Analytical methods and strategies in the study of plant polyphenolics in clinical samples
Jan
Vacek
*a,
Jitka
Ulrichová
a,
Bořivoj
Klejdus
b and
Vilím
Šimánek
a
aDepartment of Medical Chemistry and Biochemistry, Faculty of Medicine and Dentistry, Palacký University, Hněvotínská 3, 775 15, Olomouc, Czech Republic. E-mail: jan.vacek@upol.cz
bDepartment of Chemistry and Biochemistry, Mendel University of Agriculture and Forestry, Zemědělská 1, 613 00, Brno, Czech Republic
First published on 1st April 2010
Abstract
Plant phenolics are secondary metabolites that exhibit a number of physiological effects in living organisms, including humans. These compounds form an integral part of the human diet and many of them are important components of phyto-preparations. Developments in the analytical methods for certain polyphenolic compounds and their analysis in clinical samples (plasma, urine, tissues, and organs) are described. The main aim of this review is to present the application of modern methods, especially chromatographic approaches, mass spectrometry, NMR, electrochemistry, and radioactivity labelling-based methods in the research of plant polyphenolics and the study of their biological activities.
 Jan Vacek | Jan Vacek is currently an assistant of professor at the Palacky University in Olomouc under the direction of J.U. and V.S. He gained his Ph.D. in Molecular and Cell Biology at the Masaryk University in Brno. His research interests include analytical methods for phytochemical analysis and DNA analysis using chromatography/mass spectrometry and electrochemical tools. |
 Jitka Ulrichová | Jitka Ulrichová graduated from Biochemistry at the Masaryk University in Brno. Currently she is a professor of Biochemistry and the Head of the Department of Medical Chemistry and Biochemistry at the Faculty of Medicine and Dentistry at the Palacky University in Olomouc. Her major research interests include the study of experimental toxicology and biological activity of natural compounds in vitro using mammalian cell cultures. |
 Bořivoj Klejdus | Bořivoj Klejdus is professor at the Department of Chemistry and Biochemistry at the Mendel University of Agriculture and Forestry in Brno. His research is oriented towards the application of modern extraction methods for the determination of natural substances (isoflavonoids, flavonoids, lignans and other phenolics, alkaloids, steroids, plant pigments, amino acids, neurotoxins and peptides) in plant materials by LC/MS. |
 Vilím Šimánek | Vilím Šimánek works as a professor of Biochemistry at the Faculty of Medicine and Dentistry, Palacky University in Olomouc. He is co-author of more than 240 publications in scientific journals in the fields of chemistry and the biological activities of phytoceuticals, mainly alkaloids. His research interests are now focused on the study of the applications of plant polyphenolics in helping the prevention of some chronic diseases. |
1. Introduction
Plants are able to synthesize substances (secondary metabolites) as pigments, phytoalexins, or attractants for insects which play an important role in ecological interactions, e.g. biotic interactions of the plant to pathogens or animal pests.1 As has been mentioned in a large number of publications, many of these are phenols (approximately 4000 plant phenols are currently known) and many have been proven to significantly influence the physiology of those that ingest them, including humans.2–6Plant phenols are a component of the human diet and some are used as effective antioxidants in dietary supplements or as remedies in phyto-preparations.5,7,8 Phenolic substances are generally subdivided into simple phenols and polyphenols. The latter are characterized by the presence of more than one aromatic ring (phenol unit) or building block per molecule. The most significant plant phenols include: phenolic acids (hydroxyderivatives of benzoic and cinnamic acids) and their esters (chlorogenic and caftaric acids), chalcones (butein, okanin, licochalcone, and dihydrochalcones such as phloridzin), coumarins (aesculetin), flavonoids (flavonols, flavones, flavanonols, isoflavones, and/or anthocyanidins), lignans, etc. With a few exceptions, all of these substances are categorised as polyphenols, which can be found in their plant sources as free molecules (aglycones), in the form of conjugates (usually as glycosides) or even as oligomers or polymers, e.g. proanthocyanidins, lignans or lignin (Fig. 1).
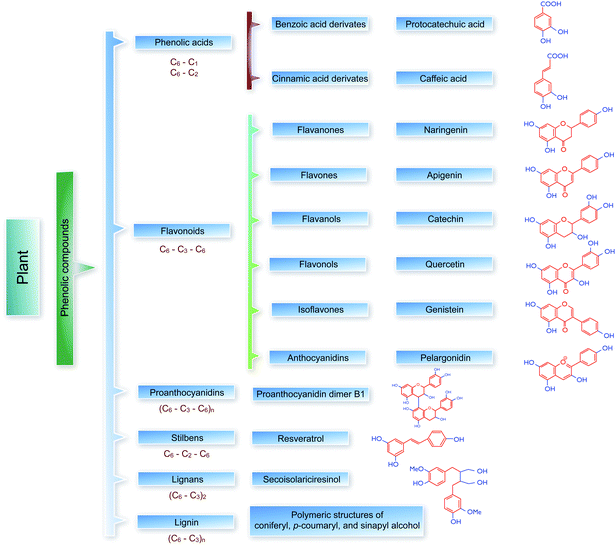 | ||
| Fig. 1 Schematic representation of selected groups, subgroups, and examples of plant phenolic compounds. | ||
The main source of the above substances is fruit and vegetables, as well as certain foodstuffs and supplements including tea, wine, olive oil, chocolate and many others (recently reviewed in Ref. 7). Epidemiological studies have shown that the consumption of plant phenols can influence the health of an individual, although the specific positive or negative effects on human physiology have not yet been determined clearly.8 A common denominator of plant phenols is their positive reaction to tests of their antioxidation capacity.9 Due to the fact that polyphenols are able to eliminate free radicals, they are accredited with cardioprotective and neuroprotective effects. Knowledge of their anti-inflammatory effects has been gained and mechanisms for incorporating polyphenols in reducing the risk of cancer are being discussed.1,8,10,11 Phenolic antioxidants are abundantly present in plant sources, usually in milligrams to grams per kilogram dry weight. To be more specific, HPLC analysis enabled 107–116 mg kg−1 of ferulic acid to be determined in grapefruit,12 and in the case of flavonoid polyphenols, levels of daidzein ranging from 18–260 mg kg−1 were found in soy.13 Apart from their antioxidative properties and connected protective effects, an estrogenic-like effect on the hormonal system of mammals was found for certain flavonoids (isoflavones) and some lignans, stilbenes and coumestrans.14 These groups of substances are designated phytoestrogens and phyto-preparations that contain them are used in complementary medicine. In closing, it needs to be noted that the physiological effects connected to the consumption of plant phenolic substances are strictly influenced by the amount and form in which they are administered.
The aim of this article is to briefly present the analytical approaches to the study of plant phenols in clinical samples of body fluids and organ tissues. To our knowledge, the above field has not yet been comprehensively described. Attention will be primarily paid to complete analytical procedures based on preparation of the sample, subsequent isolation/extraction, and determination of the substances using modern analytical methods, particularly subsequent mass spectrometry and selected spectroscopic tools. The review does not intend to give a complete overview of the topics considered.
2. Historical view
Research into natural organic compounds including phenols was initiated by C. W. Scheele and L. N. Vauquelin, who were the first to isolate gallic acid and simple organic acids (see Ref. 15 and citations therein). Important discoveries followed, hippuric acid, the final transformation product, was identified after a phenol-rich diet ingestion in horse urine.16 A significant shift in the research of plant phenols can be observed to occur at the beginning of the last century, when instruments such as the polarimeter and photometer were being introduced.17 First photometric and spectrophotometric methods enabled studies of physico-chemical properties and selective analysis of plant phenolics.18 Another great benefit was M. S. Tsvet's discovery of chromatographic separation methods,19,20 which were consequently used for analysis of selected low-molecular compounds in plant extracts and observation of their occurrence in the organisms of their consumers; the first methods based on paper chromatography were developed in the 1950s. For example, thin-layer chromatography enabled phenolic acid analysis in urine samples21 and/or applications of adsorption chromatography and paper chromatography in the study of flavonoids by S. H. Wender.22,23 Progress in chromatographic methods and their application in research into natural compounds were described in reviews.19,24As the previous century progressed, a significant development in analytical instrumentation was observed, arising from the application of chromatography (HPLC) based on silica stationary phases in combination with optical detectors, including applications aimed at fluorimetric methods.25 Flavonoid aglycones were probably studied using silica chromatographic column firstly at 1960 by R. M. Horowitz and B. Gentili.26 The second half of the 20th century, with the exception of classical chromatography approaches, radioactivity measurement (HPLC-radiocounters or microradiographic techniques) and sensitive immunological (immunohistochemical) methods were also used in the study of phenols in individual organ tissues. With the arrival of the electrospray ionisation method, it became possible to combine HPLC or other high-performance separation methods with MS.27 MS methods are applied in determining the spectra of polyphenols in biological material and were successfully used together with NMR for their structural analysis. One of the first MS characterisations of plant polyphenols was published by A. Pelter and co-workers28 and pioneer NMR studies were presented by B. Ternai and K. R. Markham; these introduced 13C-NMR into the research of flavonoidal compounds.29 The application of these analytical instruments enabled the new scientific discipline of metabolomics to arise.30 MS and NMR also greatly contributed to the study of plant polyphenol conjugates,31 to elucidate the mechanism of their metabolization and the ways in which they are excreted from the bodies of consumers.32 In January 2007, scientists completed the first draft of the human metabolome. They catalogued approximately more than 2180 endogenous metabolites, as reported in the literature.33
3. The study of the metabolization of phenols in the human body
Polyphenols ingested via food are absorbed in the digestive tract (primarily in the small and large intestine) at which they undergo chemical transformation. A partial absorption of plant phenols in the oral cavity after consumption of green tea was evidenced in the saliva of volunteers by catechin analysis. The extent of polyphenol chemical transformation in the oral cavity has not yet been precisely ascertained due to the limited number of relevant studies, nevertheless their results suggest that salivary enzymes can partially participate in glycoside hydrolysis.23–25 The key task of polyphenol absorption is deglycosylation by intestinal enzyme systems; the following enzymes participate in these hydrolysis processes: lactase phloridzine hydrolase and cytosolic β-glucosidase.10,34 A non-enzymatic decomposition of glycosides by gastric juice or a passage of free glycosides across the intestinal brush border is not likely or occurs to only a limited extent. The study of in vivo phenols absorption can be exclusively carried out using analyses of samples from patients with ileostomy (a surgically created outlet of the small intestine) where the amount of the substance absorbed can be calculated from its abundance in the diet vs. its quantity found in the ileostomy effluent.27–33 Phenolic substances that were not absorbed in the small intestine can be further metabolized by bacteria in the large intestine. There, not only deglycosylation, but also reduction and hydrolysis reactions take place, leading to the decomposition of polyphenols into individual phenol units which were found in relatively high level in feces.34Phenols, after their absorption in the digestive tract, are enzymatically metabolized in individual tissues, and substances not undergoing absorption are eliminated. In principle there is a partial formation of conjugates of phenols with glucuronic acid in intestinal mucosa, followed by conjugation reactions (glucuronidation, sulfation and methylation) to a full extent in the liver.35,36 The liver is, similarly to the case of drugs or xenobiotic metabolization, the principal organ of metabolic transformation of phenols facilitating their biological inactivation prior to their excretion from the body.14 The level of non-conjugated phenols in the body after the intake of vegetable food or phyto-preparations is generally very low. Individual metabolites and their formation can be studied by examination of the blood or plasma. Final metabolization products are then analysed in urine or feces.37–41 The metabolization process and plant phenol representation in various clinical samples is schematically shown in Fig. 2 and elaborated in Tab. 1.
| Processes in human organism | Localization (sample for analysis) | Phenol occurrence and transformations | Type of experiment and specifications | Analytical methods (phenol quantifications) |
|---|---|---|---|---|
| a To our knowledge, there are not sufficient data on analysis of these sample types in human and other mammals. b Phenol glycoside resorption is probably limited and this phenomena is still discussed. Abbreviations: n.q.: not quantified, r.q.: relative quantification (autoradiographic studies), wt: weight. | ||||
| Dietary intake and digestive tract input | Plant phenol glycosides and/or possibly aglycones in plant materials; ratio of glycosidic vs. aglycone forms is individual | Identification and quantification of phenols in food, herbs and phyto-preparations | HPLC with UV-Vis and MS (phenolic acids 10–2200 μg g−1, anthocyanins 20–7500 μg g−1, flavonols 2–1200 μg g−1, flavanones 50–685 μg g−1, isoflavones 30–1800 μg g−1, flavones 5–1850 μg g−1, all in fresh wt).10 | |
| Oral cavity (saliva) | Oral hydrolysis, partial uptake to epithelial oral cells, and phenol/salivary protein interactions. | In vitro time-course experiment of the flavonoid glycosides (e.g. quercetin-4’-glucoside) hydrolysis in saliva samples.89 | HPLC/UV-Vis (in vitro study, n.q.),89 HPLC/ED (epicatechin 1.8–7.5 μg ml−1, epigallocatechin 11.7–43.9 μg ml−1),90 modified Folin-Ciocalteau assay (96 μg ml−1 after stimulation), for details see Ref. 91. | |
| Oesophagus and stomacha | Hydrolysis and resorption of plant phenols in oesophagus and stomach is not described. | Analysis of catechin levels in saliva samples after drinking of green tea (200 ml; containing 1.2 g of tea solids).90 | ||
| Study of astringency sensation due to phenol/salivary protein interactions.91 | ||||
| Processes in small intestine and colon | Small intestine (ileostomy effluent) | Effective enzymatic hydrolysis of plant glycosides by lactase phloridzin hydrolase and cytosolic β-glucosidase and they pass from small intestine to the body of volunteers.b | Colonic availability of apple polyphenols (cinnamic acids, procyanidins, dihydrochalcones, flavonols, and flavanols) after oral intake of apple juice.92 | HPLC/UV-Vis and MS/MS (up to 33% of the oral dose was recovered in ileostomy fluids, total polyphenol amount in oral dose (1 l) was 249.9 μg ml−1).92 |
| Nonpolar and polar phenols from olive oil; in vivo and ex vivo study.93 Study of quercetin hydrolysis in ileostomy patients after ingestion of onion meal.94 | HPLC MS/MS (more than 55% of ingested dose of olive oil phenols are absorbed in ileostomy subjects.93 | |||
| HPLC/UV-Vis (19.5–35.2% of quercetin was recovered in ileostomy fluids after ingestion of 10.9–51.6 mg of quercetin flavonoids).94 | ||||
| Colon | Hydrolysis and metabolization of phenols by enteric bacteria lead to formation of less polar aglycones. Cleavage of the aglycones and formation of fermentation products which are excreted.10,95 | |||
| Resorption and circulation | Blood circulation (plasma) | Circulation of phenolics and their metabolites in blood after resorption. The analysis of plasma samples probably reflects actual phenolic status in organism. | Analysis of plasma isoflavones after 14 day in human subjects consuming 2-times per day of diet containing 20 g of isolated soy protein.57 | HPLC/MS (genistein: 142.7 pg.ml−1 and daidzein: 73.5 pg.ml−1 in plasma samples).57 |
| MS study of quercetin and its conjugates in human plasma of subjects after ingestion 0.5 g of quercetin.37 | HPLC MS/MS (quercetin aglycone in plasma: 1–8 ng ml−1).37 | |||
| Phenolic acid metabolites in rats.52 | HPLC/DAD-MS2 (danshensu, caffeic, ferulic and isoferulic acid and their metabolites were identified, n.q.).52 | |||
| Analysis of procyanidin-rich plasma which was obtained from rats 2 h after administration (by intragastric gavage) of phyto-preparation based on procyanidin in dose 1 g per kg of weight.50 | HPLC MS/MS (six metabolites were identified, catechine content in plasma samples: 247 ng ml−1).50 | |||
| Uptake and metabolization in organ tissues | Tissue metabolic transformations (liver and other tissues) | Metabolization and conjugation of phenols in liver and their uptake to organ tissues. | Genistein analysis in rats after ingestion of diets fortified with 5–500 μg ml−1 of genistein aglycone.73 | HPLC MS/MS (genistein aglycone in liver: 0.1–0.41 μg g−1).73 |
| Distribution of trans-resveratrol in mouse tissues.71 | Autoradiographic and microradiographic measurement in stomach, liver, kidney, and other (r.q.).71 | |||
| Determination of flavonol metabolites in rat tissues after oral ingestion [2-14C]quercetin-4’-glucoside].70 | HPLC-radiocounting MS/MS analysis (18 metabolites were found, only 1.5% of quercetin from ingested dose was determined in liver, r.q.).70 | |||
| Uptake of epicatechin and its metabolites identification in rat brain after administration of 100 mg kg−1 body wt/d orally for 1–10 d.41 | HPLC/UV-Vis MS/MS (epicatechin including its metabolites in rat brain tissue: ∼116 ng g−1).41 | |||
| Analysis of 14C-genistein in gut and excretory, respiratory, peripheral, and reproductive organs of rat.72 | HPLC-radiocounting MS (r.q.).72 | |||
| Determination of black and green tea polyphenols in tissue samples.65 | HPLC/ED (catechins were n.q. in real samples).65 | |||
| Excretion processes | Final metabolite excretions (urine and feces) | Final metabolite products are excreted in urine. Phenols and metabolites that were not resorbed after microbial fermentation in colon are excreted in feces. The analysis of urine samples reflects phenol elimination from organism in time. | Stereospecific analysis of naringenin in rat and human urine.62 | HPLC/UV-Vis (urine excretion profiles for naringenin and its enantiomers is presented).62 |
| Identification of phenolic compounds in human urine after oral administration of traditional herbal medicines.96 | HPLC/UV-Vis (nine phenols were identified, n.q.).96 | |||
| Flavonoid metabolites in rat urine and feces after oral administration of jujube seeds (traditional Chinese medicine).97 | HPLC/UV-Vis MSn (four and seven metabolites were found in feces and urine, respectively; n.q.).97 | |||
| NMR metabolite profiling of human feces after oral ingestion of grape juice.98 | 1H-NMR (108 metabolite profiles were recorded, phenolic metabolites were also found, n.q).98 | |||
| Study of phenol content in fecal water in volunteers with no dietary restriction.99 | GC-MS (major compound in fecal water was phenylacetic acid 479 μM; concentration of dietary polyphenols varied between 0.01 and 1.2 μM).99 | |||
| Study of phenol profile in human urine and fecal extracts of volunteers after consumption of polyphenol-rich diet100 | GC/TOF-MS (6 phenolic acids were identified in fecal extracts, n.q.100 | |||
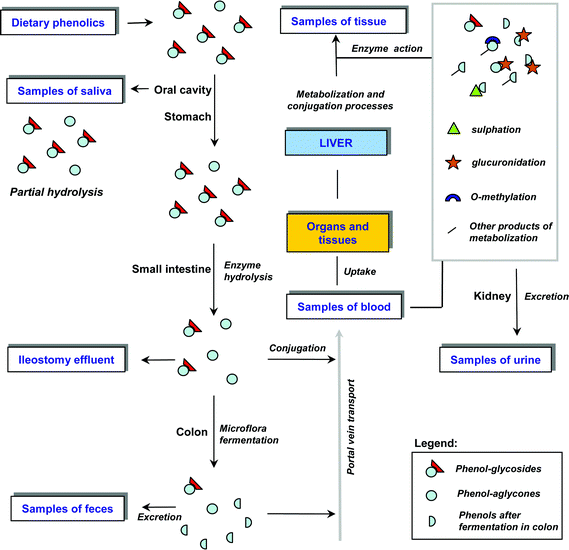 | ||
| Fig. 2 Schematic representation of different sample types in study of resorption, metabolization, and excretion of plant phenolics in mammals. For other details see section 3 and Tab. 1. | ||
4. Analysis of plant polyphenolics in clinical samples
The detection of polyphenols in clinical samples was mainly carried out in plasma and urine. Significantly fewer published results were concerned with the presence of polyphenols in organ tissues. In contrast to tissue samples, polyphenols in plasma or urine can be analysed for one individual over time and so important information can be obtained on their metabolic reactions. In terms of the methodology for the sample preparations, body fluids, in contrast to tissue samples (which need to be homogenised before analysis) present a relatively simple matrix, since 90–95% of plasma or urine is water and the remainder is comprised of inorganic ions and organic substances.35 In developing new methods of analysing metabolites in urine, the variability between various samples needs to be taken into account, since the composition of urine can change significantly on dependence on the quality and quantity of the ingested diet. Details on the stabilization of urine samples, where metabolites were examined using NMR, and suitable conditions for their storage can be found in Ref. 36.4.1. Sample preparation and hydrolysis
Basically two approaches are used in polyphenol analysis. The first examines the total polyphenol content in homogenates, in which enzymatic or chemical hydrolysis needs to be performed, during which conjugated forms are eliminated (the conjugates can form up to 95% of all polyphenols in plasma37) usually by enzyme mixture of β-D-glucuronidase and sulfatase. A sample with its conjugates removed is usually dissolved in a suitable solvent and acidified, which releases all the polyphenols that are bound to proteins. Material containing aglycones prepared in this way is usually centrifuged or filtered and the supernatant or filtrate obtained is purified or analysed directly. The second approach examines the free polyphenols or their conjugates, and so the above mentioned hydrolysis and deproteination steps are not applied. As a result, analysing the free polyphenol content is much more demanding in terms of the sensitivity of the detection equipment. Finally, polyphenols in hydrolyzed and/or non-hydrolyzed samples are often purified using a clean-up SPE procedure. SPE sorbents, usually based on reversed-phase and ion-exchange interactions have proved successful in relation to these goals.38–40 To prevent oxidation, a reducing agent such as ascorbic acid can be added to samples.41 Various approaches to the sample preparation and analysis of polyphenols are shown in Fig. 3. | ||
| Fig. 3 Simplified experimental scheme of analysis of plant phenolic compounds in clinical samples. | ||
4.2. Methods for phenol analyses in plasma and urine
The most common method for analysis plant phenols and their metabolites in body fluids is HPLC (reversed-phase columns are most often used, but more polar sorbents can also be utilised) in combination with UV-Vis DAD, ESI-MS, electrochemical or NMR analyzers.40,42,43 These methods were applied in the study of the pharmacokinetic parameters, absorption, metabolization and excretion of a whole spectrum of phenols. Simple phenolic acids,44 chalcones,45 stilbenes,46 flavonoids,47,48 flavonolignans49 and even oligomeric structures of proanthocyanidins50 have been studied (Tab. 1).For phenolic acids, HPLC was used in combination with a UV-Vis detector for the analysis of caffeic and chlorogenic acid in urine and effluents from female and male healthy ileostomy subjects.44 An ester of caffeic acid, 1,5-dicaffeoylquinic acid, and two active metabolites were analysed in the plasma of volunteers using a validated HPLC-MS/MS method.51 The metabolism of caffeic and ferulic acid was recently described in detail in rats using a method combining HPLC with a UV-Vis detector and ion-trap MS.52 Analyses were carried out on samples of plasma, urine, bile and feces purified by SPE. 19 metabolites were examined in all and their metabolic pathways were proposed and discussed. The levels of caffeic, dihydrocaffeic, ferulic, dihydroferulic and rosmarinic acids were followed in the plasma of pigs using an method connecting HPLC on-line with a system of dual-channel coulometric detector.53 For a more detailed study of the chemical properties and analysis of phenolic acids, the following review is recommended.54
Of the flavonoids, quercetin and other selected substances were analysed in the plasma of rats after the intravenous application of an extract from Ginkgo biloba using a validated HPLC-UV-Vis DAD method.55 Quercetin and its metabolites were also studied using an HPLC-MS/MS method and the profile of aglycones and conjugates contained in the plasma of volunteers during 8 h after administration was observed.37 Luteolin and apigenin, which are flavones, were also analysed.56 A validated HPLC method was used to analyse both flavones in human urine after the oral administration of tablet of Chrysanthemum morifolium extract. A range of authors have also dealt with the analysis of isoflavones. HPLC-MS methods were proposed for these analyses, and were used to examine isoflavones (daidzein and genistein) in samples of human plasma57 and other clinical material.58 In terms of more recent applications, work has been done aimed at detecting isoflavones in urine using HPLC and a coulometric detector59 or recent study where the method used to study isoflavones based on a combination of MS/MS with various efficient HPLC systems.60 Flavanones (naringenin and hesperetin) were also analysed in human plasma using an HPLC MS/MS method after administration of blood orange juice.61 It was also shown that the enantiomers of naringenin can be analysed in the biological fluids of rats or humans, where a stereospecific analysis over time was carried out with urine samples using an HPLC and UV-Vis detector.62 Anthocyanidins are another specific group of flavonoids. Among their best known members are pelargonidin, petunidin, malvidin, etc. Anthocyanins (glycosidic derivatives of anthocyanidins) found in blueberries (Vaccinium angustifolium) were analysed in a human serum that was purified with SPE. Using an HPLC method with a UV-Vis detector, 25 anthocyanins were identified in the fruits and 19 anthocyanins were determined and quantified in the serum.63 A significant number of approaches have also been developed for the analysis of catechins.64 These flavanols, abundantly present in tea, were analysed using HPLC and a coulometric detector along with theaflavins in plasma, urine, or tissue samples.65
Apart from methods aimed at monomeric polyphenols, methods were also developed to focus on the analysis of flavonolignans and oligomeric or polymeric structures of phenols. Flavonolignans such as silybin were examined in SPE extracts from human plasma66 and 125I-labelling was used to monitor silybin in the blood and selected organs of rats.67 In addition, HPLC and NMR were proposed for identifying of silybin glucuronides.49 An MS/MS method was also developed for identifying proanthocyanidins, catechin, epicatechin, dimer, and trimer in plasma samples.50 An HPLC-MS/MS method was used to study the catechins and proanthocyanidins up to trimers in rats which had been fed them in the form of a grape seed extract in their diet.68 This method was applied in the study of phenols in blood and in brain tissue.
4.3. Methods for monitoring the tissue uptake of polyphenols
When analysing tissue, the majority of developed methods were published within the framework of the distribution of polyphenols in various organs in laboratory rats and mice after their ingestion as part of their diet. In in vivo research work, polyphenols (radioactively labelled with 3H or 14C) were used and their distribution examined directly in fixated tissue using microautoradiography. In other experiments, the tissue was homogenized and the polyphenols quantified autoradiographically in the extracts obtained. Other methodological approaches were focussed on HPLC separations, where individual radioactive metabolites were identified in selected fractions (Fig. 3).This is how the distribution of [3H](-)-epigallocatechin gallate (a radioactive synthetic analogue of the polyphenol found in tea) was studied in the blood, brain, liver and other tissues of mice. In addition to classical microradiographic imaging, the radioactivity of HPLC fractions was followed off-line using a scintillation detector.69 Approaches which focus on the analysis of polyphenols without radioactive labelling have also been published. For example, epicatechin was studied in the plasma and in extracts from the brain tissue of rats, which were analysed using HPLC with a UV-Vis detector and MS/MS. Apart from epicatechins, the presence and levels of its glucuronides and sulfated or methylated forms were examined using this technique.41 Phenolic metabolites were also analysed using HPLC connected on-line with a radioactivity detector (HPLC-radiocounting) and MS/MS detector. The authors of this approach focussed on following [2-14C]quercetin-4′-glucoside. They administered a sample of this to the rats and then followed its uptake. As early as 60 min after administration, 19 metabolites exhibiting radioactivity were analysed in the intestine, plasma, liver and kidneys, which is linked to the process of their glucuronidation and formation of specific conjugates or metabolites. These conjugates were identified using MS/MS, and their highest concentrations were recorded in the intestine.70 [14C]-trans-resveratrol was also studied using radioactivity measurement.71 The total quantification of resveratrol was then conducted without differentiating between the individual metabolites for example in the intestine, liver, kidney, heart and brain.
Thanks to precise measurement of radioactive [14C]genistein, it was possible to follow the pharmacokinetic parameters of this isoflavone in rats. Knowledge was also gained with regard to the excretion of genistein, its presence in plasma and most of all its distribution in excretory organs, the respiratory system, reproductive organs, and peripheral organs.72 In addition, pharmacokinetic information on the genistein was obtained by the MS method.73 This was studied in a similar fashion in rats and was quantified in the form of aglycones in the brain, liver, thyroid gland, testes and other organs.
5. Conclusions and further research
The average intake of polyphenols in the human population approximates to 1 g day−1,74–77 while the distribution of polyphenols in body fluids and tissues is usually significantly lower due to their rapid excretion from the organism or to metabolic changes that they undergo. The overall analysis of polyphenols in body fluids (their concentration in plasma is approximately 0.5 μM after the administration of 80–100 mg of quercetin10) is influenced significantly less, due to the interaction between the analytes and the matrix, than with investigated material from tissue and organs. The concentration of polyphenols was usually in the range 30 ng to 3 μg aglycones/g tissue,10 depending on the dosage administered and a whole range of factors including the means of administration.It is to be expected that new extraction procedures, especially PSE and CO2 SFE, will be of major significance. These extraction methods have been found to be highly useful in the analysis of polyphenols in complex matrices such as plant material and their application could also contribute to an increased extraction yield in the analysis of tissues. In terms of separation methods, HPLC combined with on-line optical or MS detectors is currently king, though now also U-HPLC78 methods have been proposed, which enable analysis times to be significantly reduced.42,50,79 Two basic strategies that enable polyphenols to be detected and quantified in tissues (and also in body fluids) such as the use of radioactively labelled substances and selective analysis based on MS and NMR, could be expanded in the future by the application of immunochemical methods (these were for example elaborated in detail and utilised to study isoflavones in plasma80–82 and legumes83) and also the application of the newly introduced ionization technique DESI MS and other surface imaging mass spectrometric methods for the analysis of intact tissues.84–87 The development and application of the above methodological approaches is absolutely fundamental for a detailed understanding of the pharmacokinetic parameters and uptake of polyphenols in individual tissue types, irrespective of the fact that the level of polyphenols in tissues is not directly dependent on their current level in the bloodstream,10 which is another topic in need of detailed study.
In closing, the problems and complications connected with the analysis of polyphenols in clinical material need to be highlighted. In particular, comparing the levels of identified substances between individual studies is complex and difficult, despite the fact that many of the published methods have been validated. These differences are caused by differing methods of hydrolysis and differing approaches to handling samples before analysis, which has been covered in.48 In addition, unknown substances identified using retention times or UV-Vis absorption spectra should have been confirmed by MS because retention times of phenol glycosides are almost identical to phenol glucuronides.88 These observations will need to be taken into account in interpreting the individual studies.
List of abbreviations
HPLC – high-performance liquid chromatography, U-HPLC – ultrahigh-pressure liquid chromatography, UV-Vis DAD – ultraviolet-visible diode-array detector, MS – mass spectrometry (MS/MS – tandem MS, MSn – multi-stage MS), ESI – electrospray ionization (DESI – desorption ESI), NMR – nuclear magnetic resonance, SPE – solid-phase extraction, PSE – pressurized-solvent extraction, SFE – supercritical-fluid extraction, ELISA – enzyme-linked immunosorbent assay.Acknowledgements
This work was supported by the grant MSM 6198959216. The authors are indebted to Dr Stephen P. Hardy (Department of English and American Studies, Masaryk University, Czech Republic) for language correction.References
- M. Wink, Curr. Drug Metab., 2008, 9, 996–1009 Search PubMed.
- X. Z. Han, T. Shen and H. X. Lou, Int. J. Mol. Sci., 2007, 8, 950–988 Search PubMed.
- A. Kale, S. Gawande and S. Kotwal, Phytother. Res., 2008, 22, 567–577 CrossRef CAS.
- J. Kyle and G. Duthie, Nat. Prod. Commun., 2006, 1, 1049–1060 CAS.
- E. Middleton and C. Kandaswami, The impact of plant flavonoids on mammalian biology: implications for immunity, inflammation and cancer. In: J. B. Harborn (Ed.), The flavonoids: advances in research since, Chapman and Hall, London, UK, pp. 619–620, 1994 Search PubMed.
- A. R. Tapas, D. M. Sakarkar and R. B. Kakde, Trop. J. Pharm. Res., 2008, 7, 1089–1099 Search PubMed.
- A. Crozier, I. B. Jaganath and M. N. Clifford, Nat. Prod. Rep., 2009, 26, 1001–1043 RSC.
- J. C. Espin, M. T. Garcia-Conesa and F. A. Tomas-Barberan, Phytochemistry, 2007, 68, 2986–3008 CrossRef CAS.
- L. K. MacDonald-Wicks, L. G. Wood and M. L. Garg, J. Sci. Food Agric., 2006, 86, 2046–2056 CrossRef.
- C. Manach, A. Scalbert, C. Morand, C. Remesy and L. Jimenez, Am. J. Clin. Nutr., 2004, 79, 727–747 CAS.
- A. R. Rechner, G. Kuhnle, P. Bremner, G. P. Hubbard, K. P. Moore and C. A. Rice-Evans, Free Radical Biol. Med., 2002, 33, 220–235 CrossRef CAS.
- P. Mattila, J. Hellstrom and R. Torronen, J. Agric. Food Chem., 2006, 54, 7193–7199 CrossRef CAS.
- B. Klejdus, R. Mikelová, J. Petrlová, D. Potěšil, V. Adam, M. Stiborová, P. Hodek, J. Vacek, R. Kizek and V. Kubáň, J. Agric. Food Chem., 2005, 53, 5848–5852 CrossRef CAS.
- A. Cassidy, Int. J. Vitam. Nutr. Res., 2003, 73, 120–126 CrossRef.
- H. Cassebaum, Pharmazie., 1981, 36, 135–138 CAS.
- A. Ure, London Medical Gazette (New Series), 1841, 27(I), 73 Search PubMed.
- O. Fiehn, Study of metabolic control in plants by metabolomics, Control of Primary Metabolism in Plants (ed. W. C. Plaxton), In: Annual Plant Review, vol. 22, 2006 Search PubMed.
- J. Caldwell, In: Xenobiotic Conjugation Chemistry, G. Paulson, ACS Symposium Series, Amer. Chem. Soc., Washington, DC, 1986 Search PubMed.
- M. H. Abraham, J. Chromatogr., A, 2004, 1061, 113–114 CrossRef CAS.
- M. Tswett, Ber. Deutsch. Bot. Ges., 1906, 24, 316–323 Search PubMed.
- M. D. Armstrong, K. N. F. Shaw and P. E. Wall, J. Biol. Chem., 1956, 218, 293–303 CAS.
- T. B. Gage, C. D. Douglass and S. H. Wender, Anal. Chem., 1951, 23, 1582–1585 CrossRef CAS.
- C. H. Ice and S. H. Wender, Anal. Chem., 1952, 24, 1616–1617 CrossRef CAS.
- B. S. Patil, G. K. Jayaprakasha, K. N. C. Murthy and A. Vikram, J. Agric. Food Chem., 2009, 57, 8142–8160 CrossRef CAS.
- M. Unger, Planta Med., 2009, 75, 735–745 CrossRef CAS.
- R. M. Horowitz and B. Gentili, J. Org. Chem., 1960, 25, 2183–2187 CrossRef CAS.
- J. B. Fenn, Angew. Chem., Int. Ed., 2003, 42, 3871–3894 CrossRef CAS.
- A. Pelter, P. Stainton and M. Barber, J. Heterocycl. Chem., 1965, 2, 262 CrossRef CAS.
- B. Ternai and K. R. Markham, Tetrahedron, 1976, 32, 565–569 CrossRef CAS.
- W. B. Dunn, N. J. C. Bailey and H. E. Johnson, Analyst, 2005, 130, 606–625 RSC.
- M. Stobiecki, Phytochemistry, 2000, 54, 237–256 CrossRef CAS.
- J. K. Prasain and S. Barnes, Mol. Pharmaceutics, 2007, 4, 846–864 CrossRef CAS.
- D. S. Wishart, D. Tzur, C. Knox, R. Eisner, A. C. Guo, N. Young, D. Cheng, K. Jewell, D. Arndt, S. Sawhney, C. Fung, L. Nikolai, M. Lewis, M. A. Coutouly, I. Forsythe, P. Tang, S. Shrivastava, K. Jeroncic, P. Stothard, G. Amegbey, D. Block, D. D. Hau, J. Wagner, J. Miniaci, M. Clements, M. Gebremedhin, N. Guo, Y. Zhang, G. E. Duggan, G. D. MacInnis, A. M. Weljie, R. Dowlatabadi, F. Bamforth, D. Clive, R. Greiner, L. Li, T. Marrie, B. D. Sykes, H. J. Vogel and L. Querengesser, Nucleic Acids Res., 2007, 35, D521–D526 CrossRef CAS.
- A. J. Day, M. S. DuPont, S. Ridley, M. Rhodes, M. J. C. Rhodes, M. R. A. Morgan and G. Williamson, FEBS Lett., 1998, 436, 71–75 CrossRef CAS.
- W. C. Shiel, Webster's New World Medical Dictionary, 3rd Edition (W. C. Shiel, Ed.), Wiley Publishing Inc., 2008.
- E. J. Saude and B. D. Sykes, Metabolomics, 2007, 3, 19–27 CrossRef CAS.
- L. Wang and M. E. Morris, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2005, 821, 194–201 CrossRef CAS.
- C. D. Stalikas, J. Sep. Sci., 2007, 30, 3268–3295 CrossRef CAS.
- M. A. Rostagno, A. Villares, E. Guillamon, A. Garcia-Lafuente and J. A. Martinez, J. Chromatogr., A, 2009, 1216, 2–29 CrossRef CAS.
- J. Vacek, B. Klejdus, L. Lojková and V. Kubáň, J. Sep. Sci., 2008, 31, 2054–2067 CrossRef CAS.
- M. M. Abd El Mohsen, G. Kuhnle, A. R. Rechner, H. Schroeter, S. Rose, P. Jenner and C. A. Rice-Evans, Free Radical Biol. Med., 2002, 33, 1693–1702 CrossRef CAS.
- B. Klejdus, J. Vacek, L. Lojková, L. Benešová and V. Kubáň, J. Chromatogr., A, 2008, 1195, 52–59 CrossRef CAS.
- J. Vacek and B. Klejdus, Chromatography in the research of phenolic secondary metabolites, In: Chromatography Types, Techniques and Methods (Ed. Toma J. Quintin), Nova Science Publishers, Inc., 2010, chapter 7 Search PubMed.
- M. R. Olthof, P. C. H. Hollman and M. B. Katan, J. Nutr., 2001, 131, 66–71 CAS.
- S. C. Marks, W. Mullen, G. Borges and A. Crozier, J. Agric. Food Chem., 2009, 57, 2009–2015 CrossRef CAS.
- H. S. Lin and P. C. Ho, J. Pharm. Biomed. Anal., 2009, 49, 387–392 CrossRef CAS.
- S. B. Kim, M. J. Lee, J. I. Hong, C. Li, T. J. Smith, G. Y. Yang, D. N. Seril and C. S. Yang, Nutr. Cancer, 2000, 37, 41–48 Search PubMed.
- M. V. Martinez-Ortega, M. C. Garcia-Parrilla and A. M. Troncoso, Anal. Chim. Acta, 2004, 502, 49–55 CrossRef CAS.
- V. Křen, J. Ulrichová, P. Kosina, D. Stevenson, P. Sedmera, V. Přikrylová, P. Halada and V. Šimánek, Drug Metab. Dispos., 2000, 28, 1513–1517 CAS.
- A. Serra, A. Macia, M. P. Romero, M. J. Salvado, M. Bustos, J. Fernandez-Larrea and M. J. Motilva, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1169–1176 CrossRef CAS.
- R. L. Gu, G. F. Dou, J. Wang, J. X. Dong and Z. Y. Meng, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 852, 85–91 CrossRef CAS.
- Z. C. Zhang, M. Xu, S. F. Sun, X. Qiao, B. R. Wang, J. Han and D. A. Guo, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 871, 7–14 CrossRef CAS.
- D. Jirovský, P. Kosina, M. Myslínová, J. Stýskala, J. Ulrichová and V. Šimánek, J. Agric. Food Chem., 2007, 55, 7631–7637 CrossRef CAS.
- R. J. Robbins, J. Agric. Food Chem., 2003, 51, 2866–2887 CrossRef CAS.
- D. Q. Tang, X. X. Yin, Z. J. Zhang, Y. Y. Gao, Y. Q. Wei, Y. G. Chen and L. Han, J. Liq. Chromatogr. Relat. Technol., 2009, 32, 2065–2079.
- L. P. Li and H. D. Jiang, J. Pharm. Biomed. Anal., 2006, 41, 261–265 CrossRef CAS.
- L. Coward, M. Kirk, N. Albin and S. Barnes, Clin. Chim. Acta, 1996, 247, 121–142 CrossRef CAS.
- A. A. Franke, L. J. Custer, W. Wang and C. Y. Shi, Proc. Soc. Exp. Biol. Med., 1998, 217, 263–273 Search PubMed.
- B. Klejdus, J. Vacek, V. Adam, J. Zehnálek, R. Kizek, L. Trnková and V. Kubáň, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2004, 806, 101–111 CrossRef.
- M. I. Churchwell, N. C. Twaddle, L. R. Meeker and D. R. Doerge, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2005, 825, 134–143 CrossRef CAS.
- C. Gardana, S. Guarnieri, P. Riso, P. Simonetti and M. Porrini, Br. J. Nutr., 2007, 98, 165–172 CrossRef CAS.
- J. A. Yanez and N. M. Davies, J. Pharm. Biomed. Anal., 2005, 39, 164–169 CrossRef CAS.
- G. Mazza, C. D. Kay, T. Cottrell and B. J. Holub, J. Agric. Food Chem., 2002, 50, 7731–7737 CrossRef CAS.
- K. O. Chu, C. C. Wang, M. S. Rogers, K. W. Choy and C. P. Pang, Anal. Chim. Acta, 2004, 510, 69–76 CrossRef.
- M. J. Lee, S. Prabhu, X. F. Meng, C. Li and C. S. Yang, Anal. Biochem., 2000, 279, 164–169 CrossRef CAS.
- P. Kosina and J. Bartek, Chem. Listy, 2000, 94, 1115–1117 CAS.
- N. Skottová, Z. Svagera, R. Večeřa, K. Urbánek, A. Jegorov and V. Šimánek, Pharmacol. Res., 2001, 44, 247–253 CrossRef CAS.
- J. K. Prasain, N. Peng, Y. Y. Dai, R. Moore, A. Arabshahi, L. Wilson, S. Barnes, J. M. Wyss, H. Kim and R. L. Watts, Phytomedicine, 2009, 16, 233–243 CrossRef CAS.
- M. Suganuma, S. Okabe, M. Oniyama, Y. Tada, H. Ito and H. Fujiki, Carcinogenesis, 1998, 19, 1771–1776 CrossRef CAS.
- W. Mullen, B. A. Graf, S. T. Caldwell, R. C. Hartley, G. G. Duthie, C. A. Edwards, M. E. J. Lean and A. Crozier, J. Agric. Food Chem., 2002, 50, 6902–6909 CrossRef CAS.
- X. Vitrac, A. Desmouliere, B. Brouillaud, S. Krisa, G. Deffieux, N. Barthe, J. Rosenbaum and J. M. Merillon, Life Sci., 2003, 72, 2219–2233 CrossRef CAS.
- N. G. Coldham and M. J. Sauer, Toxicol. Appl. Pharmacol., 2000, 164, 206–215 CrossRef CAS.
- H. C. Chang, M. I. Churchwell, K. B. Delclos, R. R. Newbold and D. R. Doerge, J. Nutr., 2000, 130, 1963–1970 CAS.
- M. G. L. Hertog, P. C. H. Hollman, M. B. Katan and D. Kromhout, Nutr. Cancer, 1993, 20, 21–29 Search PubMed.
- U. Justesen, P. Knuthsen and T. Leth, Cancer Lett., 1997, 114, 165–167 CrossRef CAS.
- Y. Kikuchi, Y. Shimamura, M. Hirokado, K. Yasuda and M. Nishijima, J. Food Hyg. Soc. Jpn., 2001, 42, 122–127 Search PubMed.
- L. Sampson, E. Rimm, P. C. H. Hollman, J. H. M. de Vries and M. B. Katan, J. Am. Diet. Assoc., 2002, 102, 1414–1420 CrossRef.
- N. Wu and A. M. Clausen, J. Sep. Sci., 2007, 30, 1167–1182 CrossRef CAS.
- B. Klejdus, J. Vacek, L. Benešová, J. Kopecký, O. Lapčík and V. Kubáň, Anal. Bioanal. Chem., 2007, 389, 2277–2285 CrossRef CAS.
- O. Lapčík, R. Hampl, N. Al-Maharik, A. Salakka, K. Wähälä and H. Adlercreutz, Steroids, 1997, 62, 315–320 CrossRef CAS.
- G. J. Wang, O. Lapčík, R. Hampl, M. Uehara, N. Al-Maharik, K. Stumpf, H. Mikola, K. Wähälä and H. Adlercreutz, Steroids, 2000, 65, 339–348 CrossRef CAS.
- J. T. He, Z. H. Shi, J. Yan, M. P. Zhao, Z. Q. Guo and W. B. Chang, Talanta, 2005, 65, 621–626 CrossRef CAS.
- O. Lapčík, M. Hill, I. Černý, J. Lachman, N. Al-Maharik, H. Adlercreutz and R. Hampl, Plant Sci., 1999, 148, 111–119 CrossRef CAS.
- Z. Takats, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef CAS.
- J. Vacek, Klin. Biochem. Metab., 2006, 14, 194–195 Search PubMed.
- V. Vidová, M. Volný, K. Lemr and V. Havlíček, Collect. Czech. Chem. Commun., 2009, 74, 1101–1116 CrossRef CAS.
- J. M. Wiseman, D. R. Ifa, Y. X. Zhu, C. B. Kissinger, N. E. Manicke, P. T. Kissinger and R. G. Cooks, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18120–18125 CrossRef.
- J. P. E. Spencer, M. Abd El Mohsen and A. M. Minihane, Curr. Top. Nut. Res., 2006, 4, 187–203 Search PubMed.
- T. Walle, A. M. Browning, L. L. Steed, S. G. Reed and U. K. Walle, J. Nutr., 2005, 135, 48–52 CAS.
- C. S. Yang, M. J. Lee and L. S. Chen, Cancer Epidemiol. Biomarkers Prev., 1999, 8, 83–89 CAS.
- C. Dinnella, A. Recchia, G. Fia, M. Bertuccioli and E. Monteleone, Chem. Senses, 2009, 34, 295–304 CrossRef CAS.
- K. Kahle, M. Kraus, W. Scheppach and E. Richling, Mol. Nutr. Food Res., 2005, 49, 1143–1150 CrossRef CAS.
- M. N. Vissers, P. L. Zock, A. J. C. Roodenburg, R. Leenen and M. B. Katan, J. Nutr., 2002, 132, 409–417 CAS.
- T. Walle, Y. Otake, U. K. Walle and F. A. Wilson, J. Nutr., 2000, 130, 2658–2661 CAS.
- J. Slanina and E. Táborská, Chem. Listy, 2004, 98, 239–245 CAS.
- C. Li, M. Homma and K. Oka, J. Chromatogr., B: Biomed. Sci. Appl., 1997, 693, 191–198 CrossRef CAS.
- K. D. Bao, P. Li, L. W. Qi, H. J. Li, L. Yi, W. Wang and Y. Q. Wang, Chem. Pharm. Bull., 2009, 57, 144–148 CrossRef CAS.
- D. M. Jacobs, N. Deltimple, E. van Velzen, F. A. van Dorsten, M. Bingham, E. E. Vaughan and J. van Duynhoven, NMR Biomed., 2008, 21, 615–626 CrossRef CAS.
- A. M. Jenner, J. Rafter and B. Halliwell, Free Radical Biol. Med., 2005, 38, 763–772 CrossRef CAS.
- C. H. Grun, F. A. van Dorsten, D. M. Jacobs, M. Le Belleguic, E. J. J. van Velzen, M. O. Bingham, H. G. Janssen and J. P. M. van Duynhoven, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 871, 212–219 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |