Advances in chemical ligation strategies for the synthesis of glycopeptides and glycoproteins
Richard J. Payne*a and Chi-Huey Wongbc
aSchool of Chemistry, The University of Sydney, Sydney, Australia. E-mail: payne@chem.usyd.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 5877
bPresident, Academia Sinica, 128 Academia Road, Section 2, Nankang, Taipei, Taiwan. E-mail: chwong@gate.sinica.edu.tw; Fax: +886 2 2785 3852; Tel: +886 2 2789 9400
cDepartment of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA. E-mail: wong@scripps.edu
First published on 30th October 2009
Abstract
A number of recent advances in the chemical synthesis of glycopeptides and glycoproteins are described, with particular focus on the development of peptide ligation strategies and their implementation in the convergent assembly of complex glycopeptides. Recent applications in the synthesis of full length homogeneous glycoproteins are also highlighted.
 Richard J. Payne | Richard J. Payne was born in Christchurch, New Zealand, in 1980. He obtained his PhD from the University of Cambridge under the supervision of Professor Chris Abell and in 2006 moved to the Scripps Research Institute where he worked as a Postdoctoral Research Fellow under the guidance of Professor Chi-Huey Wong. Here, he was involved in the development of new ligation strategies for the synthesis of glycopeptides. In 2008, he was appointed as a Lecturer within the School of Chemistry at the University of Sydney. His current research interests include tuberculosis drug discovery, carbohydrate chemistry and glycopeptide synthesis. |
 Chi-Huey Wong | Dr Chi-Huey Wong is President of Academia Sinica and Professor of Chemistry at the Scripps Research Institute. He is a member of Academia Sinica, Taipei, the American Academy of Arts and Sciences, and the US National Academy of Sciences. His research interests are in the areas of bioorganic and synthetic chemistry, including the development of new synthetic chemistry based on enzymatic and chemoenzymatic reactions, synthesis of complex carbohydrates, glycoproteins and small-molecule probes for the study of carbohydrate-mediated biological recognition, development of carbohydrate microarrays for high-throughput analysis of carbohydrate–protein interactions, and drug discovery. |
1. Introduction
Protein glycosylation is a ubiquitous post-translational modification. The inherent complexity and variability of oligosaccharides allow for the introduction of an enormous level of structural diversity to proteins.1 Estimations suggest that more than fifty percent of all human proteins are glycosylated,2 a modification known to be of paramount importance for a variety of biological recognition events such as cell adhesion, cell differentiation and cell growth.3,4 In addition, aberrant glycosylation of proteins often modifies intracellular recognition and has been implicated in a number of serious illnesses including autoimmune diseases, infectious diseases and cancer.5,6 As a consequence, glycoproteins have potential utility in a clinical context for the development of therapeutics, diagnostics and vaccines.Broadly speaking, almost all native protein glycosylations can be classified into two types: O-glycosides, whereby a glycan is α- or β-linked to the hydroxyl of serine, threonine or tyrosine, or N-glycosides, in which N-acetylglucosamine is β-linked to the amide side chain of an asparagine present within an Asn-Xaa-Ser/Thr consensus sequence (Fig. 1). The oligosaccharides attached to the protein backbone are usually complex, branched structures. This is a consequence of the fact that attachment of each monosaccharide unit to the pre-existing glycan can occur at one of four hydroxyl groups with either α- or β-stereochemistry. As such, a simple library of tetrasaccharides generated combinatorially from the nine common sugar building blocks would result in over 15 million structures.7 Putting this figure in context, tetrameric peptides and nucleic acids generated in the same way (from 20 proteinogenic amino acids and four DNA bases) would give rise to 160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 256 different structures, respectively. It has been postulated that glycosylation could serve to expand the information derived from the concise human genome and facilitate biological processes exclusive to more complex organisms.8–10
000 and 256 different structures, respectively. It has been postulated that glycosylation could serve to expand the information derived from the concise human genome and facilitate biological processes exclusive to more complex organisms.8–10
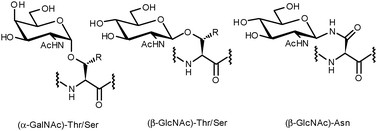 | ||
| Fig. 1 Common O- and N-linkages of glycans to the protein backbone. | ||
Despite the importance of glycoproteins in a myriad of biological processes, progress towards studying their structure and function has been agonisingly slow, due in major part to difficulties in obtaining them in homogeneous form. Obviously, if one aims to understand the role of glycosylation at a molecular level, it is imperative to have access to homogeneous glycopeptides and glycoproteins, however, this is by no means a trivial exercise. The difficulty arises from the untemplated nature of glycosylation which, unlike protein synthesis, is not under the control of a coding template but, rather, is dictated by the relative activities of a number of glycosyltransferase enzymes. For this reason the resulting glycoproteins are generally produced as heterogeneous mixtures of glycoforms which are generally inseparable by currently available chromatographic techniques. Recombinant expression cannot, in most cases, be used for the production of homogeneous glycoproteins. It is currently accepted that chemical and chemoenzymatic intervention can be used to solve the availability problem.7,10–15 In this article, we will discuss the development of a number of peptide ligation strategies and their application in the synthesis of complex, homogeneous glycopeptides. It is intended that this article complement a number of excellent reviews previously published7,10–19 and, additionally, highlight some recent breakthroughs in the area of glycoprotein total synthesis.
2. Native chemical ligation
The reliable and efficient nature of solid-phase peptide synthesis (SPPS) has provided an excellent platform for the construction of glycopeptides containing up to 40–50 amino acids. However, the linear nature of SPPS means that longer syntheses are usually plagued by truncated (uncoupled) sequences, unwanted side products and epimerisation. Furthermore, these byproducts tend to accumulate, thereby resulting in low yields and purities of the final products. Clearly this size limitation precludes the use of SPPS for the total synthesis of long glycopeptides and glycoproteins, necessitating the development of chemical ligation methods which serve to assemble these complex biomolecules in a convergent manner.By far the most efficient method for the ligation-based assembly of peptides, proteins and post-translationally modified peptides and proteins is native chemical ligation (NCL). The concept of NCL dates back to the 1950s with the pioneering work of Wieland et al.,20 however it was not until 1994 that this method gained widespread attention as an efficient method for the convergent ligation of peptides when Kent and co-workers reported its application in the synthesis of interleukin-8, a cytokine responsible for the proliferation of B cells during an immune response.21 The reaction relies on the chemoselective condensation between a peptide bearing an N-terminal cysteine residue and a peptide containing a C-terminal thioester moiety to afford an amide bond (Scheme 1). Such a ligation reaction involves a rapid, reversible transthioesterification step to generate a thioester intermediate which then undergoes an irreversible S → N acyl shift to generate a native peptide bond (Scheme 1). The scope of this method has been extensively examined, with most amino acids on the C-terminus of the peptide thioester component shown to undergo facile ligations.22 Since its inception the method has received widespread attention for the synthesis of proteins. Notably, NCL has been successfully utilised in the synthesis of over two hundred full length proteins and has been the subject of several excellent reviews.17–19,23–27
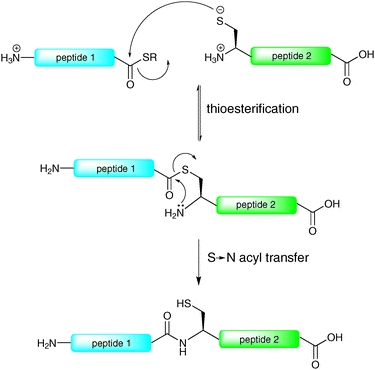 | ||
| Scheme 1 Proposed mechanism of native chemical ligation (NCL).21 | ||
NCL has also served as a useful tool for the synthesis of biologically relevant glycopeptides and glycoproteins, with the first significant example being the total synthesis of diptericin ε, an antibacterial glycopeptide containing 82 amino acids and two O-linked glycosylation sites.28 Since the primary sequence is devoid of cysteine residues, it was necessary to introduce a Gly25-Cys mutation to allow for a synthetic disconnection by NCL. It was also necessary to mutate two further residues, namely Asp29-Glu and Asp45-Glu, to enable its construction. The synthesis was initiated by SPPS of a 24-residue N-terminal glycopeptide thioester 1 and a 58-residue glycopeptide 2 containing an N-terminal cysteine residue, both fragments bearing α-N-acetylgalactosamine (α-GalNAc, also known as the TN antigen) at the two glycosylation sites (Thr11 and Thr54, Scheme 2). Owing to problems relating to thioester hydrolysis under the conditions afforded by Fmoc-strategy SPPS, peptide thioesters are traditionally produced via the Boc-strategy. However, in the context of glycopeptide thioester construction, such iterative acid deprotection conditions are usually incompatible with the labile glycosidic linkages. To circumvent this problem, Ellman’s modification of Kenner’s sulfonamide “safety-catch” linker was employed for the synthesis of the N-terminal glycopeptide thioester fragment.29,30 Upon assembly of the glycopeptide by Fmoc-strategy SPPS, the sulfonamide moiety of 3 was selectively alkylated by treatment with iodoacetonitrile in the presence of N,N-diisopropylethylamine (DIPEA) to afford resin bound 4. Thiolysis with benzyl mercaptan successfully released the fully protected glycopeptide thioester which was then treated with an acidic cocktail to remove both the side chain protecting groups and the N-terminal Boc-carbamate to afford 1. Glycopeptide thioester 1 and glycopeptide 2 were subjected to the standard NCL conditions [6 M guanidine hydrochloride (Gn·HCl), 0.1 M phosphate buffer, thiophenol (PhSH), pH 7.5] to afford, after glycan deacetylation by hydrazinolysis, synthetic diptericin ε (5) which retained antimicrobial activity despite the amino acid substitutions.
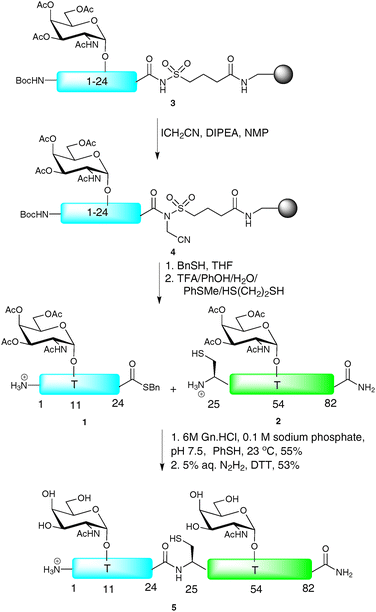 | ||
| Scheme 2 Total synthesis of the antibacterial glycopeptide diptericin εvia NCL.28 | ||
In another early example, Bertozzi and co-workers demonstrated the NCL-based synthesis of a glycoprotein with multiple O-glycosylation sites of the so-called “mucin type”.31 The target, lymphotactin (Lptn), is a 93-residue chemokine which serves as a potent chemoattractant for T cells and natural killer cells.32,33 The C-terminus of Lptn contains a mucin domain with up to eight serine and threonine O-glycosylation sites. The synthetic strategy involved the use of NCL to join two equally sized fragments: peptide thioester 6 corresponding to Lptn1–48 and glycopeptide 7 (Lptn49–93) bearing an N-terminal cysteine and eight α-GalNAc moieties (Scheme 3). As peptide thioester 6 does not possess any backbone glycosylation, it was synthesised via Boc-strategy SPPS. In contrast, glycopeptide 7 was synthesised via Fmoc-strategy SPPS incorporating the preformed glycosylamino acids into the growing chain. The two fragments were ligated under standard NCL conditions to afford the 93-amino acid glycoprotein in 38% yield after HPLC purification. The low yield in this case can be attributed to the use of a thioester containing a C-terminal valine, known to react slowly under NCL conditions owing to the sterically demanding nature of the side chain. In order to generate the native structure, the final step involved folding, achieved by incubating at pH 8 in the presence of cysteine and cystine, to afford the native glycoprotein 8 in 49% yield. Synthetic Lptn 8 was subsequently assessed for its ability to bind its cognate chemokine receptor (XCR1) expressed on human embryonic kidney cells, specifically by assessing its activation of a signal transduction cascade providing an increase in intracellular calcium concentrations. Surprisingly, 8 exhibited similar activity to its unglycosylated counterpart, suggesting that the simple glycans do not play a role in recognition and binding to the chemokine receptor. The authors suggest that the native (presumably more complex) O-linked glycans on Lptn would impart a different three-dimensional structure compared to the bridgehead monosaccharides in their synthetic version therefore modulating function in vivo.
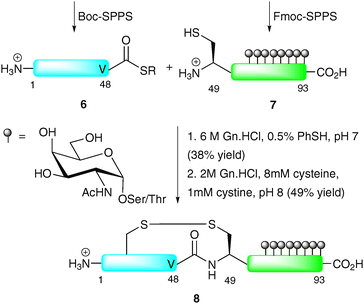 | ||
| Scheme 3 Total synthesis of lymphotactin (Lptn) by NCL.31 | ||
The examples discussed so far have demonstrated the utility of NCL for the generation of glycopeptides displaying simple O-linked monosaccharides on the peptide backbone. However, most glycoproteins require more complex glycans for optimal biological activity (vide supra). Over recent years a suite of glycosyltransferase enzymes have become available that can be used for the chemoenzymatic elaboration of glycopeptides and glycoproteins.34–42 While this represents a powerful method for the synthesis of complex glycopeptides, there are several possible drawbacks. These include the potential for incomplete glycosylation, giving rise to glycoformic mixtures which may or may not be separable by chromatography. Additionally, there are a number of glycosidic linkages which cannot be accessed because the requisite enzyme is not readily available. To alleviate these problems, many groups have decided to prepare the desired oligosaccharides synthetically. These can then be incorporated into glycopeptides in one of two ways: the first being introduction of preformed glycosylamino acids which can be used as “cassettes” directly in the SPPS of glycopeptides and glycopeptide thioesters. Alternatively, a more convergent approach, first reported by Cohen-Anisfeld and Lansbury and dubbed the “Lansbury aspartylation,” allows simple access to N-linked glycopeptides.43,44 The method relies on the direct coupling of a glycosylamine onto a polypeptide chain containing an aspartic acid residue. The reaction conditions have been optimised to minimise aspartimide side product formation and, due to the nature of the coupling conditions, extensive protection of the peptide backbone and oligosaccharide is not necessary (with the exception of other carboxylate side chains). The glycopeptides and glycopeptide thioester fragments synthesised by one of these two strategies can be implemented in NCL reactions in the normal way to generate glycopeptides and glycoproteins bearing complex glycans as defined glycoforms.
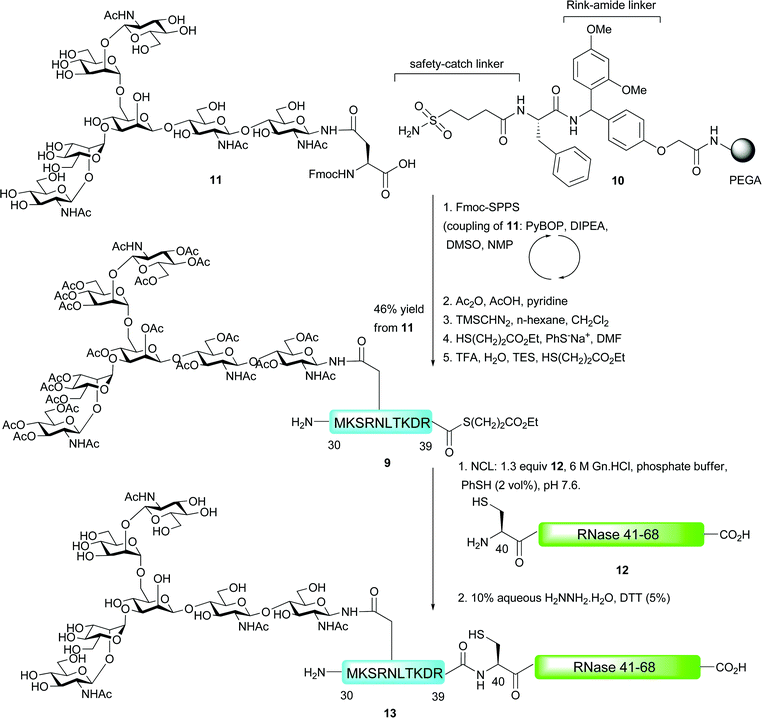
The first example of an N-linked glycopeptide bearing a complex-type glycan to be constructed by means of NCL was reported by Unverzagt and co-workers in their synthesis of a glycosylated fragment of RNase B (Scheme 4).45 Fmoc-strategy SPPS was utilised for the synthesis of glycodecapeptide thioester 9 which was assembled on PEGA resin 10 incorporating two linkers before the assembled glycopeptide. The first of these linkers was the Ellman-sulfonamide safety-catch linker discussed previously.29,30 The group also utilised a Rink amide linker which allowed for peptides to be released during peptide assembly in order to assess the efficiency of the synthesis by HPLC and LC-MS. The Fmoc-protected glycosylasparagine presenting an unprotected biantennary heptasaccharide (11) was coupled into the growing peptide chain as a “cassette” in high yield by pre-activation with 1-H-benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) and DIPEA. The authors used just 0.8 equivalents of the precious glycosylamino acid with respect to the resin bound peptide and achieved a 95% yield as determined by measuring the piperidine–fulvene adduct produced upon deprotection with piperidine–DMF. Acetylation of the oligosaccharide hydroxyls was next conducted to stabilise the glycosidic linkages to acidic cleavage conditions and to prevent unwanted acylation reactions. This was achieved using acetic anhydride, acetic acid and pyridine and allowed for selective acetylation without activating the sensitive sulfonamide linker. Following acetylation, the sulfonamide was activated by treatment with the powerful methylation reagent trimethylsilyldiazomethane followed by thiolysis with mercaptopropionic acid ethyl ester (100 equiv.) and sodium thiophenolate (3 equiv.) to release the fully protected glycopeptide thioester from the solid support. Finally, acidic cleavage of the side chain protecting groups furnished the desired glycopeptide thioester 9 in 46% yield based on the coupling of glycosylamino acid 11. Generation of the C-terminal fragment 12 bearing an N-terminal cysteine residue (RNase 41–68) was achieved via standard Fmoc-strategy SPPS and this was reacted with thioester 9 under standard NCL conditions (on an analytical scale). After reacting for 8 h, followed by in situ deacetylation by treatment with aqueous hydrazine in the presence of DTT, glycopeptide 13, corresponding to RNase 30–68, was observed by HPLC and mass spectrometry.
More recently, Danishefsky and co-workers have reported the synthesis of a protected version of the full length glycoprotein β-human follicle-stimulating hormone (β-hFSH) possessing two N-linked chitobiose disaccharide units.46 In this report the authors also describe the synthesis of a glycopeptide fragment of β-hFSH bearing an N-linked dodecasaccharide and a C-terminal thioester. It is anticipated that this fragment will be utilised for the NCL-based synthesis of a more complex glycoform of this important glycoprotein in future studies.
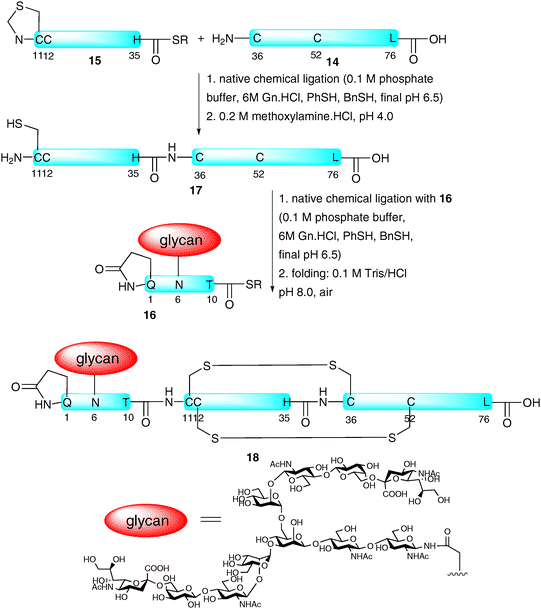
Although hundreds of proteins19,23 and a number of glycopeptides have been synthesised with the assistance of NCL-based strategies, the total synthesis of a homogeneous glycoprotein bearing a complex glycan and a native amino acid sequence remained elusive until 2008 when the groups of Dawson and Kajihara reported the preparation of a single glycoform of the 76-amino acid chemokine monocyte chemotactic protein-3 (MCP-3).47 The group chose to assemble the glycoprotein via the ligation-based assembly of three fragments. These included peptide 14 (residues 36–76) bearing an N-terminal cysteine and peptide thioester 15 (MCP-3 11–35) containing an N-terminal thiazolidine as a masked cysteine which were both prepared by Boc-strategy SPPS (Scheme 5). The final fragment was glycopeptide thioester 16 (MCP-3 1–10) containing a complex-type sialylated N-glycan. The latter fragment was prepared using both Boc- and Fmoc-strategy SPPS, the former being particularly impressive given the potential lability of glycosidic linkages under iterative treatment with trifluoroacetic acid in the Boc-deprotection steps (vide supra). The three fragments were subsequently assembled via two consecutive NCL reactions. Specifically 14 and 15 were ligated to afford MCP-3 11–76 (17) followed by ligation with 16 to furnish the full length glycoprotein in relatively high yield, further demonstrating the efficiency of the NCL reaction in the assembly of complex targets. The glycoprotein was subsequently folded to afford native MCP-3 (18) as a pure glycoform. The correct alignment of disulfide bonds was confirmed by chymotrypsin degradation studies.
Since the seminal report by Kent and co-workers,21 a number of modifications have been made to the traditional NCL reaction. For example, Boons and co-workers have carried out glycopeptide ligations within liposomes to aid in the facile construction of lipophilic glycopeptides.48,49 These liposome-mediated NCL reactions were found to increase reaction rates and provide higher yields than those conducted in aqueous buffered media.
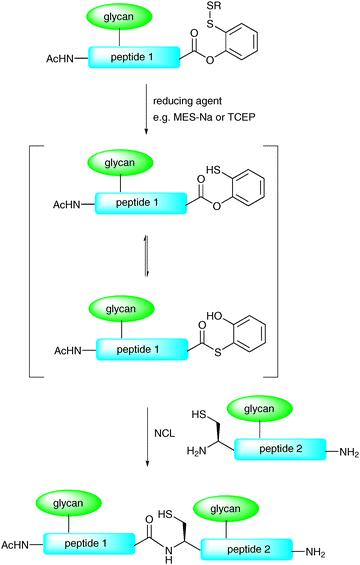
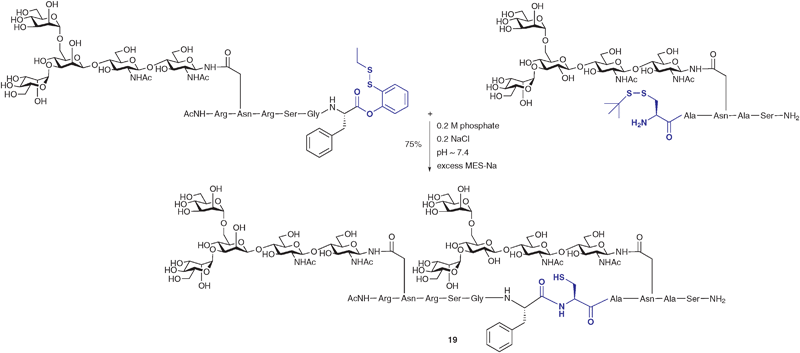
In another modification to the traditional NCL reaction, a number of groups have chosen to modify the C-terminal thioester to incorporate a masked thioester or an alternative acyl donor. For example, Danishefsky and co-workers reported a method for the synthesis and use of masked thioesters in NCL, thus overcoming some of the problems associated with lability of peptide and glycopeptide thioesters. The strategy was to synthesise a C-terminal peptide phenolic ester bearing an o-disulfide moiety which could rearrange to produce a thioester under reductive conditions (Scheme 6).50 The phenolic ester can be readily prepared by introducing a suitably protected 2-mercaptophenol unit to a peptide or glycopeptide (generated by SPPS). Upon reduction of the disulfide with a suitable reagent e.g. sodium 2-mercaptoethanesulfonate (MES-Na) or triscarboxyethyl phosphine (TCEP) a free thiol is revealed which can participate in a reversible O → S acyl shift to generate a C-terminal thioester (Scheme 6). Importantly, the thioester can be generated in situ in a ligation reaction by adding a suitable reducing agent and, in the presence of a peptide or glycopeptide bearing an N-terminal cysteine, will undergo an NCL reaction. This strategy was applied in the synthesis of glycopeptide 19 bearing two N-linked glycans including the core N-linked pentasaccharide and an unnatural version containing a bridging glucose in place of mannose (Scheme 7). Based on the early work of Bodanszky,51 Danishefsky and co-workers have also reported the use of an activated p-nitrophenyl ester as a thioester surrogate in NCL reactions.52 Recently, Blanco-Canosa and Dawson reported the preparation of C-terminal peptide isoureas which can serve as precursors to C-terminal thioesters.53 Alternatively, these can undergo thioesterification in situ and can participate in NCL reactions. Peptide isoureas could be synthesised in high yield using standard Fmoc-strategy solid-phase chemistry.53 Similarly, the cysteinyl prolyl ester has recently received attention as a masked thioester equivalent in peptide ligation reactions.54 Due to ease of preparation, these new C-terminal acyl donors and thioester precursors should find wide application in future glycopeptide and glycoprotein syntheses. It is also anticipated that further modifications to the NCL reaction will continue to be reported with a view to improving its utility in the synthesis of a host of glycopeptides and glycoproteins.
 | ||
| Scheme 7 NCL-based synthesis of a complex model glycopeptide using an o-disulfide phenolic ester as a masked thioester.50 | ||
3. Expressed protein ligation
NCL represents a powerful tool for the construction of complex glycopeptides, however for larger targets it is often desirable to produce one of the two coupling partners recombinantly, a method dubbed expressed protein ligation (EPL).55–58 Strategies for recombinant production of both C-terminal and N-terminal coupling partners have been developed in recent years. Production of the C-terminal coupling partner involves introduction of an N-terminal protease recognition sequence followed by a cysteine into a peptide or protein fragment which is expressed in bacteria.59 After cleavage of the protease recognition sequence, an N-terminal cysteine residue is unveiled which can undergo an NCL reaction with an appropriate thioester. This strategy finds application in the synthesis of glycopeptides and glycoproteins containing glycosylation at the N-terminus of the sequence. Both factor Xa and tobacco etch virus protease (TEV protease) have been used to remove the N-terminal protease cleavage sequences introduced into fusion proteins to generate fragments containing an N-terminal cysteine residue (Scheme 8).59–61 It is also possible to introduce an N-terminal affinity tag into the recombinant peptide or protein. This allows for simple purification before removal of the protease sequence to afford the desired fragment. This strategy was employed by Tolbert et al. for the synthesis of a homogeneous glycoform of interleukin-2 (IL-2), a T-cell growth factor used in the clinic for the treatment of renal cell carcinoma and metastatic melanoma.60 In this work, C-terminal fragment 20 was first expressed as a fusion protein possessing a hexahistidine tag (His-tag), the TEV protease recognition sequence (ENLYFQ) and a long C-terminal fragment of IL-2 (7–133) (Scheme 8). Following affinity purification (Ni2+) the protein was treated with TEV protease to provide 21 with an unmasked cysteine (Cys-7) residue. The protein underwent smooth NCL with a synthetic glycopeptide thioester to afford IL-2 (22) in >95% yield.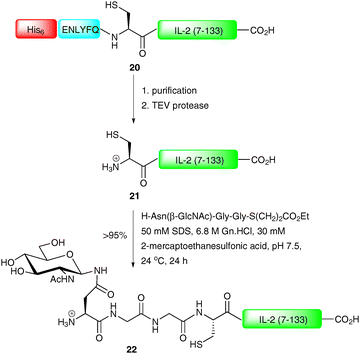
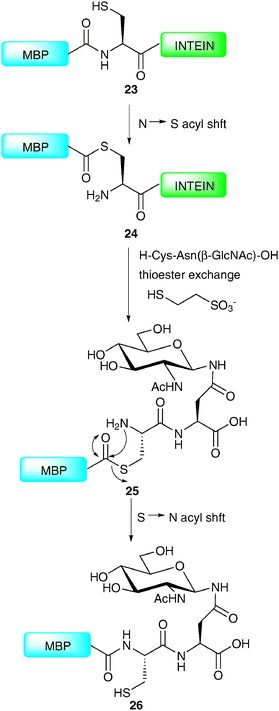
Production of an N-terminal coupling partner for use in NCL requires that the fragment be expressed bearing a C-terminal thioester. This can be achieved by taking advantage of naturally occurring self-splicing elements called inteins, analogous to introns present in nucleic acids.55–57 Inteins have the ability to catalyse their excision from a protein through a series of acyl-transfer reactions in which a cysteine thioester is a key intermediate. By introducing an affinity tag into the intein, this intermediate can be isolated, and upon exposure to an appropriate ligation partner will undergo NCL to afford the desired product. In the context of glycoprotein synthesis, this allows the semi-synthesis of proteins containing glycosylation at their C-terminus and has recently been exploited by a number of groups.62–65 The power of this technology was aptly demonstrated in the synthesis of a homogeneous glycoprotein variant of maltose-binding protein (MBP) by the Wong group (Scheme 9).62 The synthesis initially involved expression of the 392-amino acid MBP in Escherichia coli as a fusion protein to the N-terminus of an intein derived from Saccharomyces cerevisiae possessing a chitin-binding domain at the C-terminus (23). The thioester 24 formed between the N-terminal cysteine residue of the intein and the C-terminus of the MBP was purified on chitin beads before undergoing transthioesterification (thioester exchange) to generate a soluble C-terminal MBP thioester. This was reacted in situ with a glycodipeptide bearing an N-terminal cysteine, e.g. H-Cys-Asn(β-GlcNAc)-OH to afford thioester 25 which underwent an S → N acyl shift to furnish homogeneous MBP 26 bearing C-terminal glycosylation. Enzymatic elaboration of the bridgehead glycan was also demonstrated, thereby providing scope for the synthesis of more complex glycoforms.
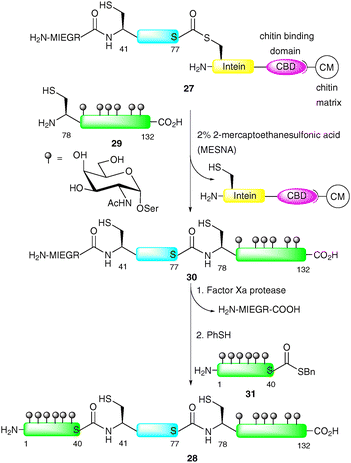
An additional advantage of EPL is that both the N-terminal and C-terminal engineering approaches to afford peptide and protein fragments are orthogonal to one another. This allows for the introduction of glycopeptide fragments at both the C- and N-termini of a suitably masked expressed protein fragment via NCL. Macmillan and Bertozzi were the first to exploit a double ligation approach in the EPL-based synthesis of three distinct glycoforms of GlyCAM-1, a glycoprotein ligand involved in leukocyte homing (Scheme 10).63 GlyCAM-1 consists of two heavily glycosylated mucin domains at the N- and C-termini which are bridged by a central unglycosylated domain. To enable the synthesis of this complex glycoprotein by EPL, three substitution mutations were made to cysteine, namely Lys-41, Gln-78 and Gln-102. The central domain (GlyCAM-1 41–77) was produced recombinantly using the IMPACT system, which relies on a pH-dependent intein cleavage (Scheme 10).66 The fusion construct 27 was designed containing a factor Xa cleavage site which served as a protecting group, masking the N-terminal cysteine (Cys-41) for a subsequent ligation. The most impressive glycoprotein synthesised in this study was 28, containing heavily glycosylated N- and C-termini. Glycopeptide 29 (GlyCAM-1 78–132) was first ligated to fusion construct 27via NCL to afford 30.64 Cys-41 was then liberated by treatment with factor Xa followed by ligation with glycopeptide thioester 31 to afford the full length 132-amino acid GlyCAM-1 (28). Upon completion of the synthesis, the internal cysteine residues were capped with iodoacetamide so as to mimic the glutamine residues found in the native glycoprotein.67 It should be noted that Imperiali and co-workers have also used a fusion protein produced via the IMPACT technology for the semi-synthesis of a glycosylated version of the bacterial immunity protein Im7.65
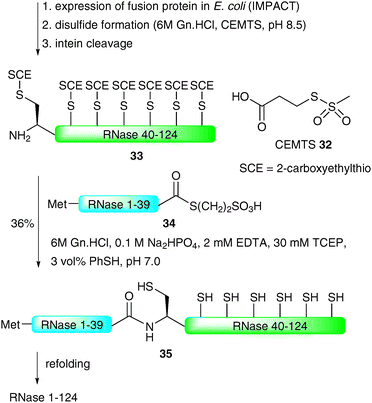
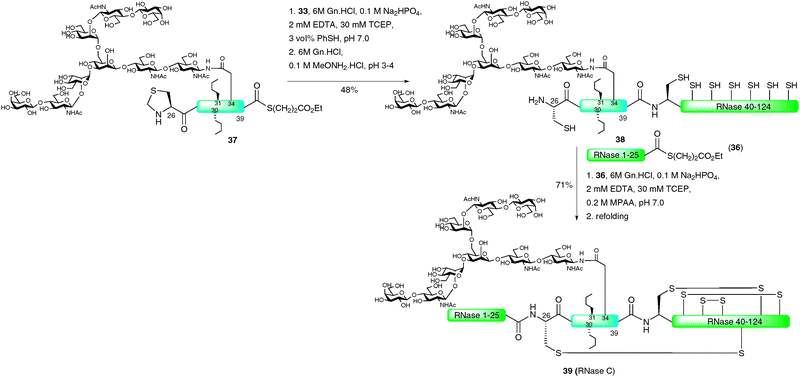
Perhaps the most impressive example of an EPL-based semi-synthesis of a full length homogeneous glycoprotein was the recently disseminated total synthesis of the 124-amino acid enzyme RNase C by the Unverzagt group.68,69 This work significantly built on previous studies by the same group in which a 38-amino acid glycopeptide fragment of RNase B (13) was prepared using a combination of SPPS and NCL assembly strategies (see Scheme 4).45 In this recent study, the group chose to utilise the commercially available IMPACT system to produce a large peptide fragment (RNase 40–124) which could subsequently be used in the NCL-based assembly of a full length protein as a single glycoform. Unfortunately, in preliminary studies, expression of the fusion protein in E. coli led to the formation of inclusion bodies and, as such, the intein did not self-cleave to afford the desired fragment bearing an N-terminal cysteine residue.68 To remedy this problem the authors developed a novel approach to facilitate solubility of the thiol rich fragment. Specifically, carboxyethylmethanethiosulfonate (CEMTS)7032 was used to chemoselectively derivatise the seven cysteine side chains as mixed disulfides under refolding conditions (Scheme 11). The disulfide-protected fragment 33 was ligated with an intein derived peptide thioester, Met-RNase1–39 (34), which, under reductive conditions (TCEP), gave the full length protein 35 in 36% yield after isolation by gel filtration. It should be noted that the ligation reaction was conducted under strictly inert conditions in a nitrogen tent (<10 ppm O2) to prevent reoxidation (which would inevitably lead to insoluble protein). The semi-synthetic protein was subsequently refolded using a glutathione redox couple under rapid dilution which produced stable RNase which exhibited enzymatic (hydrolase) activity.
With the methodology developed and applied successfully for unglycosylated RNase, the stage was set for application to the total synthesis of a single glycoform of RNase C.69 Initial studies aimed at preparing the RNase1–39 glycopeptide thioester using the solid-phase double-linker strategy previously reported by the group45 thus allowing for a two fragment–one ligation synthesis of the glycoprotein. However, the 40-amino acid fragment could not be produced in this manner thereby necessitating a further disconnection at Cys26. This in turn led to three fragments that could be assembled by two sequential NCL reactions (Scheme 12). The N-terminal peptide thioester 36 (RNase 1–25) was assembled on double-linker resin which, after activation and thiolysis under the previously developed conditions, gave the desired fragment in moderate yield (20%). In this case the authors incorporated two Ser-Ser pseudoproline dipeptide units in order to increase the yield of the solid-phase synthesis. The central fragment, a glycopeptide thioester corresponding to RNase 26–39 (37), was synthesised in a similar manner. The complex-type nonasaccharide was obtained from egg yolk and modified into the preformed Fmoc-Asn building block for incorporation into SPPS. After coupling, the free glycan hydroxyls were acetylated to prevent cross reactivity. Efficient coupling of further amino acids was maximised by the use of a Lys-Ser pseudoproline dipeptide and norleucine residues were incorporated in place of Met 30 and 31 to prevent oxidative sulfoxide formation during the synthesis. Additionally, a thiazolidine was incorporated at the N-terminus of the fragment to prevent homocoupling during the ligation reaction. Activation and thiolysis of the fully assembled glycopeptide provided thioester 37 in 18% yield. With the desired synthetic fragments in hand, the group turned to the semi-synthesis of RNase C. This involved reaction of disulfide-protected RNase 40–124 (33) with thioester 37 (RNase 26–39), containing the complex nonasaccharide, under reductive NCL conditions (TCEP) to afford the 98-amino acid RNase fragment bearing an N-terminal thiazolidine. This was subsequently unmasked with methoxyamine at pH 3–4 to generate glycopeptide 38 bearing an N-terminal cysteine residue. Ligation with peptide thioester 36 using mercaptophenyl acetic acid (MPAA) as the activating thiol under strictly anaerobic conditions afforded full length RNase C. This was refolded directly over four days and the desired glycoprotein 39 was isolated in high yield by gel filtration (71%). The synthetic enzyme displayed a similar circular dichroism spectrum to that of the native folded protein and was shown to be hydrolytically active, exhibiting 50% of the activity of RNase. Although the synthetic variant contains two methionine–norleucine mutations, this ligation-based assembly of a full length homogeneous glycoform possessing a complex glycan represents a landmark study in the field of glycoprotein synthesis. It is also possible that such a strategy will allow for more efficient routes to a number of other homogeneous glycoproteins by synthetic means in coming years.
4. Thiol auxiliary ligation
By virtue of their fast and chemoselective nature, NCL and the corresponding semi-synthetic variant (EPL) currently represent the most efficient means of constructing glycopeptides and glycoproteins by chemical synthesis. This is evident from recent application in the synthesis of complex full length homogeneous glycoproteins, especially those recently reported by the groups of Kajihara47 and Unverzagt.69 However, it should be noted that the methods suffer from one important limitation, namely the necessity for an N-terminal cysteine residue on a peptide or glycopeptide fragment. Although intimately involved in the overall structure (and function) of proteins via the formation of disulfide bonds, cysteine residues are relatively rare in protein sequences (occurrence ca. 1.7%). As such, it can often be difficult to locate a feasible ligation junction containing a native cysteine residue. Initially, this was remedied by the artificial introduction of cysteine residues to facilitate a synthesis (e.g. diptericin ε). However, a number of alternative strategies have emerged which allow for disconnections at other amino acids, thereby avoiding the introduction of a mutation. These represent complementary tools to the cysteine-based methods for the ligation-based assembly of glycopeptides and glycoproteins.Initial studies directed at overcoming the requirement of an N-terminal cysteine in NCL led to the development of thiol-based auxiliaries. These included 1-phenyl-2-mercaptoethyl auxiliaries 40 and 4171 and the 4,5,6-trimethoxy-2-mercaptobenzyl (Tmb) auxiliary 42.72 These can be attached to the N-terminus of a synthetic (glyco)peptide and are readily removed following ligation by treatment with trifluoroacetic acid (TFA) (Scheme 13). It is believed that the thiol of the auxiliary facilitates chemical ligation in an analogous manner to the side chain of a cysteine residue, as depicted for the Tmb auxiliary in Scheme 13. The utility of these auxiliaries with respect to glycopeptide synthesis was first demonstrated by Macmillan and Anderson who used both auxiliaries to synthesise fragments of the O-linked glycoprotein GlyCAM-1.73
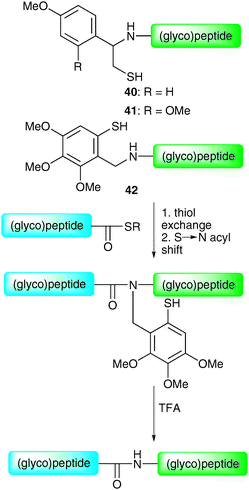 | ||
| Scheme 13 Structures and proposed mechanism of the 1-phenyl-2-mercaptoethyl auxiliaries 40 and 41 and 4,5,6-trimethoxy-2-mercaptobenzyl (Tmb) auxiliary 42.71,72 | ||
Several elaborate examples of such auxiliary-based ligations have since been reported by Danishefsky and co-workers in the construction of complex glycopeptides bearing multiple glycans.74,75 In one pertinent example the authors describe the thiol auxiliary-based ligation between glycopeptide 43 bearing a variation of the Tmb auxiliary and a masked glycopeptide thioester 44, both presenting an N-linked chitobiose glycan (Scheme 14). The masked thioester 44 was equipped with a C-terminal phenolic ester (described previously by the Danishefsky group, vide supra), which was converted to the desired thioester via an O → S acyl migration under the reducing ligation conditions. The resulting thioester underwent thioesterification with the auxiliary-containing glycopeptide 43 (after in situ reduction of the disulfide bond) followed by an S → N acyl shift to afford the desired glycopeptide 45 bearing two glycans. This example is particularly impressive given the reported difficulty of auxiliary-mediated ligations for hindered amino acids at the ligation junction.73 Following ligation, the N-terminal thiazolidine was unmasked to provide an N-terminal cysteine residue which was ligated with another complex glycopeptide thioester via a standard NCL reaction.
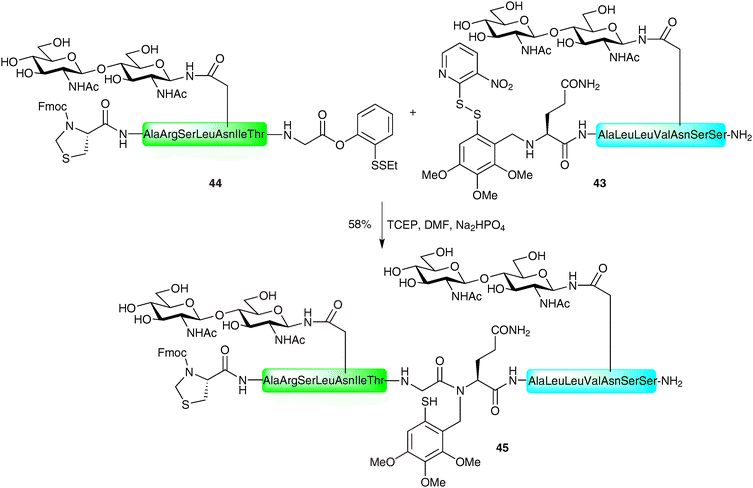 | ||
| Scheme 14 Synthesis of N-linked glycopeptide 45 using a variant of the Tmb auxiliary.74 | ||
5. Sugar-assisted ligation
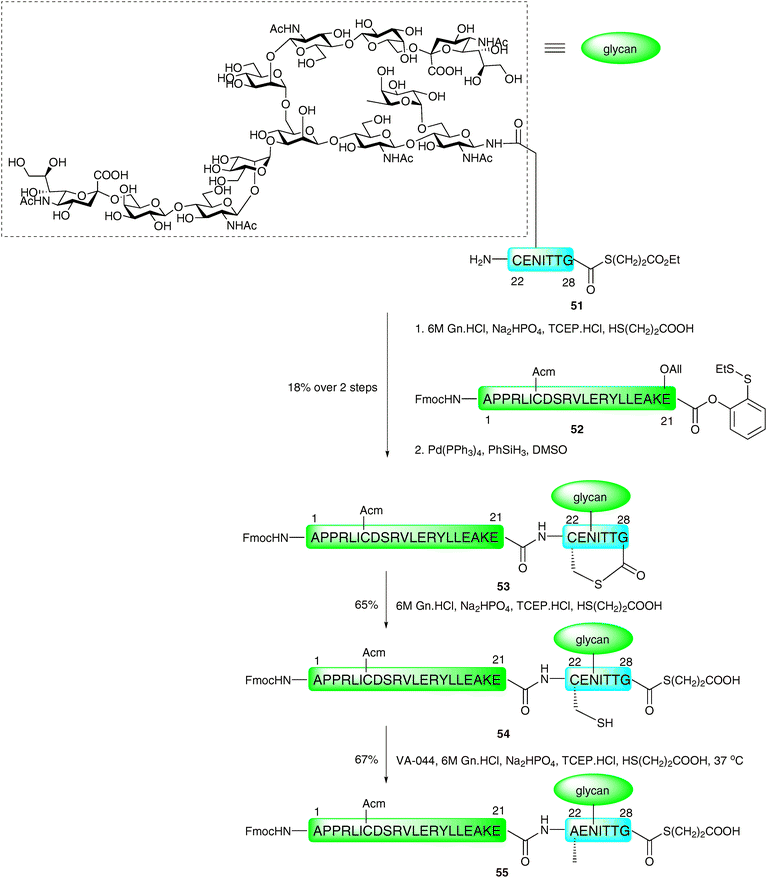
The Wong group recently reported a further modification to the thiol auxiliary approach, designed to accomplish convergent assembly of glycopeptide fragments at non-cysteinyl ligation sites.76,77 The method, referred to as sugar-assisted ligation (SAL), utilises a glycopeptide in which the carbohydrate (rather than the peptide) is derivatised with a thiol auxiliary at C-276,78 (Scheme 15A) or C-379 (Scheme 15B) to facilitate amide bond formation with a peptide thioester. The reactions were initially conducted in an aqueous buffer, similar to that employed in NCL, however, reactions were also shown to proceed smoothly in a mixed solvent system which served to protect the reactive thioester moiety from hydrolysis, thus providing improved ligation yields. The method has been shown to work well for both O- and N-linked glycopeptides and appears to tolerate a variety of amino acids at the ligation junction.76,78–80 Upon completion of ligation reactions, the thiol auxiliary at C-2 can be efficiently cleaved under reducing conditions developed by Yan and Dawson (H2/Pd on alumina or RANEY® nickel)81 to yield the native acetamide functionality (Scheme 15A).76,78,80 The C-3 ester linked auxiliary can be removed by hydrazinolysis (Scheme 15B) to furnish glycopeptides bearing native glycans in high yields.79 The proposed mechanism of SAL, depicted in Scheme 16 for a C-2 derivatised glycan, involves an initial transthioesterification step, followed by an S → N acyl shift, to afford the native peptide bond in a similar manner to that described for NCL21 and other thiol auxiliary-based approaches.71,72 The key difference is the much larger ring transition state that would be generated in the S → N acyl transfer. For this reason SAL reactions tend to be significantly slower than NCL reactions. Detailed kinetic studies have been conducted to gauge the effect of increasing the number of amino acids adjacent to the glycosylation site on the glycopeptide fragment.79,80 These showed a significant decrease in reaction rate as the proposed transition state became larger. Moreover, ligation reactions with glycopeptides containing more than three amino acids between the N-terminus and the glycosylation site had similar rates suggesting that these were no longer proceeding via an intramolecular process. Recently, SAL was successfully employed on peptides bearing more complex glycans. Specifically, both O- and N-linked glycopeptides bearing thiol auxiliary-derived mono-, di- and trisaccharides were shown to undergo SAL reactions to afford ligated products in moderate to high yields.82 Interestingly, ligations proved unsuccessful when the bridgehead sugar contained an additional glycan at the C-3 position, attributed to an increase in steric bulk at the ligation junction. The utility of SAL for the construction of more complex glycopeptides and glycoproteins was aptly demonstrated in the total synthesis of a homogeneous glycoform of diptericin ε.83 This example has set the scene for the use of SAL in the total synthesis of more complex glycopeptides and glycoproteins. Indeed, it is anticipated that SAL will serve as a useful tool either alone or in combination with other peptide ligation methods in future synthetic endeavours.77,84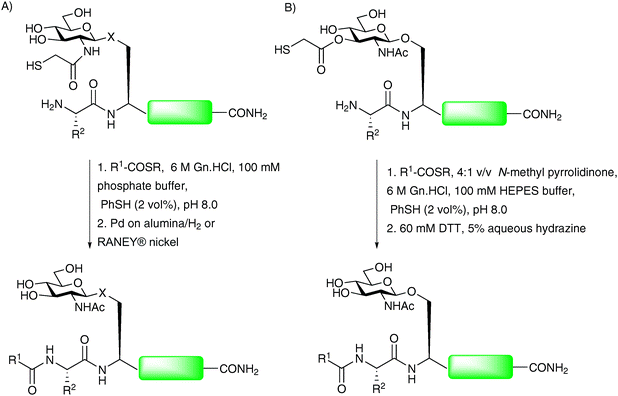 | ||
| Scheme 15 (A) Synthesis of O- and N-linked glycopeptides by sugar-assisted ligation (SAL), X = –O– or –NH(CO)–; (B) second generation SAL.76,79 | ||
 | ||
| Scheme 16 Proposed mechanism of sugar-assisted ligation (SAL).76 | ||
6. Native chemical ligation followed by desulfurisation to alanine
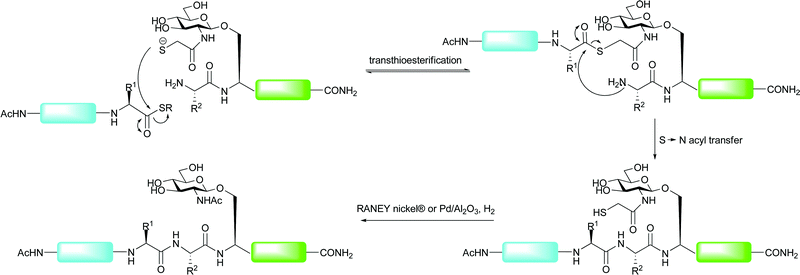
An alternative approach to expand the application of ligation methods to non-cysteinyl ligation junctions was introduced by Yan and Dawson who utilised catalytic desulfurisation methods to convert cysteine to alanine after an NCL reaction, thus allowing access to Xaa-Ala ligation sites.81 Alanine is far more ubiquitous in protein sequences when compared with cysteine and, as such, many sequences should possess a ligation junction amenable to the application of this method. Initial strategies using RANEY® nickel or Pd/Al2O3 under a H2 atmosphere enabled global desulfurisation of polypeptides containing multiple cysteine residues.81 Subsequently, a number of groups have developed desulfurisation strategies that allow for structurally important cysteine residues to remain intact upon desulfurisation. The first of these was reported by Pentelute and Kent who used RANEY® nickel to selectively desulfurise free cysteine residues to the corresponding alanine in the presence of those side chain protected with acetamidomethyl (Acm) groups.85 The Acm groups could then be removed using silver acetate. Although used in the total synthesis of a number of proteins to date, one reported drawback of the metal-based desulfurisation strategies involved the adsorption of certain peptide sequences to the metal surfaces, leading to low recovery rates.86 Additionally, such conditions have been shown to cause the reaction of methionine residues (reduced to α-aminobutyric acid) and thiazolidines, commonly used to mask the reactivity of N-terminal cysteine residues during tandem ligation reactions (vide supra).87 A recent metal-free desulfurisation strategy reported by Wan and Danishefsky aimed to remedy these problems, thus enabling the high yielding construction of complex glycopeptides.87 The method, based on an early report by Hoffmann et al.,88 utilises a trialkylphosphine in the presence of a radical initiator to induce a radical-catalysed desulfurisation. The authors chose tris(2-carboxyethyl)phosphine (TCEP) due to its tolerance to proteinogenic side chains and its ease of handling. Combined with the radical initiator 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044, 46) and t-BuSH in water or buffer, TCEP was able to desulfurise the side chain of cysteine to afford alanine in high yields for a variety of peptide model systems. Notably, the method was orthogonal to methionine residues and thiazolidine residues. This radical-based desulfurisation protocol was subsequently applied to the synthesis of an N-linked glycopeptide. The authors used a kinetically controlled NCL reaction, introduced by Kent and co-workers,89–91 to ligate glycopeptide 47 bearing a C-terminal phenolic ester with peptide 48 containing a C-terminal ethyl thioester which was left untouched under the ligation conditions (Scheme 17). This provided the 19-amino acid glycopeptide 49 containing an N-linked chitobiose core in 67% yield. A subsequent TCEP and VA-044 mediated desulfurisation of 49 in the presence of excess hydride donating thiols proceeded smoothly to afford the desulfurised glycopeptide 50 in 87% yield. This example was all the more impressive given the numerous sulfur containing residues, including methionine, an Acm-protected cysteine, a thiazolidine and a C-terminal thioester, that remained untouched over the course of the reaction.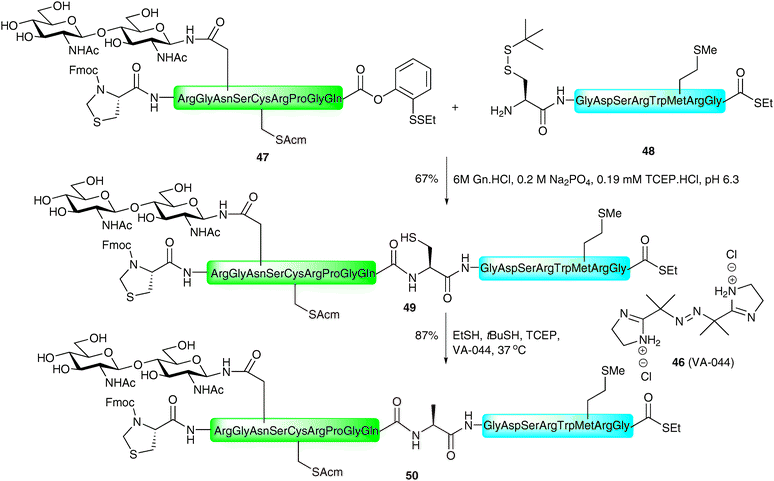 | ||
| Scheme 17 Synthesis of model glycopeptide 49 by NCL followed by metal-free desulfurisation of cysteine to alanine.87 | ||
The same authors recently reported the application of the NCL metal-free desulfurisation protocol in the synthesis of a homogeneous glycopeptide fragment of erythropoietin (EPO),92 a glycoprotein hormone produced naturally in the kidney cells and used clinically for the treatment of anaemia associated with a number of diseases.93–95 The strategy involved the synthesis of a 7-amino acid glycopeptide thioester 51via selective protecting group manipulation and introduction of the complex-type sialylated glycan via the Lansbury aspartylation (Scheme 18).43,44 This was ligated, under the standard NCL conditions, with peptide 52 corresponding to EPO1–21, containing an Acm-protected cysteine and armed with a C-terminal o-disulfide phenolic ester. Additionally, the fragment possessed an allyl-protected glutamic acid to prevent unwanted side reactions. Unfortunately, the ligation only proceeded in low yield and generated the cyclic lactone 53 after allyl ester deprotection by palladium-catalysed chemistry. The thiolactone was subsequently opened by treatment with thiopropionic acid to afford glycopeptide thioester 54 presenting a free thiol side chain which could now be desulfurised. Reaction of 54 in a reducing buffer in the presence of thiopropionic acid as a radical propagator and VA-044 gave EPO1–28 (55) in 67% yield. At this point glycopeptide 55 still contained an Acm-protected cysteine residue and a C-terminal thioester, which is amenable to further ligations, envisioned for the total synthesis of a homogeneous glycoform of EPO (see Schemes 24 and 25). Given the efficiency and chemoselectivity of these ligation–desulfurisation strategies for the generation of glycopeptides, it is anticipated that this method, and variations thereof, will continue to find wide use in the synthesis of complex glycopeptides and glycoproteins.
 | ||
| Scheme 18 Synthesis of a complex N-linked glycopeptide fragment of erythropoietin (EPO1–28) by NCL followed by metal-free desulfurisation.92 | ||
7. Native chemical ligation–desulfurisation to other proteinogenic amino acids
Recently the repertoire of NCL has been further expanded by the application of the ligation–desulfurisation strategy to other suitably thiolated amino acid side chains, a concept realised early by Yan and Dawson.81 Specifically, β- and γ-thiolated amino acids have been used to facilitate ligation reactions via an NCL-based pathway and, following desulfurisation, furnish the native amino acid side chain.7.1 Native chemical ligation at phenylalanine
The first method to be reported provides synthetic disconnections at Xaa-Phe ligation sites.96,97 Introduction of β-mercaptophenylalanine to the N-terminus of a peptide allowed for a ligation to proceed with a C-terminal peptide thioester under the standard NCL conditions (Scheme 19A). After the ligation, desulfurisation with nickel boride delivered the native phenylalanine containing peptides in moderate yields. Although the scope of the method has not yet been explored in the context of glycopeptide and glycoprotein synthesis, it is anticipated that the method will be a useful tool in this regard.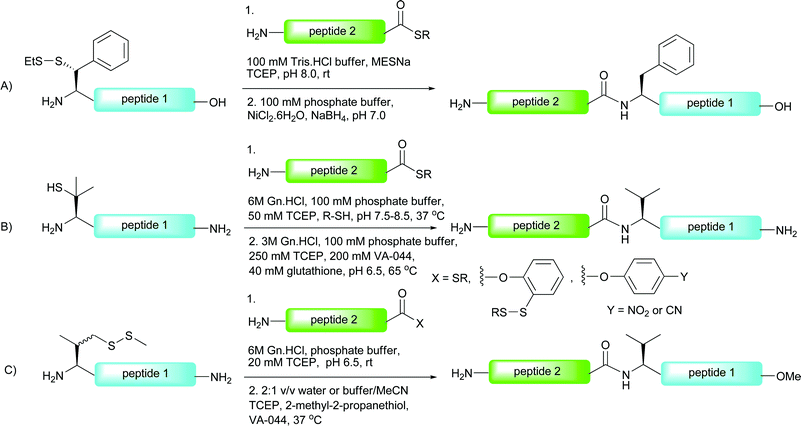 | ||
| Scheme 19 NCL-desulfurisation at (A) phenylalanine96 and (B) and (C) valine.86,98 | ||
7.2 Native chemical ligation at valine
One drawback of the phenylalanine-based NCL is the multistep synthesis necessary for the preparation of the suitably protected β-mercaptophenylalanine building block.96 This problem is alleviated in the valine variant of the NCL-desulfurisation approach reported by Seitz and co-workers.86 Here the group made use of β,β-dimethylcysteine (penicillamine), a commercially available amino acid which could be protected in one step (Boc or Trt) and incorporated into the N-terminus of a peptide using Fmoc-strategy SPPS. Under the typical aqueous conditions employed in NCL, the β-thiolate facilitated ligation reactions with peptide thioesters in high yields in most cases (Scheme 19B). The reaction proceeded smoothly even in the presence of peptide thioesters bearing sterically hindered amino acids, e.g. leucine, perhaps surprising given the expected reduction in reactivity with the sterically encumbered tertiary thiolate. Desulfurisation of the resulting ligation products was initially attempted using a large excess of RANEY® nickel in acetic acid. Unfortunately, isolated yields of the corresponding peptides were moderate in most cases and in longer peptides the product could not be removed from the metal surface. In contrast, application of a modified version of the metal-free desulfurisation method,87 introducing glutathione as a more powerful hydrogen source, facilitated clean desulfurisation reactions (Scheme 19B).In order to increase the reactivity of the β-mercaptovaline moiety in the above ligation, Danishefsky and co-workers reported the preparation of a valine building block derivatised with a more reactive primary thiol at the γ-position.98 The γ-thiol valine was generated as a mixture of diastereoisomers, however, since this stereocentre is abolished upon desulfurisation to valine it did not pose a problem in isolating peptides retaining stereochemical integrity. The authors reported significant rate enhancements with peptides bearing N-terminal γ-thiolated valine residue when compared directly against the penicillamine (β-thiol) derived peptides. It is important to note that the γ-thiol variant would proceed through a six-membered ring transition state in the S → N acyl transfer compared with a five-membered ring in the peptides bearing an N-terminal penicillamine. This suggests that the reactivity/accessibility of the thiol is an important factor for the rate of these reactions and suggests that the initial thioesterification step is rate determining. C-terminal peptide thioesters, o-thiophenolic esters, p-nitrophenyl esters and p-cyanophenyl esters were successfully employed as acyl donors in this reaction to furnish ligation products in high yields (Scheme 19C). In the context of glycopeptides, the method was implemented in the ligation of a peptide bearing an N-terminal γ-mercaptovaline (56) to a C-terminal peptide o-thiophenolic ester bearing an N-linked disaccharide (57) to generate glycopeptide 58 in 90% yield after only 30 minutes reaction time at ambient temperature (Scheme 20). Desulfurisation using the metal-free method (TCEP and VA-044)87 proceeded smoothly to afford the native glycopeptide 59 possessing a native internal valine residue in 89% yield.
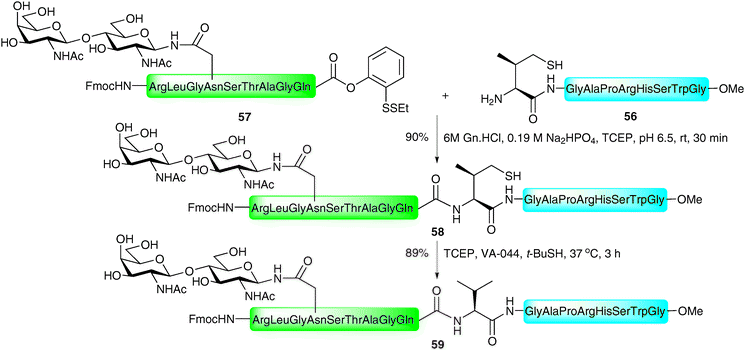 | ||
| Scheme 20 Synthesis of a model N-linked glycopeptide by NCL followed by desulfurisation at valine.98 | ||
7.3 Access to other proteinogenic amino acids at the ligation site
More recently access to Xaa-Ser ligation junctions has been realised.99 Rather than using a ligation–desulfurisation sequence, the method relies on the conversion of a cysteine residue to a native serine side chain after a traditional NCL reaction has taken place. The strategy involves an S-selective methylation of the cysteine side chain with methyl 4-nitrobenzenesulfonate followed by activation with CNBr (Scheme 21A). This activation, carried out in the presence of 80% aqueous formic acid, was proposed to induce an intramolecular attack by a neighbouring β-carbonyl oxygen thus generating an O-ester intermediate which can undergo an O → N acyl shift under slightly basic conditions to complete the conversion of cysteine to serine. Since CNBr can also react with methionine residues in a similar manner, the authors chose to oxidise the methionine side chain to the unreactive sulfoxide form.100 Reduction of the sulfoxide using NH4I, SMe2 and TFA was performed after the cysteine to serine conversion to regenerate the native methionine side chain.101 The cysteine to serine conversion was attempted on glycopeptide 60 bearing a complex-type N-linked asialooligosaccharide102 which was initially prepared by a standard NCL reaction (Scheme 21B).21 Selective methylation provided 61 which was subsequently submitted to the activation–rearrangement condition. However, during purification the authors observed broad peaks which were attributed to formylation of the free alcohols of the glycan during the CNBr–formic acid activation step. Nonetheless, deformylation could be achieved by hydrazinolysis to give the desired target 62 in good yield. The NCL-derivatisation strategy has also been successfully implemented in the synthesis of O-linked glycopeptides, namely MUC1 glycopeptides incorporating the TN antigen99 and more recently the sialyl TN antigen.103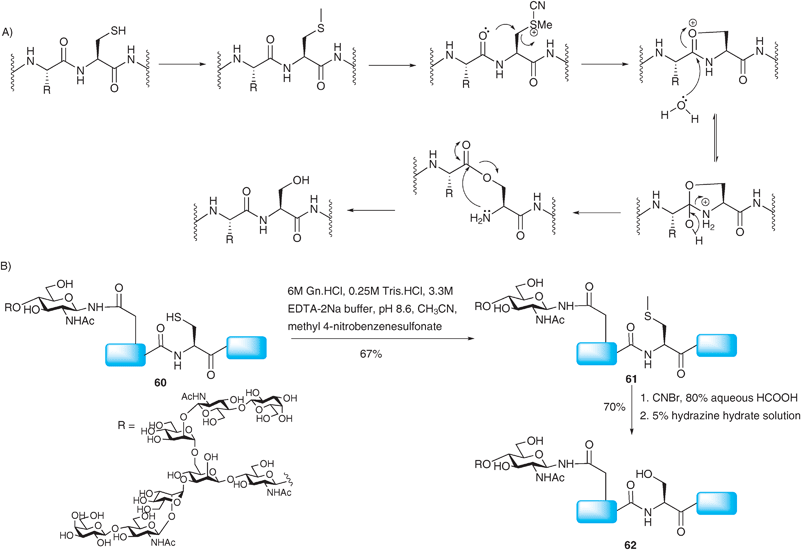 | ||
| Scheme 21 (A) Proposed mechanism for the conversion of cysteine to serine. (B) Synthesis of a model glycopeptide 62 bearing a complex N-linked glycan via NCL followed by conversion of the ligation site cysteine to serine.99 | ||
8. Thiol-free ligations
Recently, a number of cysteine and thiol-free ligation methods have been developed, aimed at facilitating the synthesis of glycopeptides and glycoproteins that could not otherwise be assembled by NCL or thiol auxiliary-based strategies. These methods all rely on the direct coupling of a peptide or glycopeptide with a peptide or glycopeptide bearing an activated C-terminus. The roots of these strategies date back to the 1970s when Kemp and co-workers used C-terminal p-nitrophenyl and N-ethylsalicylamide esters to couple two small peptide fragments in the absence of an N-terminal cysteine residue, thiol auxiliary or exogeneous coupling reagent.104,105 Reactions were conducted in dimethylformamide (DMF) or dimethylsulfoxide (DMSO) and gave ligation products in high yields. The authors reported potential problems with peptide solubility and racemisation under these conditions and, as such, it was not explored further.8.1 Thioester method
In 1981, Blake reported an improved ligation strategy which relied on the silver-ion mediated coupling of peptides with peptide thioacids.106 This procedure was further modified by Aimoto and co-workers who exploited the reactivity of peptide thioesters in combination with silver(I) and a suitable activating agent, such as 1-hydroxy-1H-benzotriazole (HOBt) or 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine (HOOBt) to facilitate ligations with the N-terminal amine of a peptide acceptor.107,108 Recently, this strategy, dubbed the “thioester method”, was exploited by Hojo and co-workers in the impressive synthesis of a 23 kDa glycoprotein representing the basal structure of MUC2, a mucin-type glycoprotein bearing multiple copies of the TN antigen.109 The synthesis involved initial preparation of a 23-amino acid glycopeptide thioester 63 bearing seven copies of the TN antigen (Scheme 22). This was constructed using a modified Fmoc protocol whereby the glycans were introduced as benzyl-protected glycosylthreonine cassettes.110 Once assembled the glycopeptide thioester was cleaved from the resin using a TFA-based cocktail and further treated with “low TfOH” to debenzylate the glycans.110–113 Glycopeptide thioester 63 was initially ligated to tripeptide 64 (TQT–NH2) using the thioester method (HOOBt, DIEA and AgCl) before treatment with piperidine to remove the N-terminal Fmoc-carbamate providing 65. This was further elaborated by five iterative ligation and deprotection cycles with 63 under the same silver-mediated coupling conditions to afford glycoprotein 66 bearing six copies of the basal MUC2 repeat. The resulting 23 kDa structure represents one of the largest glycoproteins to be synthesised by total chemical synthesis, providing encouragement for future synthetic endeavours using this ligation technology.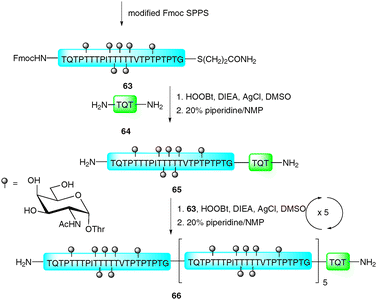 | ||
| Scheme 22 Synthesis of 23 kDa MUC2 tandem repeat glycoprotein via the silver-promoted “thioester method.”109 | ||
8.2 Phenolic ester directed amide coupling (PEDAC)
Danishefsky and co-workers have reported an extension of the “thioester method”, using the C-terminal o-disulfide phenolic ester previously developed by the group.114 Peptides bearing this entity on their C-terminus were shown to react in a facile manner with peptides containing a free N-terminal amine using the standard Aimoto conditions (AgCl, HOOBt, DIEA)108 a reaction which was termed the AgCl-assisted phenolic ester directed amide coupling (PEDAC AgCl). The efficiency of this reaction was somewhat surprising given the usual necessity for a reducing reagent to generate the reactive C-terminal thioester in situ. The authors suggest that AgCl may promote disulfide cleavage as the rate determining step of the reaction. Based on these findings, alternative conditions utilising the reducing agent tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) in place of silver(I) ions (PEDAC TCEP) also facilitated ligation reactions with C-terminal Gly- and Pro-containing peptide phenolic esters in high yields. However, both metal (AgCl) and metal-free (TCEP) methods caused epimerisation of C-terminal residues other than Gly and Pro, which may limit its use in some syntheses. Additionally, the side chains of lysine and cysteine required protection with ivDde and Acm groups, respectively, to prevent unwanted side reactions. Despite these drawbacks, the two methods do expand the number of ligation sites that can be accessed by ligation chemistry and have been used for the ligation-based assembly of several complex glycopeptides.114 A particularly impressive example is the use of the two methods in concert in the synthesis of glycopeptide 67 bearing an N-linked glycan (Scheme 23). One notable feature of this work is the fact that the target glycopeptide was prepared through unconventional N → C condensation reactions. The PEDAC TCEP method was first used to ligate glycopeptide 68 containing a C-terminal phenolic ester to peptide 69 bearing a C-terminal alkyl thioester. Under these conditions the desired glycopeptide was synthesised in 80% yield, with the alkyl thioester of 69 remaining untouched due to its low reactivity under these conditions. Peptide 70 was next coupled to the glycopeptide thioester which could now be activated using the AgCl-promoted methodology to afford the 36-amino acid glycopeptide 67 in 65% yield.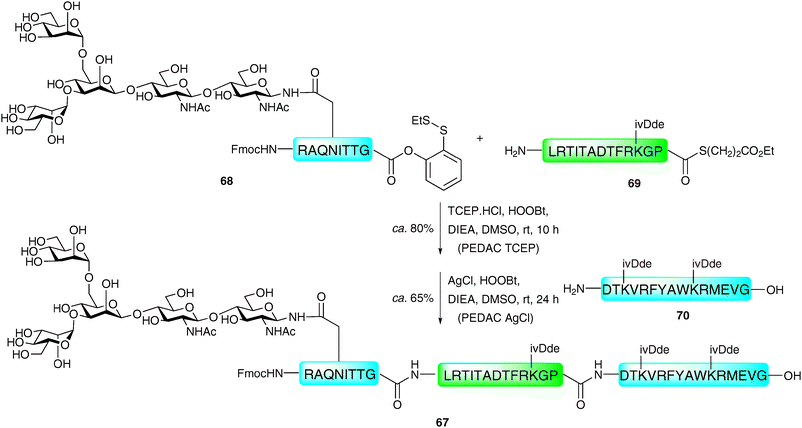 | ||
| Scheme 23 Synthesis of a model N-linked glycopeptide via sequential phenolic ester directed amide coupling reactions (PEDAC TCEP followed by PEDAC AgCl).114 | ||
The method described above was recently employed in impressive fashion for the synthesis of two complex homogeneous glycopeptide domains of EPO (Cys29–Gly77115 and Gln78–Arg166116). These were constructed bearing suitable protection and functionality with a view to future implementation in the ligation-based total synthesis of a fully glycosylated homogeneous version of the 166-amino acid glycoprotein. The synthesis of the first of these fragments (Cys29–Gly77)115 was envisaged using a three fragment, two ligation assembly in the N → C direction as described for the model glycopeptide 67 (see Scheme 23).114 The key difference was the requirement for a C-terminal thioester to remain on Gly77 to enable a subsequent ligation with the Gln78–Arg166 fragment in future synthetic endeavours. The group therefore decided to utilise three different acyl donors with tuned reactivities to achieve this unprecedented and formidable task. A model peptide, corresponding to Cys29–Pro42 bearing a C-terminal p-cyanophenolic ester,98 was first ligated to a peptide containing a C-terminal phenolic ester with an o-disulfide moiety (corresponding to Asp43–Gly57). These were reacted under the PEDAC conditions, with the omission of AgCl and TCEP to prevent reduction of the disulfide. Unfortunately, although these two fragments could be ligated, this occurred with complete hydrolysis of the masked thioester at Gly57. This hydrolysis was attributed to the steric accessibility of the glycine ester and, as such, the authors introduced an additional ortho-substituent to block both π-faces of the Gly57 carbonyl group. Under the same conditions, glycopepide 71 containing a C-terminal p-cyanophenolic ester and a complex N-linked dodecasaccharide was ligated to peptide 72, bearing the modified C-terminal phenolic ester (Scheme 24). This provided the desired glycopeptide 73 in 47% yield, and proceeded without any detectable loss of the C-terminal phenolic ester. This was then subjected to a subsequent ligation with peptide 74, corresponding to Gln58–Gly77, which contained a C-terminal glycine alkyl thioester. On this occasion, the PEDAC TCEP conditions were used to facilitate disulfide reduction and formation of the active acyl donor in situ. This provided glycopeptide 75 containing suitably protected amino acid side chains [Glu(OAllyl), Cys(Acm), Lys(ivDde)], an N-terminal thiazolidine and a C-terminal alkyl thioester in 51% yield.
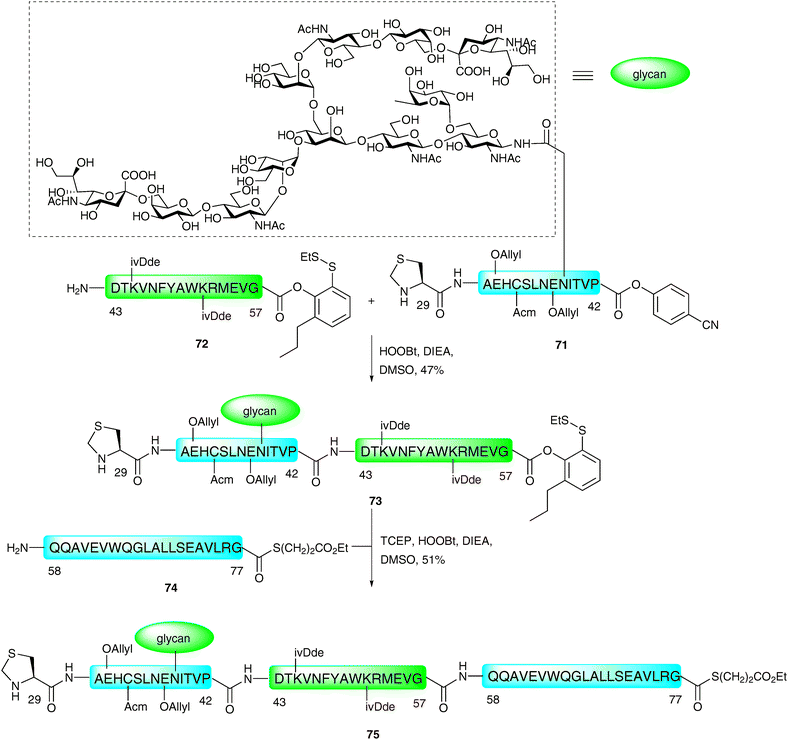 | ||
| Scheme 24 Synthesis of EPO (29–77) N-linked glycopeptide fragment by two sequential PEDAC ligations.115 | ||
The C-terminal fragment of EPO (Gln78–Arg166) was constructed by three consecutive PEDAC TCEP ligation reactions.116 Significant solubility problems were initially encountered en route to this rather hydrophobic fragment. To overcome these difficulties, secondary amino acid surrogates,117 specifically pseudoproline and Dmb-protected dipeptides, were incorporated to disrupt any secondary structures that may form during peptide assembly. The synthesis of the 88-amino acid fragment was envisaged via the assembly of two short glycopeptide fragments EPO (78–87) 76 and EPO (123–129) 77, and two long peptide fragments EPO (88–122) 78 and EPO (130–166) 79 (Scheme 25). A number of protecting group strategies were investigated, the most appropriate being the side chain protection of lysine and cysteine side chains with Alloc and Acm groups, respectively. Additionally, the N-termini of 76–78 were Fmoc protected and Asp123 was protected with a fluorenylmethyl ester (Fm) until after the first ligation reaction. In contrast to the EPO (29–77) fragment (Scheme 24), EPO (78–166) was synthesised in the traditional C → N direction. The first ligation was between glycopeptide 77, bearing a C-terminal o-disulfide phenolic ester and an unprotected glycophorin-type O-glycan (EPO123–129), and peptide 79 corresponding to EPO (130–166). This was conducted under the PEDAC TCEP conditions and, upon completion, the N-terminal Fmoc and side chain Fm ester on Asp123 were removed by treatment with piperidine in DMSO to afford the desired glycopeptide in 59% yield over the two steps. This was subsequently ligated to 34-amino acid peptide 78 which also contained a C-terminal proline o-disulfide phenolic ester and an N-terminal Fmoc group. After completion of the ligation reaction, Fmoc deprotection furnished the desired glycopeptide corresponding to EPO (88–166) in 52% yield over the two steps. The final PEDAC TCEP ligation with glycopeptide 76 containing a complex N-linked dodecasaccharide and a C-terminal proline o-disulfide phenolic ester, followed by Fmoc deprotection, furnished EPO (78–166) (80) in 50% yield. Although the Alloc and Acm side chain protecting groups could be removed at this stage (via Pd(0) and Hg(II) or Ag(I),91 respectively), these were intentionally left intact to allow for the ligation-based total synthesis of a full length homogeneous glycoform of EPO in future work. Mass spectral analysis also revealed the presence of lactones between the carboxylate of sialic acid and the 4-position of neigbouring galactose units (not shown in Scheme 25), however, these should be amenable to hydrolysis upon completion of the synthesis. The convergent assembly of the enormously complex glycopeptide fragments necessary for the total synthesis of EPO clearly demonstrates the power of the PEDAC strategy, which will undoubtedly feature in future glycoprotein syntheses where NCL cannot be employed.
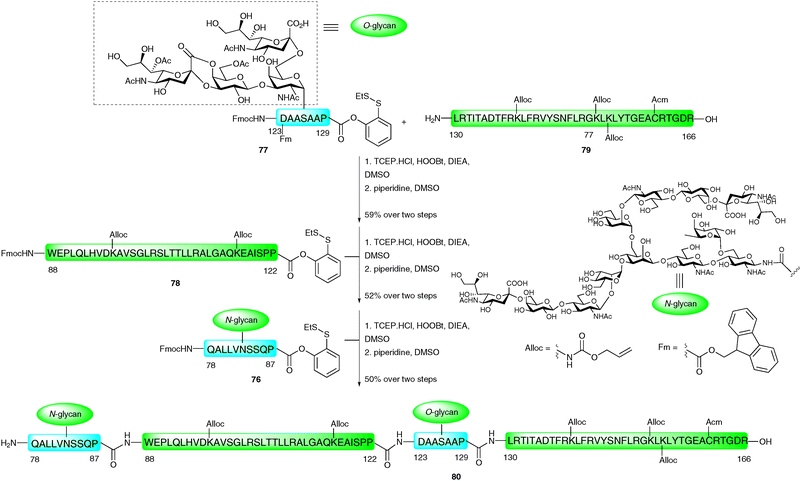 | ||
| Scheme 25 Synthesis of EPO (78–166) glycopeptide fragment containing both N- and O-linked glycans by three sequential PEDAC ligations.116 | ||
8.3 Direct aminolysis ligation
One notable drawback of the “thioester method” and the related PEDAC is epimerisation of the C-terminal residue of the acyl donating peptide fragment under the coupling conditions. As such, it is important that only non-epimerisable amino acids, namely proline and glycine, are incorporated at this position. This problem is circumvented in the direct aminolysis ligation reaction reported by Wong and co-workers.118 This method utilises a mixed solvent buffer at slightly basic pH to facilitate ligations between (glyco)peptides and (glyco)peptide thioesters without the use of the activating reagents or metal ions that are required for both the “thioester method” and PEDAC-based strategies. Importantly, the method was successfully applied to the ligation of thioesters bearing a range of C-terminal amino acid residues without any detectable epimerisation. The ligation reactions were also shown to tolerate unprotected cysteine residues in a number of model peptides, however, these reactions proceeded with varied efficiency depending on the position of the cysteine relative to the N-terminus of the peptide fragment, an observation which has been reported91 and recently investigated for NCL.119 Given the reaction mechanism (intermolecular aminolysis) it is unsurprising that lysine side chains had to be protected. A native 60 amino acid section of the cancer-associated MUC1 tandem repeat glycoprotein was subsequently synthesised using the direct aminolysis ligation method (Scheme 26). This involved initial preparation of the 20-mer tandem repeat glycopeptide thioester 81 containing a C-terminal histidine thioester and two copies of the TN antigen which was achieved by Fmoc-strategy SPPS on sulfamylbutyryl resin using the activation and thiol-release strategy described earlier.28 Trifluoroacetamide-protected glycine was incorporated as the N-terminal residue in order to prevent unwanted side reactions and to allow for subsequent ligations. Treatment of glycopeptide thioester 81 with dilute hydroxide to hydrolyse the C-terminal thioester followed by hydrazinolysis of the N-terminal trifluoroacetamide and glycan acetates furnished glycopeptide 82 in 92% yield. This was reacted with glycopeptide thioester 81 under the mixed solvent buffer conditions to afford the 40-amino acid glycopeptide, which was deprotected with hydrazine to afford 83 in 77% yield over the two steps. This was further reacted with another copy of 81 under the direct aminolysis conditions which, following hydrazinolysis, gave the 6.9 kDa hexaglycopeptide 84, containing three copies of the MUC1 repeat sequence, in 80% yield.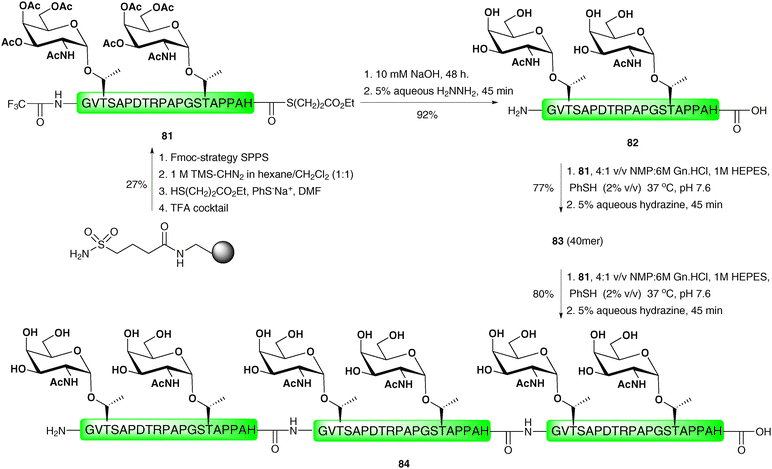 | ||
| Scheme 26 Synthesis of a 60 residue MUC1 glycopeptide via two sequential direct aminolysis ligation reactions.118 | ||
9. Conclusions
The past decade has witnessed a significant expansion in the number of ligation methods available for the convergent synthesis of complex glycopeptides and glycoproteins. The longest standing method, native chemical ligation (NCL), has proven to be extremely effective for the generation of these biomolecules and has been exploited in the total synthesis of a myriad of glycopeptides and glycoproteins. In addition to its chemoselectivity, the other desirable feature of NCL is that the requisite fragments can be expressed in bacteria, thereby making it amenable to the semi-synthetic generation of glycoproteins via expressed protein ligation (EPL). The development of thiol auxiliary-based methods and those which utilise thiolated amino acids followed by desulfurisation protocols has further expanded the number of ligation junctions that can be accessed. Furthermore, a number of thiol-free ligation methods have been developed and exploited for the synthesis of a number of complex targets. The impressive synthetic feats achieved with these latter methods highlight the promise of such strategies as tools for the convergent assembly of glycopeptides and glycoproteins. Moreover, they expand the repertoire of ligation methodology by increasing the number of ligation junctions that can be accessed by synthetic means.With the advent of this suite of ligation methods it should now be possible to disconnect the primary sequence of a glycoprotein at almost every ligation junction. For this reason it is anticipated that these methods, either alone or in combination, should aid in the rapid and efficient synthesis of an increased number of homogeneous glycopeptides and glycoproteins. Undoubtedly, success in this area would provide unparalleled opportunities for the study of these biomolecules at the molecular level, shedding light on the structural and functional implications of protein glycosylation.
Notes and references
- R. G. Spiro, Glycobiology, 2002, 12, 43–56.
- R. Apweiler, H. Hermjakob and N. Sharon, Biochim. Biophys. Acta, 1999, 1473, 4–8 CrossRef CAS.
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS.
- A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS.
- L. H. Miller, M. F. Good and G. Milon, Science, 1994, 264, 1878–1883 CrossRef CAS.
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477–488 CrossRef CAS.
- C. S. Bennett and C.-H. Wong, Chem. Soc. Rev., 2007, 36, 1227–1238 RSC.
- B. G. Davis, Science, 2004, 303, 480–482 CrossRef CAS.
- S. I. van Kasteren, H. B. Kramer, H. H. Jensen, S. J. Campbell, J. Kirkpatrick, N. J. Oldham, D. C. Anthony and B. G. Davis, Nature, 2007, 446, 1105–1109 CrossRef.
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef CAS.
- B. G. Davis, Chem. Rev., 2002, 102, 579–601 CrossRef CAS.
- M. R. Pratt and C. R. Bertozzi, Chem. Soc. Rev., 2005, 34, 58–68 RSC.
- T. Buskas, S. Ingale and G. J. Boons, Glycobiology, 2006, 16, 113R–136R CrossRef CAS.
- L. Liu, C. S. Bennett and C.-H. Wong, Chem. Commun., 2006, 21–33 RSC.
- H. Hojo and Y. Nakahara, Biopolymers, 2007, 88, 308–324 CrossRef CAS.
- A. Brik, S. Ficht and C.-H. Wong, Curr. Opin. Chem. Biol., 2006, 10, 638–644 CrossRef CAS.
- A. Dirksen and P. E. Dawson, Curr. Opin. Chem. Biol., 2008, 12, 760–766 CrossRef CAS.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS.
- S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- T. Wieland, E. Bokelmann, L. Bauer, H. U. Lang and H. Lau, Justus Liebigs Ann. Chem., 1953, 583, 129–149 CrossRef CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS.
- P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS.
- S. Kent, J. Pept. Sci., 2003, 9, 574–593 CrossRef CAS.
- D. S. Y. Yeo, R. Srinivasan, G. Y. J. Chen and S. Q. Yao, Chem.–Eur. J., 2004, 10, 4664–4672 CrossRef CAS.
- B. L. Nilsson, M. B. Soellner and R. T. Raines, Annu. Rev. Biophys. Biomol. Struct., 2005, 34, 91–118 CrossRef CAS.
- C. Haase and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 1553–1556 CrossRef CAS.
- Y. Shin, K. A. Winans, B. J. Backes, S. B. H. Kent, J. A. Ellman and C. R. Bertozzi, J. Am. Chem. Soc., 1999, 121, 11684–11689 CrossRef CAS.
- B. J. Backes, A. A. Virgilio and J. A. Ellman, J. Am. Chem. Soc., 1996, 118, 3055–3056 CrossRef CAS.
- B. J. Backes and J. A. Ellman, J. Org. Chem., 1999, 64, 2322–2330 CrossRef CAS.
- L. A. Marcaurelle, L. S. Mizoue, J. Wilken, L. Oldham, S. B. H. Kent, T. M. Handel and C. R. Bertozzi, Chem.–Eur. J., 2001, 7, 1129–1132 CrossRef CAS.
- J. A. Hedrick, V. Saylor, D. Figueroa, L. Mizoue, Y. M. Xu, S. Menon, J. Abrams, T. Handel and A. Zlotnik, J. Immunol., 1997, 158, 1533–1540 CAS.
- B. Dorner, S. Muller, F. Entschladen, J. M. Schroder, P. Frankel, R. Kraft, P. Friedl, I. Clark-Lewis and R. A. Kroczek, J. Biol. Chem., 1997, 272, 8817–8823 CrossRef CAS.
- A. Leppanen, P. Mehta, Y. B. Ouyang, T. Z. Ju, J. Helin, K. L. Moore, I. van Die, W. M. Canfield, R. P. McEver and R. D. Cummings, J. Biol. Chem., 1999, 274, 24838–24848 CrossRef CAS.
- K. M. Koeller, M. E. B. Smith, R. F. Huang and C.-H. Wong, J. Am. Chem. Soc., 2000, 122, 4241–4242 CrossRef CAS.
- K. M. Koeller and C.-H. Wong, Nature, 2001, 409, 232–240 CrossRef CAS.
- P. Sears and C.-H. Wong, Science, 2001, 291, 2344–2350 CrossRef CAS.
- A. Leppanen, S. P. White, J. Helin, R. P. McEver and R. D. Cummings, J. Biol. Chem., 2000, 275, 39569–39578 CrossRef CAS.
- C.-H. Wong, Chimia, 2009, 63, 318–326 CrossRef CAS.
- A. L. Sorensen, C. A. Reis, M. A. Tarp, U. Mandel, K. Ramachandran, V. Sankaranarayanan, T. Schwientek, R. Graham, J. Taylor-Papadimitriou, M. A. Hollingsworth, J. Burchell and H. Clausen, Glycobiology, 2006, 16, 96–107 CAS.
- N. Bezay, G. Dudziak, A. Liese and H. Kunz, Angew. Chem., Int. Ed., 2001, 40, 2292–2295 CrossRef CAS.
- M. Fumoto, H. Hinou, T. Ohta, T. Ito, K. Yamada, A. Takimoto, H. Kondo, H. Shimizu, T. Inazu, Y. Nakahara and S. I. Nishimura, J. Am. Chem. Soc., 2005, 127, 11804–11818 CrossRef CAS.
- S. T. Cohen-Anisfeld and P. T. Lansbury, J. Org. Chem., 1990, 55, 5560–5562 CrossRef CAS.
- S. T. Cohen-Anisfeld and P. T. Lansbury, J. Am. Chem. Soc., 1993, 115, 10531–10537 CrossRef.
- S. Mezzato, M. Schaffrath and C. Unverzagt, Angew. Chem., Int. Ed., 2005, 44, 1650–1654 CrossRef CAS.
- P. Nagorny, B. Fasching, X. C. Li, G. Chen, B. Aussedat and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 5792–5799 CrossRef CAS.
- N. Yamamoto, Y. Tanabe, R. Okamoto, P. E. Dawson and Y. Kajihara, J. Am. Chem. Soc., 2008, 130, 501–510 CrossRef CAS.
- S. Ingale, T. Buskas and G. J. Boons, Org. Lett., 2006, 8, 5785–5788 CrossRef CAS.
- S. Ingale, M. A. Wolfert, J. Gaekwad, T. Buskas and G. J. Boons, Nat. Chem. Biol., 2007, 3, 663–667 CrossRef CAS.
- J. D. Warren, J. S. Miller, S. J. Keding and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6576–6578 CrossRef CAS.
- M. Bodanszky, Nature, 1955, 175, 685 CrossRef CAS.
- Q. Wan, J. Chen, Y. Yuan and S. J. Danishefsky, J. Am. Chem. Soc., 2008, 130, 15814–15816 CrossRef CAS.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS.
- T. Kawakami and S. Aimoto, Tetrahedron, 2009, 65, 3871–3877 CrossRef CAS.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS.
- K. Severinov and T. W. Muir, J. Biol. Chem., 1998, 273, 16205–16209 CrossRef CAS.
- T. C. Evans, J. Benner and M. Q. Xu, Protein Sci., 1998, 7, 2256–2264 CrossRef CAS.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef.
- D. A. Erlanson, M. Chytil and G. L. Verdine, Chem. Biol., 1996, 3, 981–991 CrossRef CAS.
- T. J. Tolbert, D. Franke and C.-H. Wong, Bioorg. Med. Chem., 2005, 13, 909 CrossRef CAS.
- V. Muralidharan and T. W. Muir, Nat. Methods, 2006, 3, 429–438 CrossRef CAS.
- T. J. Tolbert and C.-H. Wong, J. Am. Chem. Soc., 2000, 122, 5421–5428 CrossRef CAS.
- D. Macmillan and C. R. Bertozzi, Tetrahedron, 2000, 56, 9515–9525 CrossRef CAS.
- D. Macmillan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2004, 43, 1355–1359 CrossRef CAS.
- C. P. R. Hackenberger, C. T. Friel, S. E. Radford and B. Imperiali, J. Am. Chem. Soc., 2005, 127, 12882–12889 CrossRef CAS.
- T. C. Evans, Jr., J. Benner and M.-Q. Xu, J. Biol. Chem., 1999, 274, 3923–3926 CrossRef CAS.
- G. G. Kochendoerfer, S. Y. Chen, F. Mao, S. Cressman, S. Traviglia, H. Y. Shao, C. L. Hunter, D. W. Low, E. N. Cagle, M. Carnevali, V. Gueriguian, P. J. Keogh, H. Porter, S. M. Stratton, M. C. Wiedeke, J. Wilken, J. Tang, J. J. Levy, L. P. Miranda, M. M. Crnogorac, S. Kalbag, P. Botti, J. Schindler-Horvat, L. Savatski, J. W. Adamson, A. Kung, S. B. H. Kent and J. A. Bradburne, Science, 2003, 299, 884–887 CrossRef CAS.
- C. Piontek, P. Ring, O. Harjes, C. Heinlein, S. Mezzato, N. Lombana, C. Pohner, M. Puettner, D. V. Silva, A. Martin, F. X. Schmid and C. Unverzagt, Angew. Chem., Int. Ed., 2009, 48, 1936–1940 CrossRef CAS.
- C. Piontek, D. V. Silva, C. Heinlein, C. Pohner, S. Mezzato, P. Ring, A. Martin, F. X. Schmid and C. Unverzagt, Angew. Chem., Int. Ed., 2009, 48, 1941–1945 CrossRef CAS.
- R. Wynn and F. M. Richards, Methods Enzymol., 1995, 251, 351 CAS.
- P. Botti, M. R. Carrasco and S. B. H. Kent, Tetrahedron Lett., 2001, 42, 1831–1833 CrossRef CAS.
- J. Offer, C. N. C. Boddy and P. E. Dawson, J. Am. Chem. Soc., 2002, 124, 4642–4646 CrossRef CAS.
- D. Macmillan and D. W. Anderson, Org. Lett., 2004, 6, 4659–4662 CrossRef CAS.
- B. Wu, J. H. Chen, J. D. Warren, G. Chen, Z. H. Hua and S. J. Danishefsky, Angew. Chem., Int. Ed., 2006, 45, 4116–4125 CrossRef CAS.
- J. H. Chen, G. Chen, B. Wu, Q. Wan, Z. P. Tan, Z. H. Hua and S. J. Danishefsky, Tetrahedron Lett., 2006, 47, 8013–8016 CrossRef CAS.
- A. Brik, Y. Y. Yang, S. Ficht and C.-H. Wong, J. Am. Chem. Soc., 2006, 128, 5626–5627 CrossRef CAS.
- A. Brik and C.-H. Wong, Chem.–Eur. J., 2007, 13, 5670–5675 CrossRef CAS.
- A. Brik, S. Ficht, Y. Y. Yang, C. S. Bennett and C.-H. Wong, J. Am. Chem. Soc., 2006, 128, 15026–15033 CrossRef.
- S. Ficht, R. J. Payne, A. Brik and C.-H. Wong, Angew. Chem., Int. Ed., 2007, 46, 5975–5979 CrossRef CAS.
- R. J. Payne, S. Ficht, S. Tang, A. Brik, Y. Y. Yang, D. A. Case and C.-H. Wong, J. Am. Chem. Soc., 2007, 44, 13527–13536 CrossRef.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS.
- C. S. Bennett, S. M. Dean, R. J. Payne, S. Ficht, A. Brik and C.-H. Wong, J. Am. Chem. Soc., 2008, 130, 11945–11952 CrossRef CAS.
- Y. Y. Yang, S. Ficht, A. Brik and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 7690–7701 CrossRef CAS.
- L. C. Hsieh-Wilson, Nature, 2007, 445, 31–33 CrossRef.
- B. L. Pentelute and S. B. H. Kent, Org. Lett., 2007, 9, 687–690 CrossRef CAS.
- C. Haase, H. Rohde and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef CAS.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS.
- F. W. Hoffmann, R. J. Ess, T. C. Simmons and R. S. Hanzel, J. Am. Chem. Soc., 1956, 78, 6414–6414 CrossRef CAS.
- D. Bang, B. L. Pentelute and S. B. H. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985–3988 CrossRef CAS.
- V. Y. Torbeev and S. B. H. Kent, Angew. Chem., Int. Ed., 2007, 46, 1667–1670 CrossRef CAS.
- T. Durek, V. Y. Torbeev and S. B. H. Kent, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4846–4851 CrossRef CAS.
- C. Kan, J. D. Trzupek, B. Wu, G. Chen, Z. P. Tan, Y. Yuan and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 5438–5443 CrossRef CAS.
- W. Jelkmann, Physiol. Rev., 1992, 72, 449–489 CAS.
- W. Jelkmann, Intern. Med., 2004, 43, 649–659 Search PubMed.
- T. B. Drueke, F. Locatelli, N. Clyne, K. Eckardt, I. C. Macdougall, D. Tsakiris, H. Burger and A. Scherhag, N. Engl. J. Med., 2006, 355, 2071–2084 CrossRef CAS.
- D. Crich and A. Banerjee, J. Am. Chem. Soc., 2007, 129, 10064–10065 CrossRef CAS.
- P. Botti and S. Tchertchian, Side chain extended ligation, WO Pat./2006/133962 Search PubMed.
- J. Chen, Q. Wan, Y. Yuan, J. Zhu and S. J. Danishefsky, Angew. Chem., Int. Ed., 2008, 47, 8521–8524 CrossRef CAS.
- R. Okamoto and Y. Kajihara, Angew. Chem., Int. Ed., 2008, 47, 5402–5406 CrossRef CAS.
- R. Kaiser and L. Metzka, Anal. Biochem., 1999, 266, 1–8 CrossRef CAS.
- C. P. R. Hackenberger, Org. Biomol. Chem., 2006, 4, 2291–2295 RSC.
- Y. Kajihara, A. Yoshihara, K. Hirano and N. Yamamoto, Carbohydr. Res., 2006, 341, 1333–1340 CrossRef CAS.
- R. Okamoto, S. Souma and Y. Kajihara, J. Org. Chem., 2009, 74, 2494–2501 CrossRef CAS.
- D. S. Kemp, S. L. H. Choong and J. Pekaar, J. Org. Chem., 1974, 39, 3841–3847 CrossRef CAS.
- D. S. Kemp, Z. W. Bernstein and G. N. McNeil, J. Org. Chem., 1974, 39, 2831–2835 CrossRef CAS.
- J. Blake, Int. J. Pept. Protein Res., 1981, 17, 273–274 CAS.
- S. Aimoto, N. Mizoguchi, H. Hojo and S. Yoshimura, Bull. Chem. Soc. Jpn., 1989, 62, 524–531 CrossRef CAS.
- S. Aimoto, Biopolymers, 1999, 51, 247–265 CrossRef CAS.
- H. Hojo, Y. Matsumoto, Y. Nakahara, E. Ito, Y. Suzuki, M. Suzuki and A. Suzuki, J. Am. Chem. Soc., 2005, 127, 13720–13725 CrossRef CAS.
- H. Hojo, E. Haginoya, Y. Matsumoto, Y. Nakahara, K. Nabeshima, B. P. Toole and Y. Watanabe, Tetrahedron Lett., 2003, 44, 2961–2964 CrossRef CAS.
- Y. Takano, M. Habiro, M. Someya, H. Hojo and Y. Nakahara, Tetrahedron Lett., 2002, 43, 8395–8399 CrossRef CAS.
- Y. Takano, N. Kojima, Y. Nakahara and H. Hojo, Tetrahedron, 2003, 59, 8415–8427 CrossRef CAS.
- Y. Takano, H. Hojo, N. Kojima and Y. Nakahara, Org. Lett., 2004, 6, 3135–3138 CrossRef CAS.
- G. Chen, Q. Wan, Z. P. Tan, C. Kan, Z. H. Hua, K. Ranganathan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 7383–7387 CrossRef CAS.
- Y. Yuan, J. Chen, Q. Wan, Z. P. Tan, G. Chen, C. Kan and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 5432–5437 CrossRef CAS.
- Z. P. Tan, S. Y. Shang, T. Halkina, Y. Yuan and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 5424–5431 CrossRef CAS.
- T. Haack and M. Mutter, Tetrahedron Lett., 1992, 33, 1589–1592 CrossRef CAS.
- R. J. Payne, S. Ficht, W. A. Greenberg and C.-H. Wong, Angew. Chem., Int. Ed., 2008, 47, 4411–4415 CrossRef CAS.
- C. Haase and O. Seitz, Eur. J. Org. Chem., 2009, 2096–2101 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |