Method validation for the quantification of As, Cd, Hg and Pb in blood by ICP-MS for monitoring purposes
Sonia
D'Ilio
*,
Costanza
Majorani
,
Francesco
Petrucci
,
Nicola
Violante
and
Oreste
Senofonte
Istituto Superiore di Sanità, Department of Environment and Primary Prevention, Viale Regina Elena 299, Rome, Italy. E-mail: sonia.dilio@iss.it; Fax: +39 4990 2011; Tel: +39 4990 2252
First published on 21st October 2010
Abstract
Medical laboratories dedicated to the quantification of trace and ultra trace elements in human biological fluids should be accredited according to the ISO standards 15189![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2007. The use of validated methods for elements determination in blood becomes crucial for laboratories involved in medical analysis. The toxicological importance of As, Cd, Hg and Pb is well known and internationally recognized. These trace elements may be accumulated in human body through the exposure to pollutants in the environment as a result of uncontrolled anthropogenic activities. The internal levels of these elements can be estimated by the collection of blood samples from groups of selected population (biomonitoring).
:2007. The use of validated methods for elements determination in blood becomes crucial for laboratories involved in medical analysis. The toxicological importance of As, Cd, Hg and Pb is well known and internationally recognized. These trace elements may be accumulated in human body through the exposure to pollutants in the environment as a result of uncontrolled anthropogenic activities. The internal levels of these elements can be estimated by the collection of blood samples from groups of selected population (biomonitoring).
A fast and simple method to allow routine analysis of hundreds of blood samples was developed and fully validated. Arsenic, cadmium, mercury and lead were quantified in blood by quadrupole inductively coupled plasma mass spectrometry (Q-ICP-MS). A high throughput open digestion procedure was applied. Trueness, repeatability, within-laboratory reproducibility and standard measurement uncertainty were evaluated as method characteristics. The limits of quantification of the method were: 2.3 (As) μg l−1, 0.10 (Cd) μg l−1, 0.66 (Hg) μg l−1 and 0.56 (Pb) μg l−1. The method can be applied in the following ranges of concentration: 5–120 (As) μg l−1, 0.5–32 (Cd) μg l−1, 2.5–160 (Hg) μg l−1, 10–640 (Pb) μg l−1.
Introduction
Medical laboratories dedicated to the quantification of trace and ultra trace elements in biological fluids are preferably expected to be accredited according to ISO standards 15189:20071 (specifying general requirements for quality management of medical laboratories) rather than to ISO 17025:2005.2 The use of validated method for elements determination in blood becomes crucial for a laboratory involved in medical analysis. A correct interpretation of the results and the toxicological findings in routine analysis may be affected by unreliable data influencing patient's health.3
The accumulation of potentially dangerous elements in human body has been mainly monitored by blood and urine. The estimation of human internal exposure can be assessed by the analysis of blood from a number of selected participants (biomonitoring).4–6 The elements As, Cd, Hg and Pb are trace elements that can be commonly found in the environment but also in food. They are often present as pollutants in the environmental compartments (i.e., natural waters, soils, air) as a result of uncontrolled anthropogenic activities.7–9
The two inorganic arsenic forms (arsenite and arsenate) are both toxic to humans. Ingestion through food and water is the main pathway of As into the organism. In humans, high doses and/or long exposures to arsenic can lead to undesirable effects, such as, severe gastrointestinal toxicities, cardiac arrhythmias, development of malignancies, diabetes and even death.10–14 The inhalation of cadmium particles or fumes during industrial operations is the main source of exposure of this toxic element that can be also found in cigarette smoke. Cadmium may cause severe damages to lung, liver, kidney, testis, brain and even to placenta.7,15–19 Whole blood is a good indicator of a recent cadmium exposure due to its half life in this fluid (few months).20 Both elemental (as a vapour) and methyl mercury are toxic, in particular they are considered as neurotoxic substances.21,22 Methyl mercury is the form of primary concern for public health, it is usually assimilated from seafood and shellfish together with some contaminated fresh water fish. Once ingested, it is distributed to tissues, kidney and brain, where it can be metabolized to inorganic mercury.7,8,22,23 A past large-scale source of exposure to lead was the organolead added to petrol and lead in old paint, at present, the switch to unleaded petrol have changed the environmental trend for lead pollution. This element can still cause adverse effects on health, especially for the most vulnerable young children. Lead tends to deposit in soft tissues and accumulate in bone, moreover, it can be transferred across the placenta to the fetus and, during lactation, to the nursing infant. Most of information on human exposure to lead is based on blood levels.7,24–26
On the basis of the toxicity information on those four elements and their abundance in the environment, many biomonitoring programs are followed worldwide in population-based surveys designed as a basis for protecting public health. These studies require the use of sensitive and accurate analytical techniques for the quantification of trace and ultra trace elements, such as, inductively coupled plasma mass spectrometry (ICP-MS), which is well known for its high sample throughput and performance.8,17,26–32 Other techniques have been considered eligible for the analysis of blood, in particular, total reflection X-ray fluorescence (TXRF),33,34 atomic absorption spectrometry (AAS)33,35,36 and hydride generation atomic fluorescence spectrometric (HG-AFS).25
There is an increasing demand for analytical methods for the quantification of trace elements in biological fluids validated in compliance with international standards. Method validation ensures the reliability of the results and more importantly that there is a degree of confidence in data produced.37–39 In this paper, a method for the simultaneous quantification of As, Cd, Hg and Pb in whole blood by ICP-MS was developed and fully validated for monitoring purposes according to ISO standard 15189:2007 and U.S. Food and Drug Administration guidance.1,40
Experimental
The method was developed and validated on two different Reference Materials (RMs), namely, Clinchek® Whole Blood Level I (RECIPE Chemicals, Munich, Germany) and Seronorm® Trace Elements Whole Blood Level 1 (SEROAS, Billingstad, Norway). Both of them were reconstituted and stored by strictly following the instruction for use reported on the certificate. In particular, the two RMs were added with the appropriate volume of ultrapure deionized water with a specific resistivity of 18 MΩ cm (Easy Pure, PBI International, Milan, Italy), shaken by means of an orbital shaker (IKA Labortechnik, model KS 125 basic, Staufen, Germany) for three hours at 400 laps/min and kept in a refrigerator at +4 °C.A simple open acid digestion procedure was applied to all blood samples. A high throughput (48 positions available) digestion system, with a corrosion resistant treated graphite block plus an advanced composites, was employed to avoid any metals contamination (ModBlock™ Digestion System, CPI International, Amsterdam, The Netherlands). Proper disposable metal-free polypropylene sample tubes of 70 ml of capacity (50 ml ± 0.25 ml) were used in the digestion. These samples tubes were previously decontaminated by a six-time rinse with ultrapure deionized water and left in a clean area to be dried overnight.
A blood portion of 1 ml was used for each sample tube and added with 2 ml of 67–69% Super Pure HNO3 (Romil, Cambridge, Great Britain) and 1 μg l−1 of Rh as the internal standard. The mixture was left in closed vessels under a hood overnight to allow the pre-digestion step. The next day, samples were added with 1 ml of 30% Super Pure H2O2 (Romil, Cambridge, Great Britain) and digested at a temperature of 60 °C for three hours. The final digested solutions were quantitatively transferred in polypropylene Falcon® tubes (Becton Dickinson Labware, Franklin Lakes, NJ, USA) of 15 ml with ultrapure deionized water up to a volume of 10 ml, stored at −20 °C and analyzed with no further dilution. Empty sample tubes filled with the digestion mixture were subjected to the same digestion procedure in order to evaluate any possible residue contamination of As, Cd, Hg and Pb (reagents blanks).
The quantification of the four elements was carried out by means of an inductively coupled plasma mass spectrometer equipped with dynamic reaction cell (Elan DCR II, Perkin Elmer SCIEX, Norwalk, CT, USA) operating in standard mode. Data acquisition parameters and instrumental setting for the ICP-MS are shown in Table 1. A two-minute washing with deionized water was done after each sample run to prevent possible memory effects occurring in the spray chamber.
| Spectrometer | Elan DRC II (Perkin Elmer SCIEX, USA) |
| RF power/W | 1350–1400 |
| Gas flow rates/l min−1 | Plasma, 15; Auxiliary, 1.0; Nebulizer, daily optimized |
| Sample uptake rate | 18–20 rpm |
| Sample introduction | Meinhard nebulizer with cyclonic spray chamber |
| Interface | Pt sampler and skimmer cones |
| Extraction lens voltage | optimized for maximum I (24Mg, 115In, 238U) |
| Acceptance limits for optimization solution | 10 μg l−1 Mg > 6000 cps; 1 μg l−1 In > 30000 cps; 1 μg l−1 U > 20000 cps; mass 220 < 2 cps; Ba++/Ba < 3%; CeO/Ce < 3% |
| Analytical masses | 75As, 114Cd, 35Cl, 37Cl, 202Hg, 206Pb, 207Pb, 208Pb, 103Rh, 82Se, 118Sn |
| Dwell time/ms | 50–100 |
| Replicates | 3–5 |
The elements As, Cd, Hg and Pb were quantified by the addition calibration approach to compensate for matrix effects and any fluctuations of the analytical signals were corrected by the use of the internal standard (rhodium). Single element 1000 mg l−1 stock standard solutions of As, Cd, Hg, Pb and Rh in 2% (v/v) HNO3 were utilized in the study (Spex Industries Inc., Edison, NJ, USA). A standard solution of chlorine at about 1000 mg l−1 from NaCl Suprapur powder (Merck, Darmstadt, Germany) was required for the interferences study. Calibrants solutions were freshly prepared each day of analysis.
Results and discussion
Interference study
Elements determination by quadrupole ICP-MS is recognized to be affected by spectral and non-spectral interferences (also known as matrix interferences). The former usually produce an increase or decrease of the signal of the analyte of interest (e.g., a plasma-based, polyatomic and doubly-charged ion or a neighboring element in the mass spectrum), while, the latter can produce a reduction or an enhancement in the analyte signal depending on sample introduction system, sample transportation and ionization in plasma.41 For the above reasons, an interference study was carried out on the selected isotopes 75As, 114Cd, 202Hg, 206Pb, 207Pb and 208Pb before validating the method.Arsenic determination normally suffers the overlapping of the typical molecular ion 40Ar35Cl. This interference can be solved either by using an appropriate reaction gas (reaction cell technology) or by applying corrective factors. The use of mathematical equations is always recommended whenever the level of the interfering element produces an interference signal ≤ 20% of the total signal.42 For this study, it was necessary to estimate the content of chlorine in digested samples and the formation of the argon molecular ion in the analytical conditions in use.
A previous study reported the use of oxygen (as the reaction gas) for the determination of arsenic in blood samples digested by close microwave (MW) digestion system.28 Since the hot plate open digestion was employed in the present method, there was the need to measure and compare the residue amount of 35Cl in samples digested by both procedures (open and close). The analysis revealed a triple signal of chlorine in samples treated with MW procedure comparing to the signal obtained with samples treated by open digestion. The lower level of chlorine found together with the negligible formation of the argon-based interference allowed the use of the spectrometer in standard mode with no mathematical correction. In any case, the signal intensity of arsenic was checked for the contribution of ArCl by analyzing the masses 77 (40Ar37Cl) and 82Se in each run of analysis. The two equations applied were those reported in a previous study.42
Cadmium at mass 114 was chosen as the most abundant isotope (28.73%). This isotope may be interfered by ions argide, oxides and isobaric interferences. Normally, argon-based interferences are easy to remove by the use of the DRC technology, however, at mass 114 the formation of the molecular ions 40Ar74Se and 40Ar74Ge can be considered negligible; in fact, the abundance of 74Se is very low (0.9%) and germanium can be found in blood only at ultra trace level. Oxides, like MoO, RuO, ZrO, are the potential interferences on 114Cd that can not be removed with the technology used in this study. Nevertheless, the formation of these oxides is rather small, due to the low content of these three elements in blood, and it can be further reduced below 3% by optimizing the instrumental conditions for oxides formation (calculated on CeO/Ce ratio). As for the isobaric interferences, a mathematical correction (−0.027250 · 118Sn) was applied to solve the potential interference of 114Sn on the analyte.
No major interferences occurred on lead and mercury isotopes. Since in the natural state the abundance of isotopes of lead is not uniform, it is a common practice to use the correction for the sum of the isotopes 206Pb, 207Pb and 208Pb instead of the signal of the most abundant isotope 208Pb (52.4%). Three isotopes of mercury could be selected on the basis of their abundance, namely, 200Hg (23.13%), 202Hg (29.80%), 201Hg (13.22%), but only the most abundant isotope was preferred (202Hg).
Method validation
Validation can be defined as “the confirmation by examination and the provision of objective evidence that the particular requirements for a specific intended use are fulfilled”.2 Method validation in analytical chemistry is considered one of the technical parts of the comprehensive scheme of quality assurance. Single-laboratory method validation is considered correct in several circumstances, among these, when the feasibility of the method is ensured before approaching an expensive formal collaborative trial, data from a collaborative trial are not available or conducting a collaborative study is impossible.43In this study, the following method characteristics were selected and evaluated for the in-house method validation: selectivity/specificity, instrumental/method detection (LoDs) and quantification limits (LoQs), trueness, repeatability, within-laboratory reproducibility, working concentration range (linearity range), standard measurement uncertainty, stability study.
An internal quality control procedure was adopted by quantifying the blanks level (residue contamination) and controlling any possible instrumental drift in each run of the analysis. The blanks were prepared by adding 5 ml of high purity deionized water to empty BD Vacutainer® Blood Collection Tube containing Heparin (Becton, Dickinson and Company, NJ, USA), subsequently, 1 ml of this solution was subjected to the whole digestion procedure. The levels of arsenic, cadmium and mercury in all blanks were below the limits of quantification, while, for lead a maximum value of one-tenth the amount in blood (about 0.100 μg l−1) was considered acceptable. The instrumental drift was checked by running a single matrix-matched standard at known concentration every 10 samples. A corrective action (repetition of the calibration curve) was taken whenever a deviation in concentration of the reference standard was found more than 8%.
Selectivity/specificity
Both terms describe the extent to which a method uniquely reacts to a selected element. This topic is treated in the previous section of “Interferences study”.Limits of detection and quantification
Limits of detection (LoDs) and quantification (LoQs) were evaluated by using the common approach of 3σ and 10σ. The instrumental/method LoDs and LoQs were determined by running 10 unspiked samples of a pool of reagents blanks/digested blood and 10 spiked samples with 5 μg l−1 of As and Pb, 0.050 μg l−1 of Cd and 0.500 μg l−1 of Hg. A dilution factor of 10 was taken into account for the final computation. The results are in Table 2.| Method characteristics | As | Cd | Hg | Pb |
|---|---|---|---|---|
| Concentration (μg l−1) | ||||
| Instrumental LoD | 0.079 | 0.013 | 0.100 | 0.030 |
| Instrumental LoQ | 0.262 | 0.043 | 0.334 | 0.101 |
| Method LoD | 0.696 | 0.030 | 0.197 | 0.168 |
| Method LoQ | 2.3 | 0.100 | 0.657 | 0.560 |
| Working concentration range | 5–120 | 0.5–32 | 2.5–160 | 10–640 |
| As | Cd | Hg | Pb | ||
|---|---|---|---|---|---|
| Level of addition in blood (μg l−1) | |||||
| 50 | 10 | 1.0 | 2.0 | 25 | |
| (%) | |||||
| Recovery | 96 | 105 | 102 | 108 | 108 |
| Trueness | — | — | 97 | 110 | 95 |
| Repeatability (CV) | 4.9 | 5.5 | 3.4 | 8.0 | 3.1 |
| Within-laboratory reproducibility (CV) | 9.6 | 8.6 | 7.3 | 9.3 | 11.4 |
| Expanded measurement uncertainty (f = 2) | 24 | 23 | 19 | 28 | 23 |
Trueness
Trueness is commonly termed as “closeness of agreement between the average of an infinite number of replicate measured quantity values and a reference quantity value”. Measurement trueness is inversely related to systematic measurement error, but is not related to random measurement error.44,45 Trueness is quantitatively stated in terms of bias, usually, a smaller bias indicates good trueness. The bias is classically calculated by comparing the response of the method to a RM with a known value assigned.43Trueness was assessed by using the RM Clinchek® Whole Blood Level I and the recovery. Since this RM reported a reference value in blood only for Cd (1.3 ± 0.4 μg l−1), Hg (3.8 ± 1.2 μg l−1) and Pb (81 ± 15 μg l−1), the trueness for As was estimated by the use of the recovery at two different levels of additions (10 and 50 μg l−1 in blood) on this material. Four series of ten independent blood samples were digested and analyzed in four different days over a month (this was also done for the recovery of arsenic at two levels of concentration). Moreover, a test for the recovery for Cd, Hg and Pb was carried out on ten samples in one run on the RM Seronorm® Whole Blood Level 1, the levels of the additions in blood were the following: Cd 1.0 μg l−1, Hg 2.0 μg l−1 and Pb 25.0 μg l−1. This last reference material was chosen for the recovery for its lower content of the elements (As 1.8 μg l−1, Cd 0.74 μg l−1, Hg 2.2 μg l−1, Pb 27.6 μg l−1). All additions were made before the digestion.
The trueness is usually referred as a bias and it was assessed by dividing the detected mean concentration by the certified value and multiplying by 100. The recovery was calculated as a percentage: recovery % = (100 · concentration found)/spiked concentration. A bias of ± 10% was considered acceptable for the trueness and an acceptance range of 90–110% was selected for the recovery.
The results for trueness and mean recovery fell in the pre-established acceptance range confirming the suitability of the method to produce reliable data (see Table 2).
Working concentration range
This characteristic is critical to establish method applicability and verify to which extent the analytical response is linear as a function of concentration. This response was checked by a seven-point calibration curve assuming as adequate a linear regression coefficient r2 > 0.999. The linearity ranges were appropriate to the levels of these elements commonly found in blood (Table 2).Repeatability and within-laboratory reproducibility
The repeatability is the precision under repeatability conditions, that are the conditions where independent test results are obtained with the same method on the same sample in the same laboratory by the same operator using the same equipment in a short interval of time. While, the within-laboratory reproducibility is the precision obtained in the same laboratory under predetermined conditions over long time intervals.The intra-day repeatability and within-laboratory reproducibility were calculated as coefficient of variation in percent (CV %) of ten samples analyzed in one simple run and four independent sets of ten samples analyzed in four different days, respectively. For arsenic, this was done at the two levels of concentration. As concerns the within-laboratory reproducibility, the analytical conditions were modified over a month of measurements, in particular, operators and pipettes were changed.
The results are in Table 2. All CV% were always under 20%, this is realistic for the concentrations quantified.
Stability study
An unsatisfactory stability of the analyte in sample during the step of storage may produce significant deviation in the results.Analytes stability in whole blood was determined by evaluating the recovery, after two freeze and thaw cycles, of ten spiked human bloods samples from a pool of randomly selected samples from patients participating in a monitoring program. Samples, kept frozen at −20 °C, were left at room temperature for 24 h and pooled. Then, ten aliquots were spiked, refrozen for 48 h, thawed and subsequently analyzed for the content of the four elements. The recovery for all elements fell in the established acceptance range of 90–100% confirming their stability in whole blood.
The stability of the analytes in the digested solutions was also tested after two freeze and thaw cycles at a storage temperature of −20 °C. Ten spiked digested blood solutions (from the above mentioned pool) were frozen for 24 h, thawed and then analyzed. The results for the recoveries were consistent to the values reported in Table 2.
Measurement uncertainty
A laboratory has to demonstrate the quality of the results produced and its fitness for purpose by giving a measure of the confidence that can be placed on the result. Measurement uncertainty is defined by the VIM3 as “Non-negative parameter characterizing the dispersion of the quantity values being attributed to a measurand, based on the information used”.44Since the RM Clinchek® Whole Blood offers only a control range of concentrations for the elements analyzed, no value for the reproducibility was available. Then, the standard measurement uncertainty associated to the recovery was employed in the final results. In this study, only the largest and most significant contributions to the combined measurement uncertainty (uc) were estimated according to Eurachem/Citac guide: the standard uncertainty associated to the recovery (urec), the preparation of the standard solutions (us) and the within-laboratory reproducibility of the measurements (um).46,47
To the aim of the validation, the following procedure was adopted: urec = rsd/n1/2 where, rsd is the ratio between the standard deviation of the mean recovery and the mean recovery value and n is the number of spiked samples; the uncertainty us associated with the preparation of the standard solutions was considered by the sum of the contributes of pipettes volume and stock solutions:
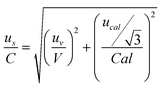
and the expanded measurement uncertainty (U) with a coverage factor of 2 (k), which considers a normal distribution of measurements with a 95% confidence level:

| U = uc(x)·k |
Table 3 shows the percent contribution of each single factor to the combined measurement uncertainty, as for arsenic only the high level is represented. As expected, the main contribution for all elements is due to the within-laboratory reproducibility of the measurements. Data for the expanded measurement uncertainty is displayed in Table 2.
| As (high level) | Cd | Hg | Pb | |
|---|---|---|---|---|
| Uc | (%) | |||
| Urec | 4 | 16 | 3 | 0.2 |
| Us | 41 | 37 | 52 | 3.8 |
| Um | 55 | 47 | 45 | 96 |
Conclusions
This paper described method development and validation for the quantification of As, Cd, Hg and Pb in blood by ICP-MS according to common international standards.1,40,46,47 The overall performance of the present method demonstrate that the requirements of those standards are fulfilled. The LoQs allow the quantification at low levels. Mercury presents the highest intra-day repeatability (CV, 8.0%), but this is probably due to major fluctuations of the signal that may be ascribable to small memory effects, distinctive for this element. The results for trueness and recovery always fall in the pre-established acceptance range (90–110%), this outcome is particularly successful considering the open digestion procedure applied. Additionally, the analysis of reagent blanks confirms that there was no cross contamination during the digestion.The precision of the present method is in agreement with those found by a Brazilian study for As, Cd and Pb in blood by ICP–MS (5–10%)29 and by a Spanish research on As, Cd and Hg in blood by AAS (5–8.2%).16 Different concentrations of Pb were analyzed obtaining an inter-day reproducibility between 2 to 14%, that is also consistent to the present results.38
This validated method was successfully applied to two monitoring programs of potentially exposed Italian population requiring the analysis of hundreds of blood samples. Elevated sensitivity and high sample throughput (typical of ICP-MS), together with the good performance of the method, offer the possibility of a fast and accurate quantification of As, Cd, Hg and Pb in human blood for monitoring purposes.
References
- ISO standards, ISO 15189:2007 Ed.2. Medical laboratories - Particular requirements for quality and competence.
- Uni Cei EN ISO/IEC 17025:2005. General requirements for the competence of testing and calibration laboratories.
- F. T. Peters, H. D. Olaf and F. Musshoff, Forensic Sci. Int., 2007, 165, 216–224 CrossRef CAS.
- K. Sexteon, L. L. Needham and J. L. Pirkle, Am. Sci., 2004, 92, 38–45.
- National Center for Environmental Health, Division of Laboratory Sciences. Third National Report on Human Exposure to Environmental Chemicals. Center for Disease Control and Prevention, Atlanta, GA (USA); NCEH Pub. No. 05-0570, 2005, pp.487 Search PubMed.
- Technical Guidance Document on Risk Assessment. European Commission - Joint Research Centre, Institute for Health and Consumer Protection, European Chemicals Bureau (ECB). Office for official publications of the European Communities, Luxembourg; TGD part I, 2003, pp.311 Search PubMed.
- Joint WHO/Convention Task Force on the Health Aspects of Air Pollution. Health risks of heavy metals from long-range transboundary air pollution. World Health Organization, WHO Regional Office for Europe, Copenhagen (Denmark); E91044, 2007, pp.144 Search PubMed.
- W. McKelvey, R. Charon Gwynn, N. Jeffery, D. Kass, L. E. Thorpe, R. K. Garg, C. D. Palmer and P. J. Parsons, Environ. Health Perspect., 2007, 115, 1435–1441 CAS.
- M. M. Mumtaz, P. Ruiz and C. T. De Rosa, Toxicol. Appl. Pharmacol., 2007, 223, 104–113 CrossRef CAS.
- L. C. Platanias, J. Biol. Chem., 2009, 284, 18583–18587 CrossRef CAS.
- T. Chin-Hsiao, Toxicol. Appl. Pharmacol., 2009, 235, 338–350 CrossRef.
- J. C. States, S. Srivastava, Y. Chen and A. Barchowsky, Toxicol. Sci., 2009, 107, 312–323 CAS.
- T. S. Y. Choong, T. G. Chuah, Y. Robiah, F. L. Gregory Koay and I. Azni, Desalination, 2007, 217, 139–166 CrossRef CAS.
- B. Curros-Gontad, M. C. Barciela-Alonso, M. D. Bujan-Villar, E. Pena-Vázquez, P. Herbello-Hermelo and P. Bermejo-Barrera, Eur. Food Res. Technol., 2008, 227, 1165–1172 CrossRef CAS.
- M. Méndez-Armenta and C. Ríos, Environ. Toxicol. Pharmacol., 2007, 23, 350–358 CrossRef CAS.
- F. Gil, L. F. Capitán-Vallvey, E. De Santiago, J. Ballesta, A. Pla, A. F. Hernández, M. Gutiérrez-Bedmar, J. Fernández-Crehuet, J. Gómez, O. López-Guarnido, L. Rodrigo and H. Villanueva, Sci. Total Environ., 2006, 372, 49–57 CrossRef CAS.
- C. E. Khassouani, R. Soulaymani, Y. Mauras and P. Allain, Clin. Chim. Acta, 2000, 302, 155–160 CrossRef CAS.
- L. Chen, T. Jin, B. Huang, G. Nordberg and M. Nordberg, Toxicol. Appl. Pharmacol., 2006, 215, 93–99 CrossRef CAS.
- M. Ikeda, Z.-W. Zhang, S. Shimbo, T. Watanabe, H. Nakatsuka, C.-S. Moon, N. Matsuda-Inoguchi and K. Higashikawa, Sci. Total Environ., 2000, 249, 373–384 CrossRef CAS.
- L. Farzin, M. Amiri, H. Shams, M. A. Ahmadi Faghih and M. E. Moassesi, Biol. Trace Elem. Res., 2008, 123, 14–26 CrossRef CAS.
- S. Bose-O'Reilly, B. Lettmeier, R. Matteucci Gothe, C. Beinhoff, U. Siebert and G. Drasch, Environ. Res., 2008, 107, 89–97 CrossRef.
- S. S. Vupputuri, M. P. Longnecker, J. L. Daniels, X. Guo and D. P. Sandler, Environ. Res., 2005, 97, 195–200 CrossRef CAS.
- K. L. Caldwell, M. E. Mortensen, R. L. Jones, S. P. Caudill and J. D. Osterloh, Int. J. Hyg. Environ. Health, 2009, 212, 588–598 CrossRef CAS.
- R. L. Jones, D. M. Homa, P. A. Meyer, D. J. Brody, K. L. Caldwell, J. L. Pirkle and M. J. Brown, Pediatrics, 2009, 123, e376–e385 CrossRef.
- J.-H. Wang, Y.-L. Yu, Z. Du and Z.-L. Fang, J. Anal. At. Spectrom., 2004, 19, 1559–1563 RSC.
- A. Bazzi, O. J. Nriagu and A. M. Linder, J. Environ. Monit., 2008, 10, 1226–1232 RSC.
- I. Rodushkin, F. Ödman, R. Olofsson and M. D. Axelsson, J. Anal. At. Spectrom., 2000, 15, 937–944 RSC.
- S. D'Ilio, N. Violante, M. Di Gregorio, O. Senofonte and F. Petrucci, Anal. Chim. Acta, 2006, 579, 202–208 CrossRef.
- J. A. Nunes, B. L. Batista, J. L. Rodrigues, N. M. Caldas, J. A. G. Neto and F. Barbosa Jr., J. Toxicol. Environ. Health, Part A, 2010, 73, 878–887 Search PubMed.
- J. P. Goullé, L. Mahieu, J. Castermant, N. Neveu, L. Bonneau, G. Lainé, D. Bouige and C. Lacroix, Forensic Sci. Int., 2005, 153, 39–44 CrossRef CAS.
- P. Heitland and H. D. Köster, J. Trace Elem. Med. Biol., 2006, 20, 253–262 CrossRef CAS.
- M. H. M. Chan, I. H. S. Chan, A. P. S. Kong, Risa Osaki, Robert C. K. Cheung, Chung S. Ho, Gary W. K. Wong, Peter C. Y. Tong, Juliana C. N. Chan and Christopher W. K. Lam, Pathology, 2009, 41, 467–472 CrossRef CAS.
- T. Martinez, J. Lartigue, P. Avila-Perez, G. Zarazua, M. Navarrete, S. Tejeda and A. Ramírez, J. Radioanal. Nucl. Chem., 2004, 259, 511–514 CrossRef CAS.
- A. Khuder, M. A. Bakir, J. Karjou and Kh.-M. Sawan, J. Radioanal. Nucl. Chem., 2007, 273, 435–442 CrossRef CAS.
- J.-Y. Son, J. Lee, D. Paek and J.-T. Lee, Environ. Res., 2009, 109, 738–744 CrossRef CAS.
- O. S. Ertas and H. Tezel, J. Pharm. Biomed. Anal., 2004, 36, 893–897 CrossRef CAS.
- M. Castelli, B. Rossi, F. Corsetti, A. Mantovani, G. Spera, C. Lubrano, L. Silvestroni, M. Patriarca, F. Chiodo and A. Menditto, Microchem. J., 2005, 79, 349–355 CrossRef CAS.
- C. Bonnefoy, A. Menudier, C. Moesch, G. Lachâtre and J.-M. Mermet, J. Anal. At. Spectrom., 2002, 17, 1161–1165 RSC.
- S. Izquierdo Álvarez, M. L. Calvo Ruata, J. M. González López, A. García de Jalón Comet and J. F. Escanero Marcén, J. Trace Elem. Med. Biol., 2007, S121, 26–28 CrossRef.
- Guidance for Industry, Bioanalytical Method Validation. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM), USA.U.S. FDA Guide, 2001 Search PubMed.
- S. D'Ilio, F. Petrucci, O. Senofonte and N. Violante, Study of oxide-based interferences on trace elements determination in ICP-MS, in: J. K. Lang (Ed.). Handbook on mass spectrometry: instrumentation, data and analysis, and applications, Nova Science Publishers, Inc., New York, 2009, 4th Quarter pp. 201–224 Search PubMed.
- S. D'Ilio, F. Petrucci, M. D'Amato, M. Di Gregorio, O. Senofonte and N. Violante, Anal. Chim. Acta, 2008, 624, 59–67 CrossRef CAS.
- M. Thompson, S. L. R. Ellison and R. Wood, Pure Appl. Chem., 2002, 74, 835–855 CrossRef CAS.
- Joint Committee for Guides in Metrology. International vocabulary of metrology-Basic and general concepts and associated terms (VIM3), 3rd ed. JCGM 200, 2008 Search PubMed.
- International vocabulary of metrology. Basic and general concepts and associated terms; (VIM)ISO/IEC Guide 99, 2007 Search PubMed.
- EURACHEM/CITAC Guide CG 4. Quantifying Uncertainty in Analytical Measurement; 2nd edition, 2000 Search PubMed.
- Joint Committee for Guides in Metrology. Evaluation of measurement data - Guide to the expression of uncertainty in measurement, JCGM 100: 2008 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |