticle surfaces. Fitzmaurice et al. have described the surface attachment of an electronically switchable tripodal [2]rotaxane on TiO2 nanoparticles (Fig. 10). Cyclic voltammetry analyses of tripodal [2]rotaxanes immobilised on TiO2 NPs have revealed the complicated nature of the underlying redox response in this system. Four oxidation and reduction steps observed during cyclic voltammetry experiments were associated with the interactions of the crown ether with sequential oxidation and reduction of the viologen groups. These redox processes and the change in electrostatic associations that accompany them have been identified as the main driving force for translocation of the macrocycle along the axle component.52,53
The switching characteristics of bistable [2]catenanes and pseudorotaxanes immobilized on a variety of noble metal (i.e., Au, Pt, Pd) NPs have been recently reported by Stoddart and co-workers.54 Cyclic voltammetry of pseudorotaxane functionalised NPs in solution has revealed a rather complex underlying redox response in these systems. In the case of bistable [2]catenane functionalized AuNPs (Fig. 11), five resolved oxidation and reduction steps have been associated with electrostatic interactions between the TTF/DNP containing axle and the CBPQT4+ macrocycle. In modulating these interactions through the addition of chemical redox species, such as iron(II) perchlorate and ascorbic acid, circumrotation of the macrocycle can be triggered.
In utilising the ability to trigger motion in nanoparticle confined rotaxanes, Stoddart et al. have also developed a series of “nanoscopic valves” by tethering bistable [2]rotaxanes onto silica nanoparticles. When the macrocycle lies close to the particle surface the surface pores are effectively “closed”. The pH or redox triggered macrocycle vectorial motion facilitates an “opening” of these pores and subsequent release of trapped “guest molecules”.55,64–67
Surface confined interlocked assemblies for sensing applications
The application of host–guest recognition to highly specific analyte detection is well established. In an extension of the well understood principles behind much of this work, one may exploit the unique interlocked three dimensional host cavity for guest recognition and seek to utilise the uniquely switchable characteristics of rotaxanes/catenanes as a transduction mechanism (that is to use analyte recognition to trigger motion). As a prelude to such work we have recently demonstrated that the anions used to template an interlocked surface assembly may be removed to leave a rotaxane or catenane with a highly specific vacant interlocked host cavity. In cases where the assembly possesses one or more redox groups, standard voltammetric methodologies can be used in demonstrating subsequent highly specific anion recognition.42,46In one example, an anion binding macrocycle containing an appended ferrocene redox centre and an ion paired axle component (also redox active) were mixed in solution to form a chloride anion templated pseudorotaxane (Fig. 17) assembly.42 The two component nature of the rotaxane SAM subsequently formed by axle chemisorption was confirmed by grazing FTIR and square wave voltammetry (the latter resolving and integrating macrocycle and axle confined redox waves) (Fig. 18).
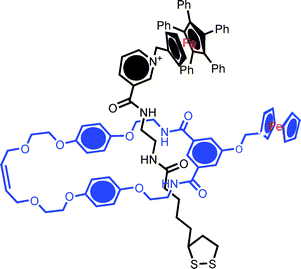 | ||
Fig. 17 Example of a redox active rotaxane assembled by anion templation. Gold electrodes were immersed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ferrocene appended isophthalamide macrocycle and a pentaphenylferrocene pyridinium nicotinamide chloride ion-pair thread in chloroform solution (shifting the equilibrium in favour of rotaxane formation). Hydrogen bond donor based anion recognition in the central (“three dimensional”) cavity is communicated by cathodic perturbation of the electrochemical signals. :1 mixture of ferrocene appended isophthalamide macrocycle and a pentaphenylferrocene pyridinium nicotinamide chloride ion-pair thread in chloroform solution (shifting the equilibrium in favour of rotaxane formation). Hydrogen bond donor based anion recognition in the central (“three dimensional”) cavity is communicated by cathodic perturbation of the electrochemical signals. | ||
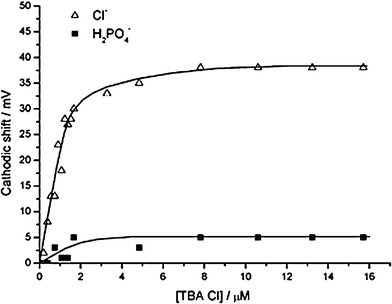 | ||
| Fig. 18 Sensing function performed by electrochemically tagged rotaxane immobilized SAM on a gold surface. The graph compares the voltammetric response of the bisferrocene functionalised rotaxane SAM to chloride and dihydrogen phosphate ions (in acetonitrile). (© Royal Society of Chemistry, reprinted with permission).42 | ||
Upon subsequent chloride anion template removal, the resulting SAM confined interlocked assemblies contain a unique topologically-defined hydrogen bond donating binding domain which exhibits a high degree of selectivity for the templating halide anion. The stoppering ferrocene moiety in this system lies too far away from the receptive hydrogen bonding site to be responsive but the macrocycle redox wave is observed to shift cathodically in the presence of micromolar levels of chloride. Significantly, the ferrocene appended macrocycle’s electrochemical response to dihydrogen phosphate, acetate or hydrogen sulfate observed prior to threading is switched off and the chloride response conversely amplified, a switch assigned to an exclusion of the larger (even if more basic) anion from the interlocked binding domain. The comparative voltammetric responses to chloride and dihydrogen phosphate (in acetonitrile) are shown in Fig. 18. Preliminary electrochemical experiments with these and related interlocked SAMs demonstrated an ability to selectively detect chloride in a 100-fold excess of competing dihydrogen phosphate. It is worth noting that this, to the best of our knowledge, constitutes the first reported example of a surface assembled interlocked receptor being applied as a sensory tool.
Somewhat analogous to this has been a demonstration by us of the use of SPR to detect anions that template macrocycle threading over electrode-confined axles. Though the anions themselves are of too low molecular weight to be normally detectable by SPR, their binding at a receptive axle is amplified by the templated threading of suitable macrocycles from solution (generating a detectable change in surface dielectric).46 Significantly, negligible threading/dielectric response is observed on axle SAM exposure to macrocycle in the absence of specific anions.
More recently we have reported the sensing capabilities of anion templated indolocarbazole containing pseudorotaxane on silica NPs (Fig. 12).47 Silica NPs are optically transparent, non-quenching and can be used in solution phase optical detection/switching applications that retain the advantages of immobilized surface receptors. The sensitivity of the axle fluorescence spectrum to threading can be utilised indirectly in the sensing of anions which template this process. Specifically, threading induced bathochromic shifts are observed to be fully consistent with the relative hydrogen bonding affinity of anions and 1H NMR titration studies carried out on analogous particles.47
Conclusions
The surface assembly of increasingly complex molecular assemblies on optically or electrochemically/electrically addressable surfaces lies central to many fields of chemistry. From a host–guest sensing perspective, the advantages of tethering the former to a transducing surface (such as that presented by an electrode) are considerable. In addition to the possibility of generating a robust, multiplexable and renewable host surface capable of operation in a variety of fluid media (static or flowing) there exists considerable evidence that the thermodynamics of host–guest association are favoured at such interfaces.43,50,68 The marriage of these interactions with interfacial chemistry is a powerful one in facilitating the generation of increasingly complex multifunctional assemblies. Though the analysis of these surfaces is often hampered by coverage and crystallinity issues, their potential application in the development of nanoscale motors, energy/data storage (triggered readable changes in bistable films) and sensing remain as broad as scientists dare imagine. This feature article has attempted to summarise relevant work in the area, highlighting typical characteristics and areas of recent development.Acknowledgements
The authors acknowledge financial support from the EPSRC (EP/D008417/1, EP/E033962/1, EP/F011504/1).Notes and references
- A. H. Flood, R. J. A. Ramirez, W.-Q. Deng, R. P. Muller, W. A. Goddard, III and J. F. Stoddart, Aust. J. Chem., 2004, 57, 301–322 CrossRef CAS.
- V. Balzani, A. Credi and M. Venturi, ChemPhysChem, 2008, 9, 202–220 CrossRef.
- I. Willner, B. Basnar and B. Willner, Adv. Funct. Mater., 2007, 17, 702–717 CrossRef CAS.
- H. Tian and Q.-C. Wang, Chem. Soc. Rev., 2006, 35, 361–374 RSC.
- V. Balzani, A. Credi, B. Ferrer, S. Silvi and M. Venturi, Top. Curr. Chem., 2005, 262, 1–27 CAS.
- A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 433–444 CrossRef CAS.
- C. A. Nijhuis, B. J. Ravoo, J. Huskens and D. N. Reinhoudt, Coord. Chem. Rev., 2007, 251, 1761–1780 CrossRef CAS.
- A. Ulman, J. Mater. Educ., 1989, 11, 205 Search PubMed.
- A. B. Braunschweig, B. H. Northrop and J. F. Stoddart, J. Mater. Chem., 2006, 16, 32–44 RSC.
- A. Coskun, S. Saha, I. Aprahamian and J. F. Stoddart, Org. Lett., 2008, 10, 3187–3190 CrossRef CAS.
- J. S. Hannam, T. J. Kidd, D. A. Leigh and A. J. Wilson, Org. Lett., 2003, 5, 1907 CrossRef CAS.
- C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303 CrossRef CAS.
- O. Lukin and F. Vögtle, Angew. Chem., Int. Ed., 2005, 44, 1456–1477 CrossRef CAS.
- G. Brancato, F. Coutrot, D. A. Leigh, A. Murphy, J. K. Y. Wong and F. Zerbetto, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4967–4971 CrossRef CAS.
- J.-P. Collin, V. Heitz and J.-P. Sauvage, Top. Curr. Chem., 2005, 262, 29–62 CAS.
- M. D. Lankshear and P. D. Beer, Acc. Chem. Res., 2007, 40, 657–668 CrossRef CAS.
- M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211–225 RSC.
- R. A. Bissell, E. Cordova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef.
- C. P. Collier, J. O. Jeppesen, Y. Luo, J. Perkins, E. W. Wong, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2001, 123, 12632–12641 CrossRef CAS.
- T. Lu, L. Zhang, G. W. Gokel and A. E. Kaifer, J. Am. Chem. Soc., 1993, 115, 2542–2543 CrossRef CAS.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS.
- B. Champin, P. Mobian and J.-P. Sauvage, Chem. Soc. Rev., 2007, 36, 358–366 RSC.
- J. P. Sauvage, Acc. Chem. Res., 1990, 23, 319–327 CrossRef CAS.
- G. Kaiser, T. Jarrosson, S. Otto, Y.-F. Ng, A. D. Bond and J. K. M. Sanders, Angew. Chem., Int. Ed., 2004, 43, 1959–1962 CrossRef CAS.
- R. Vilar, Eur. J. Inorg. Chem., 2008, 357–367 CrossRef CAS.
- M. J. Chmielewski, J. J. Davis and P. D. Beer, Org. Biomol. Chem., 2009, 7, 415–424 RSC.
- L. M. Hancock and P. D. Beer, Chem.–Eur. J., 2009, 15, 42–44 CrossRef CAS.
- M. D. Lankshear and P. D. Beer, Coord. Chem. Rev., 2006, 250, 3142–3160 CrossRef CAS.
- K.-Y. Ng, V. Felix, S. M. Santos, N. H. Rees and P. D. Beer, Chem. Commun., 2008, 1281–1283 RSC.
- B. X. Colasson, C. Dietrich-Buchecker, M. C. Jimenez-Molero and J.-P. Sauvage, J. Phys. Org. Chem., 2002, 15, 476–483 CrossRef CAS.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS.
- E. Coronado, P. Gavina and S. Tatay, Chem. Soc. Rev., 2009, 38, 1674–1689 RSC.
- N. Weber, C. Hamann, J.-M. Kern and J.-P. Sauvage, Inorg. Chem., 2003, 42, 6780–6792 CrossRef CAS.
- L. Raehm, J.-M. Kern, J.-P. Sauvage, C. Hamann, S. Palacin and J.-P. Bourgoin, Chem.–Eur. J., 2002, 8, 2153–2162 CrossRef CAS.
- J.-M. Kern, L. Raehm and J.-P. Sauvage, C. R. Acad. Sci., Ser. IIc: Chim., 1999, 2, 41–47 CrossRef CAS.
- E. Coronado, A. Forment-Aliaga, P. Gavina and F. M. Romero, Inorg. Chem., 2003, 42, 6959–6961 CrossRef CAS.
- C. De Nadai, C. M. Whelan, C. Perollier, G. Clarkson, D. A. Leigh, R. Caudano and P. Rudolf, Surf. Sci., 2000, 454–456, 112–117 CrossRef CAS.
- A. L. Vance, T. M. Willey, T. van Buuren, A. J. Nelson, C. Bostedt, G. A. Fox and L. J. Terminello, Nano Lett., 2003, 3, 81–84 CrossRef CAS.
- H. Azehara, W. Mizutani, Y. Suzuki, T. Ishida, Y. Nagawa, H. Tokumoto and K. Hiratani, Langmuir, 2003, 19, 2115–2123 CrossRef CAS.
- S. S. Jang, Y. H. Jang, Y.-H. Kim, W. A. Goddard, III, A. H. Flood, B. W. Laursen, H.-R. Tseng, J. F. Stoddart, J. O. Jeppesen, J. W. Choi, D. W. Steuerman, E. DeIonno and J. R. Heath, J. Am. Chem. Soc., 2005, 127, 1563–1575 CrossRef CAS.
- S. R. Bayly, T. M. Gray, M. J. Chmielewski, J. J. Davis and P. D. Beer, Chem. Commun., 2007, 2234–2236 RSC.
- P. D. Beer, J. J. Davis, D. A. Drillsma-Milgrom and F. Szemes, Chem. Commun., 2002, 1716–1717 RSC.
- P. D. Beer, M. R. Sambrook and D. Curiel, Chem. Commun., 2006, 2105–2117 RSC.
- M. J. Chmielewski, L. Zhao, A. Brown, D. Curiel, M. R. Sambrook, A. L. Thompson, S. M. Santos, V. Felix, J. J. Davis and P. D. Beer, Chem. Commun., 2008, 3154–3156 RSC.
- L. Zhao, J. J. Davis, K. M. Mullen, M. J. Chmielewski, R. M. J. Jacobs, A. Brown and P. D. Beer, Langmuir, 2009, 25, 2935–2940 CrossRef CAS.
- L. Zhao, K. M. Mullen, M. J. Chmielewski, A. Brown, N. Bampos, P. D. Beer and J. J. Davis, New J. Chem., 2009, 33, 760–768 RSC.
- M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul, A. R. Cowley, F. Szemes and M. G. B. Drew, J. Am. Chem. Soc., 2005, 127, 2292–2302 CrossRef CAS.
- B. Huang, S. M. Santos, V. Felix and P. D. Beer, Chem. Commun., 2008, 4610–4612 RSC.
- D. P. Cormode, J. J. Davis and P. D. Beer, J. Inorg. Organomet. Polym. Mater., 2008, 18, 32–40 CrossRef CAS.
- K. Nikitin, E. Lestini, M. Lazzari, S. Altobello and D. Fitzmaurice, Langmuir, 2007, 23, 12147–12153 CrossRef CAS.
- B. Long, K. Nikitin and D. Fitzmaurice, J. Am. Chem. Soc., 2003, 125, 5152–5160 CrossRef CAS.
- B. Long, K. Nikitin and D. Fitzmaurice, J. Am. Chem. Soc., 2003, 125, 15490–15498 CrossRef CAS.
- R. Klajn, L. Fang, A. Coskun, M. A. Olson, P. J. Wesson, J. F. Stoddart and B. A. Grzybowski, J. Am. Chem. Soc., 2009, 131, 4233–4235 CrossRef CAS.
- T. D. Nguyen, Y. Liu, S. Saha, K. C. F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626–634 CrossRef CAS.
- G. Fioravanti, N. Haraszkiewicz, E. R. Kay, S. M. Mendoza, C. Bruno, M. Marcaccio, P. G. Wiering, F. Paolucci, P. Rudolf, A. M. Brouwer and D. A. Leigh, J. Am. Chem. Soc., 2008, 130, 2593–2601 CrossRef CAS.
- J. Berna, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Perez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef CAS.
- E. Katz, O. Lioubashevsky and I. Willner, J. Am. Chem. Soc., 2004, 126, 15520–15532 CAS.
- Y. B. Zheng, Y.-W. Yang, L. Jensen, L. Fang, B. K. Juluri, A. H. Flood, P. S. Weiss, J. F. Stoddart and T. J. Huang, Nano Lett., 2009, 9, 819–825 CrossRef CAS.
- H.-R. Tseng, D. Wu, N. X. Fang, X. Zhang and J. F. Stoddart, ChemPhysChem, 2004, 5, 111–116 CrossRef CAS.
- E. Katz, L. Sheeney-Haj-Ichia and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 3292–3300 CrossRef CAS.
- E. Katz, R. Baron, I. Willner, N. Richke and R. D. Levine, ChemPhysChem, 2005, 6, 2179–2189 CrossRef CAS.
- K. Kim, W. S. Jeon, J.-K. Kang, J. W. Lee, S. Y. Jon, T. Kim and K. Kim, Angew. Chem., Int. Ed., 2003, 42, 2293–2296 CrossRef CAS.
- K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y.-W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382–2383 CrossRef CAS.
- R. Hernandez, H.-R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370–3371 CrossRef CAS.
- S. Angelos, Y.-W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344–11346 CrossRef CAS.
- S. Angelos, Y.-W. Yang, K. Patel, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222–2226 CrossRef CAS.
- J. J. Davis and P. D. Beer, The Encyclopedia of Nanoscience and Nanotechnology, Marcel Dekker, 2004, pp. 2477–2492 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |