Spot test of urinary protein using Erythrosin B and a membrane film
Emiko
Kaneko
*a,
Hiroko
Yasuda
b,
Asami
Higurashi
b and
Hajime
Yoshimura
b
aNew Industry Creation Hatchery Center, Tohoku University, Sendai 980-8579, Japan. E-mail: emiko@sda.att.ne.jp
bShino-Test Co., 2-29-14 Oonodai Sagamihara, Kanagawa 229-0011, Japan
First published on 20th May 2010
Abstract
Point-of-care testing is currently one of the subjects of growing interest in analytical chemistry. Elevated levels of urinary protein imply renal failure, which is one of the world's biggest public health problems. In spite of the urgent necessity for a screening test of protein in urine, there are no reports of a simple yet sensitive method for its detection. In this study, we developed a new visual method, using Erythrosin B and a cellulose acetate membrane film as the substrate for a new spot test of urinary protein in the presence of poly(ethylene glycol) (PEG). The noteworthy point of our work is that when a drop of dye–protein solution containing PEG is set on a membrane film, a red ring-shaped stain of the dye-bound protein is formed on the film surface. PEG plays a significant role in eliminating the reagent blank, thus providing a clear contrast. Measurements taken using a dynamic light scattering particle size analyzer indicated that the underlying mechanism of this contrast is brought about by the different sizes of the excess dye and dye–protein particles. The visual detection limit is 0.5 mg dm−3 for human serum albumin (HSA), the main protein in urine. Our visual method is sufficiently sensitive to detect urinary protein even for healthy subjects, providing a higher sensitivity than test strips by a factor of 60–200. When 0.15 cm3 of urine is used to prepare 10 cm3 of sample solution, the practical detection threshold is 30 mg dm−3 in urine using a 67× dilution factor. The proposed method will be useful as a simple, rapid, and cost-effective screening test for the diagnosis of renal failure at an early stage.
Introduction
The development of various simple, inexpensive analytical methods which do not require sophisticated instruments, and do not create hazardous waste is needed in the environmental and bioanalytical fields. The objective of this study is to develop a simple yet sensitive test for the detection of urinary protein for the non-invasive diagnosis of renal failure, and also for the clinical monitoring of patients with renal problems. It is a well known fact that diabetes mellitus is often associated with renal failure. In particular, type 2 (adult-onset) diabetes mellitus exacts a huge toll in public health expenditure as well as in human suffering. The number of cases worldwide is estimated at more than 150 million.1 A number of recommendations have been published on the need to prevent diabetic nephropathy, and microalbuminuria has long been accepted as an early indicator.2–4To date, several methods have been used for the detection of urinary protein. A comparison of a number of different techniques was outlined by Sperlingova et al.5 In spite of incremental improvements in the boiling method, the sulfosalicylic acid method, spectrophotometry based on a dye-binding reaction, and the Lowry method, the application of all these methods for early diagnosis is limited due to their lack of sensitivity. Test strips impregnated with a pH indicator dye are the most frequently used method for the mass screening of urinary protein. However, like most other dye-binding assays, a significant drawback of the dip strip test is its insufficient sensitivity. Depending on the type of strip used, the detection threshold is in the range of 30–100 mg dm−3. In addition to the lack of sensitivity, test strips are far from perfect because of matrix interference. Although the threshold of urinary protein concentrations for early diagnosis is still under discussion, a rapid test to detect protein at concentrations of less than 30 mg dm−3 is needed. There are several approaches based on a dot-blotting technique using silver,6 and a filtration-based staining method using a membrane filter and Coomassie Brilliant Blue (CBB).7 However, these methods are both time consuming and tedious. Using an immunoassay also presents limitation due to the time required to carry out the color development and washing steps, and the high cost involved. Since Pasternack et al. published articles on their resonance light scattering technique for the detection of chromophore aggregates8 there have been numerous publications on the analytical applications of dispersed micro-particles for protein and other trace elements.9 Recently, Laiwattanapaisal et al. reported a simple and sensitive sequential injection analysis of microalbuminuria with fluorescent detection using Albumin Blue-580.10 However, neither the light scattering nor the fluorescent detection methods are applicable to Point-of-care testing. More sensitive screening tests are required to pre-select the samples for early diagnosis, requiring neither sophisticated instrumentation nor laboratory skill.
In our previous work, we have reported a sensitive spectrophotometric method for human serum albumin (HSA) using Erythrosin B and TritonX-100.11 Earlier work by Soedjak showed that Erythrosin B is excellent for the spectrophotometric determination of protein in aqueous solution at levels as low as 2 mg dm−3, using a dye-binding reaction at 90–95 °C.12 The spectrophotometry was markedly improved in our previous work, offering a detection limit of 0.06 mg dm−3 at room temperature.11
Here we describe the new screening test we developed for urinary protein. The rapid visual detection of trace protein was achieved by using Erythrosin B and a membrane film as the substrate for a new spot test in the presence of poly(ethylene glycol) (PEG). Although there were numerous publications on the conventional spot test in the twentieth century since it was developed by Feigl and Anger,13 the application of these methods is limited due to insufficient sensitivity. There has never been a spot test for protein determination reported. The noteworthy point of our method is the formation of a sharp red ring of dye-bound protein on a cellulose acetate membrane film, while eliminating the reagent blank using PEG to provide a clear contrast. The method reported here enables a highly sensitive and cost-effective protein measurement for mass screening despite being composed of simple and easy operations.
Experimental
Materials
A dye solution containing 6 × 10−5 mol dm−3 Erythrosin B (KANTO CHEMICAL CO., INC.) and 1.2% poly(ethylene glycol) 10000 (PEG 10000, Fluka) was prepared by dissolving the reagents in water and stored in an amber bottle to protect the dye from degradation from light exposure. The pH buffer solution used was 1 mol dm−3 citric acid–KOH (pH 3.5). Solutions of HSA were prepared by dissolving the HSA reagent (SIGMA) in water. The cellulose acetate membrane film (pore size: 3 μm) was purchased from ADVANTEC. For cross-validation, the Micro TP-test WAKO (Wako Pure Chemical Industries) was employed for spectrophotometric measurements based on the pyrogallol red–molybdenum (PR–Mo) method.14 All other reagents used were of guaranteed reagent grade.Apparatus
Spectrophotometric measurements were made using a DV-800 spectrophotometer (Beckman Coulter, Inc.). Particle size measurements were carried out using a dynamic light scattering particle size analyzer LB 550 (HORIBA, Ltd.). The urine samples were also analyzed using an automatic analyzer 7170S (Hitachi Ltd.) with the PR–Mo kit described in the Materials section.Typical procedure
A typical procedure is as follows: a sample solution of 0.15 cm3 is pipetted into a 25 cm3 plastic bottle like those used for eyedrops. The following solutions are added to the sample: 8.35 cm3 of the dye solution containing 6 × 10−5 mol dm−3 Erythrosin B and 1.2% PEG 10000 and 1.5 cm3 of the pH buffer solution (pH 3.5). After letting the solution stand for 5 min, a drop of the mixture (approximately 0.02 cm3) is set on a membrane film. Then a visual determination of the resulting stain is carried out.Results and discussion
Characteristics of Erythrosin B
Erythrosin B (HA−, Scheme 1) shows an absorption maximum at 535 nm in aqueous solution. The acid dissociation constants of Erythrosin B was determined as 3.20 for pKa1 and approximately 6 for pKa2 from the spectrophotometric data shown in Fig. 1a. In solutions with pH values below 3, protonation takes place, yielding precipitation of the neutral dye, H2A, at concentrations higher than 2 × 10−6 mol dm−3. | ||
| Scheme 1 Equilibrium of Erythrosin B species. | ||
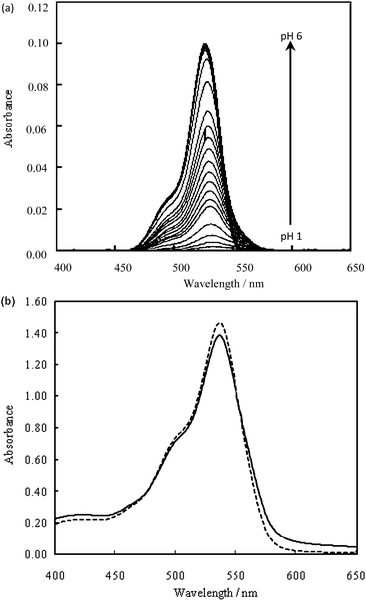 | ||
| Fig. 1 Absorption spectra of Erythrosin B in water at different pH values (a), and effect of HSA on the absorption spectrum of Erythrosin B (b). (a) Erythrosin B: 1.2 × 10−6 mol dm−3; temperature: 20 °C; ionic strength: 0.1 (Na2SO4); (b) Erythrosin B: 4.2 × 10−5 mol dm−3; pH: 3.5; PEG 10000: 1%; dashed line: reagent blank; solid line: with 0.6 mg dm−3 HSA. | ||
The absorption spectra for Erythrosin B in solutions with pH values of 3.5 are shown in Fig. 1b together with the typical spectrum in the presence of HSA. The protein-bound dye also shows an absorption maximum at 535 nm under analytical conditions. We found that the slight spectral change for HSA shown in Fig. 1b is markedly enhanced, and the reagent blank is eliminated in our spot test, as described below.
Optimization of spot test conditions
The analytical procedure for the HSA test was optimized with respect to the selection of the substrate, pH, reaction time, and solution compositions. Interactions of the dye–protein solutions with several commercially available membrane films have been investigated. Among them, the cellulose acetate membrane film described in the Experimental section was chosen as a primer since it is the best membrane film for visual detection testing, providing a clear ring formation of the dye-bound protein, as shown in Fig. 2. A reaction time of 5 min at room temperature was chosen for the clear ring formation prior to the spot test. When a drop of sample solution is set on the film, the solution is absorbed by the film, creating a ring of the dye-bound protein with a diameter of approximately 8 mm on the film surface. The concentrations of HSA in the 10 cm3 sample solutions and the visual results (−, +, ++) are given in Fig. 2 under the resulting stains. The stain with a red ring (perimeter) indicates an HSA level of more than 0.5 mg dm−3. Conversely, no red ring is observed for the faint pink stains when the HSA level is less than 0.3 mg dm−3. The intensity of the red ring increases with increasing HSA concentration in the range of 0.3–0.6 mg dm−3, and remains constant at more than 0.6 mg dm−3 HSA. | ||
| Fig. 2 Photographs of the spots for detecting HSA. Membrane filter: cellulose acetate (pore size: 3.0 μm); standing time: 5 min; spotting volume: 0.02 cm3; the other conditions are the same as those in Fig. 1(b); the HSA concentrations are given as the values in the final solutions of 10 cm3. | ||
The stains of the dye-bound protein are strongly dependent on pH. It is well recognized that an anionic dye binds to the positively charged site of a protein molecule under acidic conditions through electrostatic and hydrophobic interactions. At pH values higher than 4, no red ring forms, indicating that the dye does not associate with protein at higher pH values. At pH values lower than 3.2, the intensity of the red stain is vivid for the reagent blank. Based on these data, a pH value of 3.5 was adopted.
No clear contrast of the stains shown in Fig. 2 was obtained without PEG 10000 in the sample solution. In this work, PEG 10000 plays a significant role in eliminating the red color of the stains for the reagent blank, affecting the particle size as described below. In the presence of PEG, the excess dye is absorbed through to the reverse side of the membrane film, leaving a very faint pinkish stain on the surface of the film. Without the use of PEG, the stain of the reagent blank is a vivid red, making it impossible to detect the dye-bound protein. Note that the naked eye is remarkably sensitive, able to detect concentration levels as low as 0.5 mg dm−3 HSA.
Particle size distribution
The particle formation of dye-bound HSA was investigated using a dynamic light scattering particle size analyzer. Fig. 3a and 3b show that PEG has a significant effect on the particle size distribution. The addition of PEG resulted in a decrease in the particle sizes of the free dye from 100–1000 nm to lower than 200 nm (Fig. 3b). However, the particle sizes of the dye-bound HSA were 200–2000 nm in the presence of 0.6 mg dm−3 HSA under analytical conditions (Fig. 3a(D)). Fig. 3c shows that the sizes of the dye-bound HSA rapidly grow to 600 nm in size on average in the first five minutes, and remain almost constant from 7 to 12 min. No time dependence was observed for the reagent blank (figure not shown). | ||
| Fig. 3 Particle size distribution and passage integration of Erythrosin B–HSA (a), the reagent blank with 1% PEG 10000 and without PEG (b), and the effect of standing time on the particle size of Erythrosin B–HSA (c). Erythrosin B: 4.2 × 10−5 mol dm−3; pH: 3.5; PEG 10000: 1%; A: reagent blank; B: 0.3 mg dm−3 HSA; C: 0.5 mg dm−3 HSA; D: 0.6 mg dm−3 HSA. | ||
Consequently, the effect of PEG provides a clear contrast between the reagent blank and the dye-bound HSA particles clearly visible in the red ring on the membrane film. Based on these results, the underlying mechanism of the red ring formation on the membrane film at HSA concentrations of higher than 0.5 mg dm−3 is explained by the aggregation of the dye–HSA. When a drop of the sample solution is set on a cellulose acetate membrane film, the solution spreads through the capillaries of the film. Then, the aggregate is trapped by the film at the circumference and a red ring-shaped stain of the coagulated Erythrosin B–protein is formed on the film surface.
Sensitivity
The visual detection limit is a concentration of 0.5 mg dm−3 of HSA in the final volume of 10 dm3 (Fig. 2). When 0.15 cm3 of urine is used to prepare 10 cm3 of sample solution, the practical detection threshold is 30 mg dm−3 in urine using a 67× dilution factor. The concentration of HSA can be determined by visual comparison with a previously prepared standard series of 0.3, 0.5, and 0.6 mg dm−3. One of the advantages of the spot test reported here is that it can be applied to the visual screening of increased urinary protein at relatively low levels.Reported values of HSA in normal urine are less than 30 mg dm−3,2 while the HSA concentration in the early morning urine of patients with renal failure increases to 30–300 mg dm−3.3 Our visual method is sufficiently sensitive to detect HSA in urine even for healthy subjects. The sensitivity of our test is superior to conventional spectrophotometric methods by a factor of 4–20 and to test strips by a factor of 60–200, depending on the test.
Selectivity
The effect of foreign substances, such as creatinine, creatine, urea, uric acid, and hippuric acid, sodium chloride, potassium chloride and ascorbic acid on HSA detection was tested. None of these substances had any significant effect. The high sensitivity of the proposed method provides the following practical advantages: only a very small amount (0.15 cm3) of urine sample is required, which makes it much easier to use in the clinical field, and the high dilution factor (×67) effectively removes the matrix interferences. It should be pointed out that the dye-binding method will not work with undiluted urine concentrate, and will give a false-positive or -negative result due to the complicated matrix of urine.The interaction of Erythrosin B and several proteins has been investigated using spectrophotometry.11 It was observed that transferrin, immunoglobulin G (IgG), β2-microglobulin and Bence Jones protein increased the absorbance signal to an appreciable extent (30–90%, compared to HSA). These proteins also create a red ring in this test. Since there is hardly any excretion of these proteins in normal urine, a positive result caused by these proteins is also an indication of renal failure or other sickness.
These results indicate that this method is essentially free from urinary matrix interference and would be suitable as a screening test for renal failure.
Application
Having established suitable conditions to visually detect urinary protein, the method was applied to 73 urine samples from healthy subjects. The spot tests of 16 samples from 73 healthy subjects tested showed negative results using this method with a detection threshold of 30 mg dm−3 (Fig. 4a). Although there is no consensus regarding the optimal threshold of urinary protein for early diagnosis, the high sensitivity (down to 0.5 mg dm−3) of this method allows the detection threshold to be significantly lowered by decreasing the dilution factor of urine when a lower ON/OFF threshold is required. | ||
| Fig. 4 Analytical result of healthy subjects (a), and comparison to PR–Mo spectrophotometry (b). Dashed line: detection limit of the PR–Mo method (37 mg dm−3). The urine samples were collected from 73 healthy volunteers (age: 28–60). The spot test conditions are the same as those in Fig. 2. The histogram in Fig. 4b shows the number of the subjects whose protein was not detectable when using the PR–Mo method. | ||
Although it is well understood that there are significant differences between the protein values depending on the analytical method employed, all the urine samples were also analyzed by the PR–Mo method, one of the most frequently used methods for the determination of protein. However, the PR–Mo spectrophotometry lacks the required sensitivity for the measurements of urinary protein of healthy subjects. The detection limit of the PR–Mo method using the test kit described in the Materials section is reportedly 37 mg dm−3. The values measured using the PR–Mo kit and an automatic analyzer were summarized in Fig. 4b together with the results of the spot test. The protein concentrations of 49 samples from 73 healthy subjects, however, were not detectable when using the PR–Mo method (the histogram in Fig. 4b) because of the poor sensitivity of the conventional method. Among the 22 subjects who showed ++ (higher than 40 mg dm−3) results in the spot test, only the 15 samples which showed protein concentrations above 38 mg dm−3 were detected using the PR–Mo assay.
Conclusion
The method reported here enables a highly sensitive and cost-effective protein measurement even though it is composed of simple and easy operations, requiring neither complex apparatus nor skilled laboratory support. We believe that the proposed method can be used both for the long awaited need for the mass-population screening of renal failure and diabetic nephropathy, and, also for the daily monitoring of patients.Acknowledgements
The authors wish to acknowledge Mr Tetsuya Anada of Shino-Test Co. for his technical advice for this work. This study was supported by Japan Science and Technology Agency.References
- J. Diamond, Nature, 2003, 423, 599 CrossRef CAS.
- E. Mogensen, New Engl. J. Med., 1984, 310, 356.
- C. E. Mogensen, W. F. Keane, P. H. Bennett, G. Jerums, H.-H. Parving, P. Passa, M. W. Steffes, G. E. Striker and G. C. Viberti, Lancet, 1995, 346, 1080 CrossRef CAS.
- B. T. Doumas and T. Peters, Clin. Chim. Acta, 1997, 258, 3 CrossRef CAS.
- I. Sperlingova, L. Dabrowska, M. Tichy and J. Kucera, Fresenius J. Anal. Chem., 1998, 361, 756 CrossRef CAS.
- K. Matsuda, N. Hiratsuka, Y. Kurihara and K. Shiba, J. Clin. Lab. Anal., 2001, 15, 171 CrossRef CAS.
- D. Acharya, D. Saha, D. Roy, P. Jain and T. K. Dhar, Anal. Biochem., 2008, 379, 121 CrossRef CAS.
- R. F. Pasternack, C. Bustamante, P. J. Collings, A. Giannento and E. Gibbs, J. Am. Chem. Soc., 1993, 115, 5393 CrossRef CAS; R. F. Pasternack and K. F. Schaefer, Inorg. Chem., 1994, 33, 2062 CrossRef CAS; R. F. Pasternack and P. J. Collongs, Science, 1995, 269, 935 CrossRef CAS; J. Parkash, J. H. Robblee, J. Agnew, E. Gibbs, P. Collings, R. F. Pasternack and J. C. Paula, Biophys. J., 1998, 74, 2089 CrossRef CAS.
- C. Q. Ma, K. A. Li and S. Y. Tong, Anal. Biochem., 1996, 239, 86 CrossRef CAS; C. Z. Huang, K. A. Li and S. Y. Tong, Anal. Chem., 1996, 68, 2259 CrossRef CAS; Z. Chen, W. Ding, F. Ren, J. Liu and Y. Liang, Anal. Chim. Acta, 2005, 550, 204 CrossRef CAS; H. Zhong, J. J. Xu and H. Y. Chen, Talanta, 2005, 67, 749 CrossRef CAS; Z. L. Jiang, S. J. Sun, C. Y. Kang, X. Lu and J. Lan, Anal. Bioanal. Chem., 2005, 381, 896 CrossRef CAS.
- W. Laiwattanapaisal, U. Kunanuvat, W. Intharachuti, C. Chinvongamorn, S. Hannongbua and O. Chailapakul, Talanta, 2009, 79, 1104 CrossRef CAS.
- J. Isoe and E. Kaneko, Chem. Lett., 2006, 35, 922 CrossRef CAS.
- H. S. Soedjak, Anal. Biochem., 1994, 220, 142 CrossRef CAS.
- F. Feigl and V. Anger, Spot Test in Inorganic Analysis, Elsevier, Amsterdam, 1972 Search PubMed.
- Y. Fujita, I. Mori and S. Kitano, Bunseki Kagaku, 1983, 32, E379–E386.
| This journal is © The Royal Society of Chemistry 2010 |