
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu-catalyzed carboboration of acetylene with Michael acceptors†
Tairan
Cheng
,
Boxiang
Liu
,
Rui
Wu
and
Shifa
Zhu
 *
*
Key Lab of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: zhusf@scut.edu.cn
First published on 8th June 2022
Abstract
A copper-catalyzed three-component carboboration of acetylene with B2Pin2 and Michael acceptors is reported. In this reaction, a cheap and abundant C2 chemical feedstock, acetylene, was used as a starting material to afford cis-alkenyl boronates bearing a homoallylic carbonyl group. The reaction was robust and could be reliably performed on the molar scale. Furthermore, the resulting cis-alkenyl boronates could be converted to diverse functionalized molecules with ease.
Copper-catalyzed syn-1,2-borofunctionalization of alkynes has become a powerful and practical strategy for the installation of both boron and other functional groups across the C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond in excellent regio- and stereoselectivity.1 The general mechanism is that the borofunctionalization is initiated by the syn-addition of nucleophilic Cu-Bpin across the CC bond to generate the key intermediate species borylated alkenyl copper, followed by the interception of electrophiles to produce multi-substituted alkenyl boronates (Scheme 1a). So far, different electrophiles, such as H+, halides (including alkyl, allyl, aryl and alkynyl halides) and CO2, have been successfully and efficiently employed to intercept the putative borylated alkenyl copper.2–4 In sharp contrast, as excellent electrophiles, Michael acceptors (acrylate, vinyl ketone, acrylonitrile etc.) have rarely been employed to capture the in situ generated borylated alkenyl copper intermediate. This is not because Michael acceptors are not capable of reacting with the nucleophilic alkenyl copper, but because the electron-deficient Michael acceptors are typically more reactive than the electron-rich alkynes toward the borylcupration of Cu-Bpin species5 (Scheme 1b). To circumvent this challenge, Lin and Tian reported a Cu-catalyzed asymmetric borylative cyclization of cyclohexadienone-containing 1,6-enynes through an intramolecular Michael addition.6 In this reaction, the authors smartly leveraged the steric hindrance from the quaternary carbon of C1 to suppress the undesired borylcupration of cyclohexadienone (Scheme 1c). Recently, by using the same strategy, Carretero and Mauleón also reported an intramolecular borylative cyclization of 1,6-enynes with a β,β-disubstituted acrylate fragment to provide densely functionalized pyrrolidines.7 Despite these elegant studies, due to the competition between two different borylcupration pathways, copper-catalyzed carboboration of alkynes through intermolecular Michael addition is still challenging and remains unknown.
C bond in excellent regio- and stereoselectivity.1 The general mechanism is that the borofunctionalization is initiated by the syn-addition of nucleophilic Cu-Bpin across the CC bond to generate the key intermediate species borylated alkenyl copper, followed by the interception of electrophiles to produce multi-substituted alkenyl boronates (Scheme 1a). So far, different electrophiles, such as H+, halides (including alkyl, allyl, aryl and alkynyl halides) and CO2, have been successfully and efficiently employed to intercept the putative borylated alkenyl copper.2–4 In sharp contrast, as excellent electrophiles, Michael acceptors (acrylate, vinyl ketone, acrylonitrile etc.) have rarely been employed to capture the in situ generated borylated alkenyl copper intermediate. This is not because Michael acceptors are not capable of reacting with the nucleophilic alkenyl copper, but because the electron-deficient Michael acceptors are typically more reactive than the electron-rich alkynes toward the borylcupration of Cu-Bpin species5 (Scheme 1b). To circumvent this challenge, Lin and Tian reported a Cu-catalyzed asymmetric borylative cyclization of cyclohexadienone-containing 1,6-enynes through an intramolecular Michael addition.6 In this reaction, the authors smartly leveraged the steric hindrance from the quaternary carbon of C1 to suppress the undesired borylcupration of cyclohexadienone (Scheme 1c). Recently, by using the same strategy, Carretero and Mauleón also reported an intramolecular borylative cyclization of 1,6-enynes with a β,β-disubstituted acrylate fragment to provide densely functionalized pyrrolidines.7 Despite these elegant studies, due to the competition between two different borylcupration pathways, copper-catalyzed carboboration of alkynes through intermolecular Michael addition is still challenging and remains unknown.
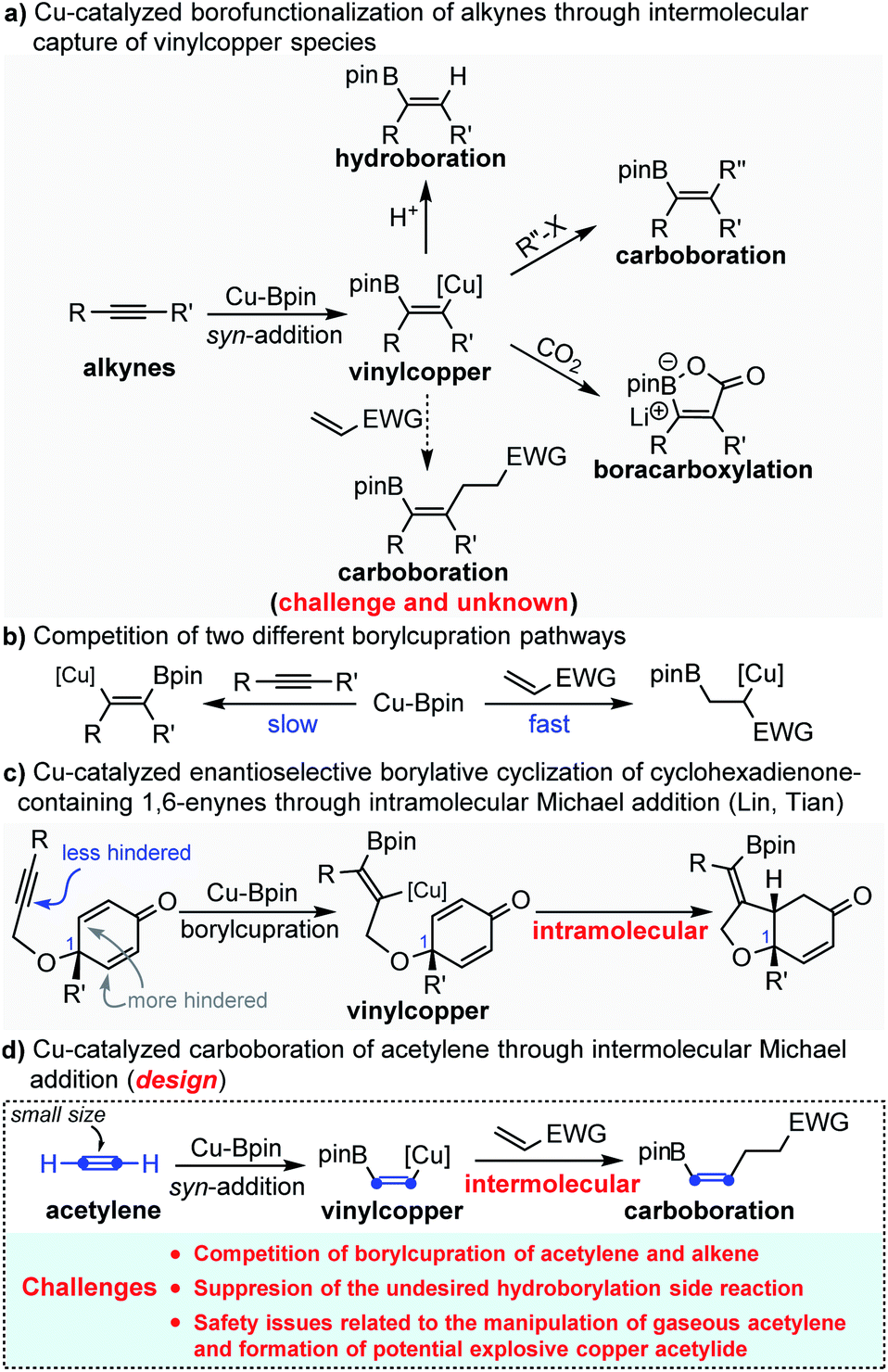 | ||
| Scheme 1 Cu-catalyzed borofunctionalization of alkynes. | ||
Acetylene, with the molecular formula C2H2, is the simplest and smallest alkyne.8 Due to its structural simplicity and high reactivity, acetylene represents a unique C2 alkenyl building block for organic synthesis through addition of its triple bond.9 Highly industrially important vinyl-containing monomers, such as vinyl ether,10 vinyl amine,11 vinyl chloride,12 acrylic acid and its derivatives,13 are being synthesized in millions of tons per year globally. However, in fine chemistry, catalytic protocols directly incorporating acetylene into high value-added chemicals are limited. In fact, while phenylacetylene acts as a model substrate in many state-of-the-art catalytic systems concerning alkyne transformations, acetylene is usually neglected in the substrate scope studies due to its gaseous nature and explosive hazard.9i,h This obscures the true reactivity of acetylene and makes the acetylene chemistry lag behind in the progress of modern catalytic alkyne chemistry.
Inspired by the elegant studies of Lin, Tian, Carretero and Mauleón, in which a steric hindered Michael acceptor was used to suppress the undesired conjugate borylation of Cu-Bpin, we envisioned that the small size of acetylene might also enable the borylcupration of the CC bond to outcompete the borylcupration of the Michael acceptor, thus allowing the Cu-catalyzed carboboration of acetylene through the challenging intermolecular Michael addition (Scheme 1d). However, the application of acetylene in the copper-catalyzed carboboration reaction through intermolecular Michael addition might encounter the following potential challenges: (1) competition between two different borylcupration processes; (2) suppression of the undesired hydroborylation side reaction; (3) safety issues related to the manipulation of acetylene gas and formation of the potentially explosive copper acetylide.
With this above design in mind, we initially investigated the competition of the borylcupration reaction among different types of alkynes. As shown in Scheme 2, when equal amounts of acetylene, phenylacetylene and 1-pentyne were subjected to the typical borylcupration catalytic system with 1.0 equivalent of B2pin2 as the limiting reagent and IMesCuCl as the catalyst, an unexpected reaction selectivity was observed. The hydroborylation product of acetylene (A1) was observed in 62% yield, which is about 8 times higher than that of phenylacetylene (A2). In addition, no corresponding hydroborylation product of 1-pentyne (A3) was detected by NMR. The competition results obviously indicated that the borylcupration of acetylene is much faster than that of both aryl alkynes and alkyl alkynes. More importantly, by comparison of the hydroborylation of acetylene and 1-pentyne, we can draw the conclusion that the steric hindrance of the substituent does have a significant impact on the borylcupration of the CC bond by taking into account that both of them are unactivated alkynes.
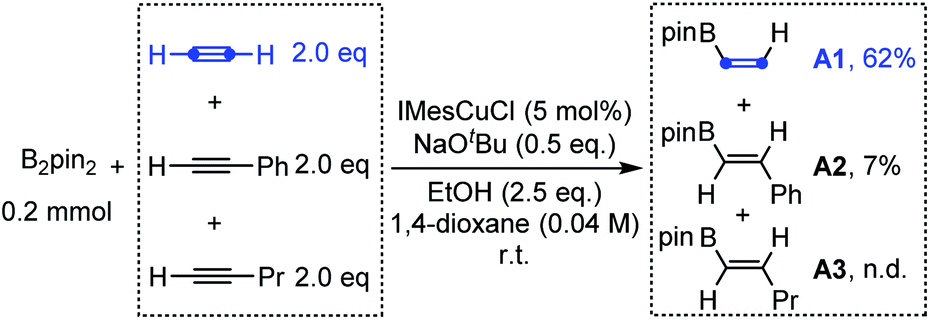 | ||
| Scheme 2 The competition of borylcupration among different alkynes. | ||
Encouraged by the above results, we further investigated Cu-catalyzed three-component carboboration of different alkynes in the presence of B2pin2 and butyl acrylate 1a with IMesCuCl as the catalyst and NaOtBu as the base. As shown in Table 1, when the substituted alkynes, phenylacetylene and 1-pentyne, were used as substrates, the desired three-component carboboration product 2 could not be detected (entries 1–4), even with a large excess of alkynes (12.5 equivalents, entries 2 and 4). The hydroborylation reactions of alkynes and butyl acrylate always dominated, giving the corresponding products A and B. However, the distribution of A and B clearly indicated that the hydroborylation of electron-deficient butyl acrylate (Michael acceptor) is much faster than that of substituted alkynes, which echoes their borylcupration reactivities (Scheme 1b). In sharp contrast, the desired three-component carboboration product 2a could be successfully generated when acetylene was applied as the alkyne component (entries 5 and 6). The carboboration product 2a could be produced in 38% yield even with 1.0 equivalent of acetylene, which is equal to the yield of hydroborylation product B (entry 5). When the amount of acetylene was further increased up to 12.5 equivalents (the gaseous acetylene was supplied with a balloon), the hydroborylation of butyl acrylate was completely suppressed with no byproduct B detected, furnishing the desired carboboration product 2a in 62% yield (entry 6). Taken together, the application of acetylene, due to its small size, could enable the realization of the syn-1,2-carboboration of the CC bond through an intermolecular Michael addition, producing the highly useful alkenyl boronates.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | x | A | B | 2 |
| a The reaction was conducted with a balloon containing acetylene; the saturated solution of acetylene in 1,4-dioxane was about 0.50 M (about 12.5 eq.). | |||||
| 1 | Ph | 1.0 | 14% | 72% | n.d. |
| 2 | 12.5 | 34% | 40% | n.d. | |
| 3 | Pr | 1.0 | n.d. | 65% | n.d. |
| 4 | 12.5 | 8% | 65% | n.d. | |
| 5 | H | 1.0 | 9% | 38% | 2a (38%) |
| 6a | 12.5 | 22% | n.d. | 2a (62%) | |
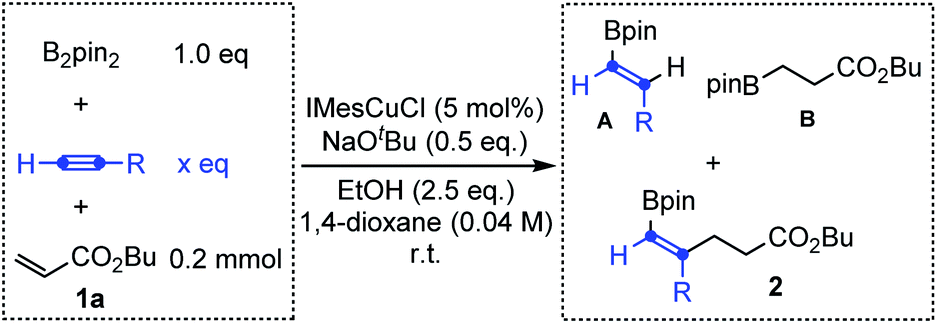
With the initial success achieved, we then systematically screened the reaction conditions and the optimal reaction conditions were established by using 5 mol% IMesCuCl as the catalyst, 0.5 equivalents of NaOtBu as the base and 2.5 equivalents of EtOH as the proton source in a solution of 1,4-dioxane (0.04 M) under a 1 atm acetylene atmosphere at room temperature. The desired alkenyl boronate 2a could be obtained in 90% NMR yield and 72% isolated yield (see the ESI for details).†
With the optimized reaction conditions in hand, we then explored the generality of this Cu-catalyzed carboboration of acetylene. As shown in Scheme 3, this reaction has a very broad substrate scope and excellent functional group tolerance. Different acrylates, thioacrylates, vinyl ketones, 1,3-dienyl ketone, acrylonitrile and vinyl sulphones with rich functional groups could be applied as effective substrates for this carboboration reaction. The carboboration of acetylene with acrylates was first demonstrated (2a–2m). Simple butyl, ethyl and phenyl acrylate could be transformed to target boronates 2a, 2b and 2c in 72%, 67% and 99% yields, respectively. As a base stronger than NaOH, NaOtBu has usually been considered to cause poor functional group tolerance. However, the introduction of SN2-sensitive functional groups such as methylsulfonyloxy, epoxy and alkyl bromide caused no deleterious effect on the reaction efficiency giving the desired products 2e, 2f, and 2i in >82% yields. The most unexpected thing was that even after a deliberately lengthened reaction time (22 hours), product 2j which contains an iodoalkyl group could still be isolated in 76% yield. In addition, no protodeborylation byproduct was observed. Markedly, besides the SN2-sensitive functional groups, the carbon–carbon triple bond was also found to be compatible, furnishing the desired products containing terminal alkynes in excellent yields (2g–2h). This was consistent with the results observed in Table 1 that the borylcupration process prefers acetylene substantially to other substituted alkynes. Free hydroxy and nitrile groups have no obvious negative effects on the reaction efficiency (2d and 2l). The carbon–silicon bond was also tolerable leading to the desired product 2k in 61% yield. Though acrylamide was not an efficient substrate to capture the β-boryl alkenyl copper intermediate, thioacrylate could be used to furnish the product 2m in 44% yield.
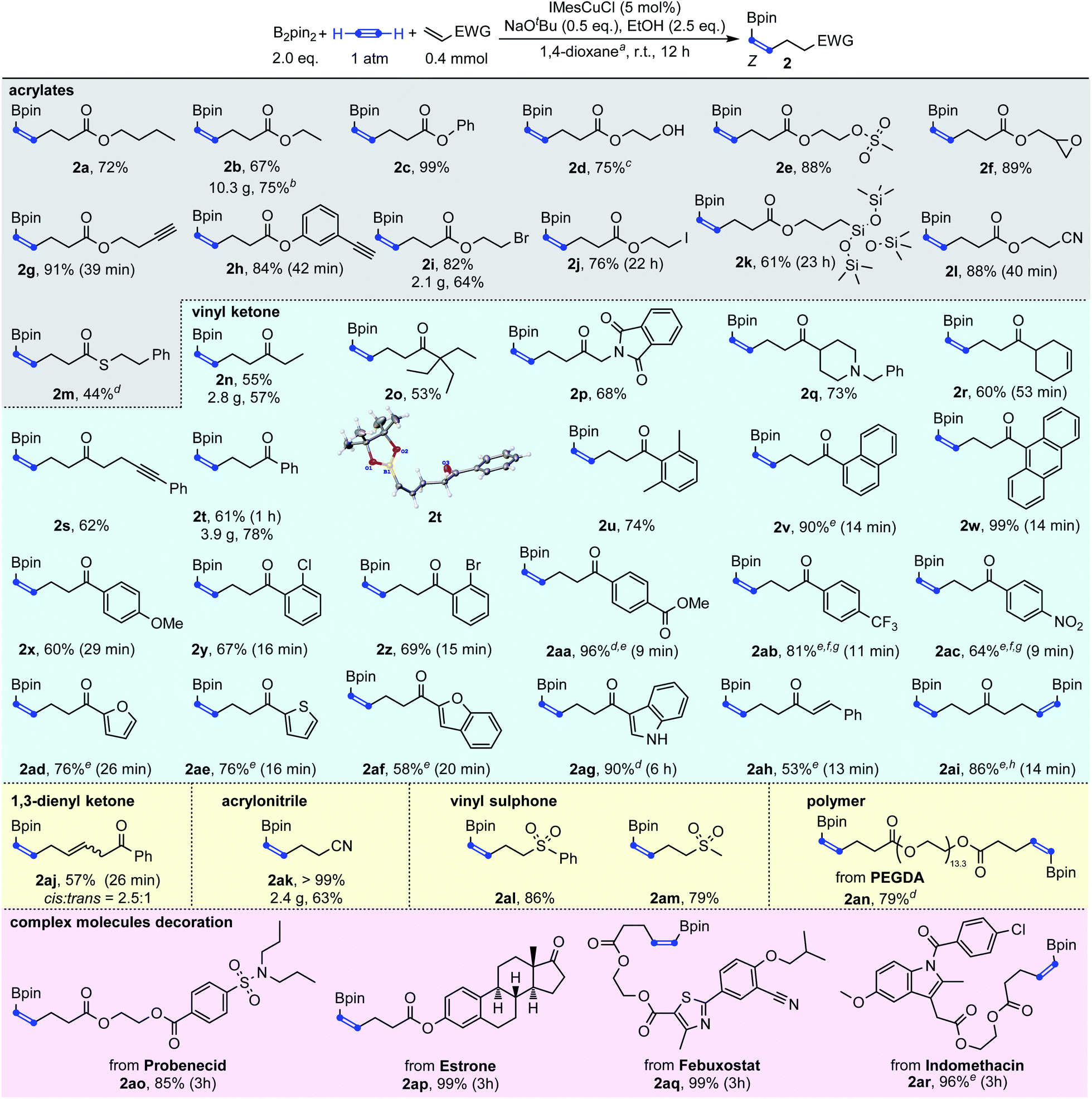 | ||
| Scheme 3 Cu-catalyzed carboboration of acetylene with B2Pin2 and Michael acceptors. a0.4 mmol of Michael acceptor in 10 mL 1,4-dioxane; bthe purity of the product was 93% by weight and the reported yield is based on the pure product; cwith 2.0 eq. EtOH; dwith 10 mol% IMesCuCl; esubstrate diluted with 1,4-dioxane or toluene and added dropwise for about 2 min; fwith 15 mol% IMesCuCl; gwith 2.5 eq. MeOH instead of EtOH; hsubstrate (1.0 mol L−1 in pentane) was added dropwise. | ||
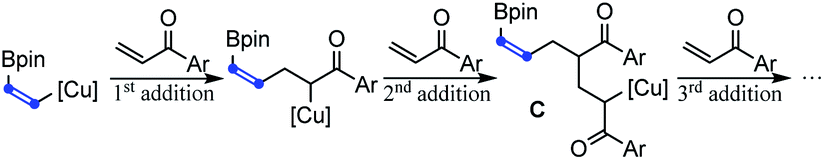
In addition to acrylate derivatives, vinyl ketones were also suitable substrates for this transformation. Simple ethyl and phenyl vinyl ketones gave 2n and 2t in 55% and 61% yields, respectively. Replacing the ethyl group in 2n with sterically bulkier triethylmethyl (2o) made no significant difference in yield. The functional groups of imide (2p), tertiary amine (2q), and internal alkyne (2s) were all well-tolerated, leading to the products in good yields. Besides alkyl vinyl ketones, aryl vinyl ketones were also suitable for this reaction (2t–2ag). The steric hindrance of the aryl group was found to have a beneficial effect on the reaction results. For example, the simple phenyl vinyl ketone gave the product 2t only in 61% yield, but the yields for bulkier 2,6-dimethyl phenyl, 2-naphthalenyl and 9-anthracenyl vinyl ketones increased substantially to 74%, 90% and 99% for 2u, 2v and 2w, respectively. The abnormal steric effects on products' yields were at least partially due to the suppression of the undesired multi-Michael addition byproduct C by the steric hindrance of the aryl ring.14 A variety of substitutions on the aryl ring are compatible. Both the electron-donating methoxyl group and weakly withdrawing halogen atoms did not affect the reaction (2x–2z). Strongly withdrawing groups (COOMe, CF3 and NO2) also succeeded through system acidity increase,7 reaction time control and slow addition (2aa, 2ab and 2ac). Michael acceptors derived from heteroaromatic rings such as furan thiophene, benzofuran and indole performed equally well (2ad, 2ae, 2af, and 2ag). Interestingly, when alkenyl vinyl ketone and divinyl ketone served as substrates, the reactions only selectively transformed the unsubstituted vinyl groups, while leaving the substituted alkenyl group intact (2ah and 2ai). An interesting feature of the transformation of aryl and alkenyl substituted vinyl ketones was the high reaction rate. Except 1ag whose carbonyl group was deactivated by indole nitrogen, from 1v to 1aj, all reactions were typically completed in less than 30 minutes.
Besides the acrylate and vinyl ketone-based Michael acceptors, 1,3-dien-1-yl ketone could be transformed to skipped diene 2aj in 57% yield. Acrylonitrile 1ak and vinyl sulphones 1al and 1am were also proved to be competent coupling partners, leading to the corresponding products in up to quantitative yields. Interestingly, polyethyleneglycol diacrylate (PEGDA), a commercially available polyethylene glycol-based material that is used as a prepolymer solution that can be used in the formation of a cross-linked polymeric system, could also be decorated with borylalkenyl groups on both ends with 79% yield under the standard catalytic conditions (2an). The reactions were easily scaled-up to the gram scale and to even more than 10 grams with good yields (2i, 2n, 2t, 2ak and 2b).
Given the high functional group tolerance obtained by this methodology, four acrylates derived from probenecid, estrone, febuxostat and indomethacin were subjected to the standard reaction conditions. To our delight, products 2ao–2ar were produced in excellent to quantitative yields, which further demonstrated the robustness of this methodology. Pitifully only Michael acceptors with an unsubstituted vinyl group were compatible in this reaction.
To further showcase the robustness of this carboboration, a large-scale preparation was also tested. Large-scale synthesis has a special meaning for this transformation, because many substrates of this reaction, such as acetylene, acrylates and vinyl ketones, are bulk industrial raw materials and the products, cis-alkenyl boronates, are also important and useful synthetic intermediates.3b,15 As shown in Scheme 4, an efficient molar-scale synthesis of cis-alkenyl boronate 2b using ethyl acrylate as the Michael acceptor has been successfully realized under slightly modified reaction conditions. 260 grams of light brown product boronate 2b could be distilled from the reaction mixture with 88% yield (based on the pure product) and 86% purity after a simple work-up procedure. The purity could be further improved by a further distillation process. Fortunately, no Z/E isomerization was found during the distillation with the temperature up to 200 °C.
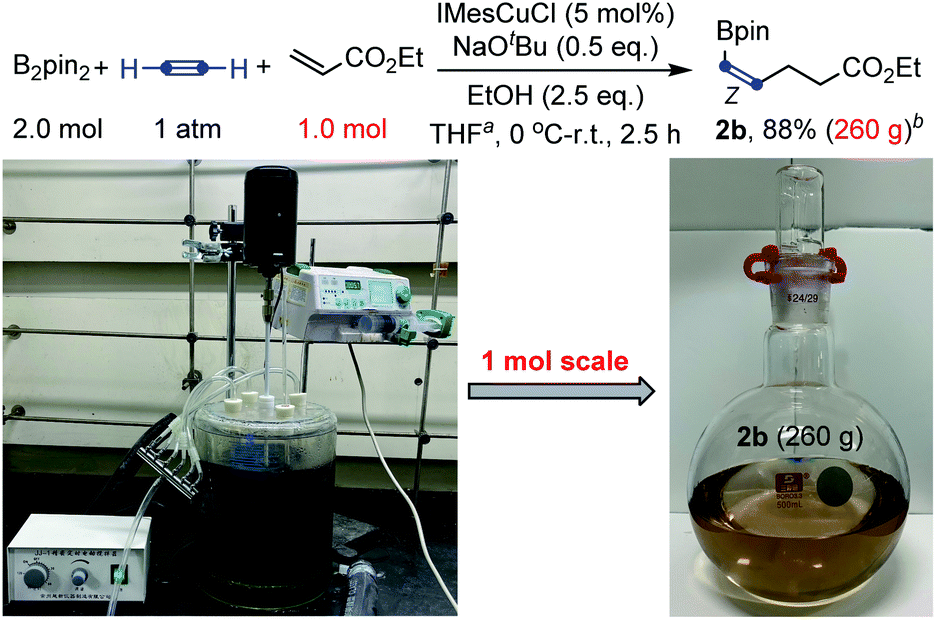 | ||
| Scheme 4 Large-scale synthesis. aTHF (3.0 L). bThe purity of the product 2a is 86% after simple distillation and the reported yield is based on the pure product. | ||
To illustrate the synthetic potential of these structurally simple but functional group-rich molecules, a series of transformations of cis-alkenyl boronates 2b and 2t were then performed (Scheme 5). First the pinacolatoboron group could transform into the phenyl group by Suzuki coupling (product 3) and the azide group by copper catalysis (product 4) with full retention of the cis double bond configuration. The boron group could be removed by copper catalyzed protodemetalation (product 5) and a formyl group was introduced successfully by direct oxidation of sodium peroxyborate (product 6). Epoxidation of the double bond could be achieved (product 7) once the boron atom is transferred to other oxidation insensitive groups, such as phenyl. Wittig reaction was found to be compatible with the existence of pinacol boron (product 8). Finally, a rhodium-catalyzed allylation tandem intramolecular transesterification reaction with aldehyde forming a five-membered lactone structure was also achieved with good diastereoselectivity and yield (product 9). In general, the three functional groups in the products (pinacol boronate, carbon–carbon double bond and ketone/ester group) could transform individually or synergistically to form other useful structures.
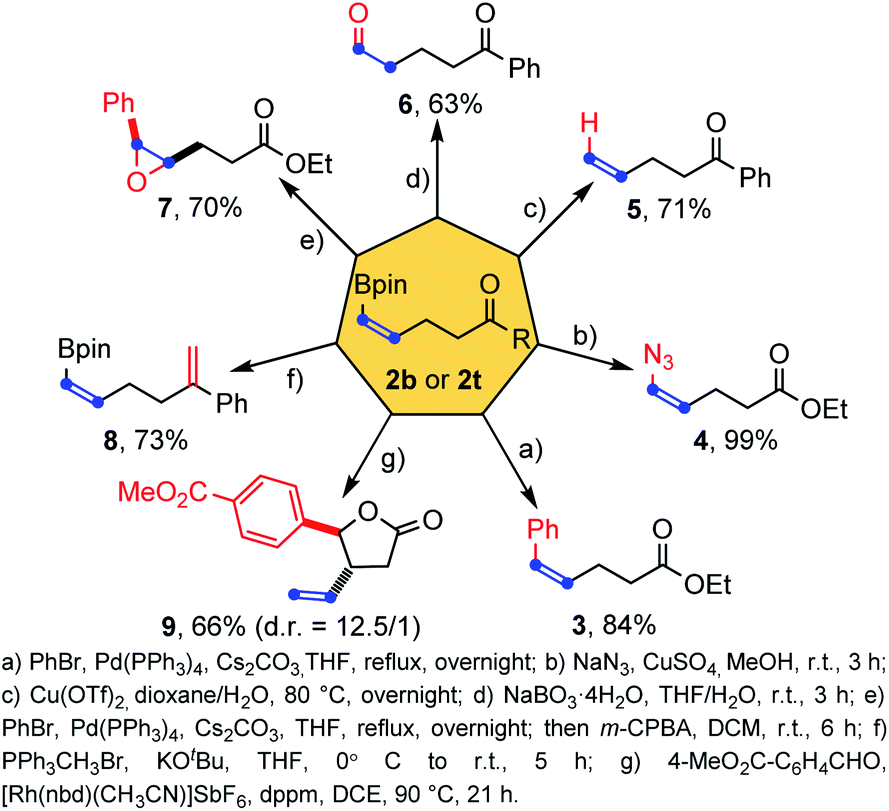 | ||
| Scheme 5 Synthetic applications of products.16 | ||
Conclusions
In conclusion, we have developed a copper-catalyzed three-component carboboration of acetylene with B2Pin2 and Michael acceptors. In this reaction, the cheap and abundant C2 chemical feedstock acetylene was used as the starting material to afford the homoallylic carbonyl group-containing cis-alkenyl boronates in a regiospecific manner. The reaction has high robustness and can be easily and reliably performed on the molar scale. In this reaction, acetylene does not solve the problems encountered with other alkynes, but in turn, the general problems of substituted alkynes may not definitely confine acetylene.17 Acetylene is acetylene and it is unique in nature. The small size of the acetylene molecule accelerates the rate of borylcupration and also facilitates the following C–C bond formation process through intermolecular Michael addition, which is typically challenging for substituted alkynes. Moreover, the resulting homoallylic carbonyl group-containing cis-alkenyl boronates could be converted to diverse functionalized molecules with ease. It is expected that this unique protocol may inspire and prompt chemists to further explore novel catalytic systems using acetylene as the C2 building block toward the synthesis of valuable chemicals.Data availability
Experimental procedures, NMR, IR, HRMS spectra, crystallographic data of 2t and some unsuccessful trials are provided in the ESI.†Author contributions
Conceptualization, funding acquisition, resources and supervision were done or provided by S. Z.; project administration, data curation, investigation and formal analysis were done by T. C.; validation was done by B. L.; writing was done by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22071062, 21871096) and Guangdong Science and Technology Department (2018B030308007).Notes and references
-
(a) A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS
; (b) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed
- For copper catalyzed hydroboration of alkynes:
(a) W. G. Woods, I. S. Bengelsdorf and D. L. Hunter, J. Org. Chem., 1966, 31, 2766–2768 CrossRef CAS
- For copper catalyzed carboboration of alkynes:
(a) R. Alfaro, A. Parra, J. Alemán, J. L. G. Ruano and M. Tortosa, J. Am. Chem. Soc., 2012, 134, 15165–15168 CrossRef CAS PubMed
- For copper catalyzed boracarboxylation of alkynes: L. Zhang, J. Cheng, B. Carry and Z. Hou, J. Am. Chem. Soc., 2012, 134, 14314–14317 CrossRef CAS PubMed
- The conjugate borylation of (2E)-2-nonen-7-ynal was found to be preferred to the borylation of the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond: H. Wu, S. Radomkit, J. M. ÓBrien and A. H. Hoveyda, J. Am. Chem. Soc., 2012, 134, 8277–8285 CrossRef CAS PubMed
C bond: H. Wu, S. Radomkit, J. M. ÓBrien and A. H. Hoveyda, J. Am. Chem. Soc., 2012, 134, 8277–8285 CrossRef CAS PubMed - P. Liu, Y. Fukui, P. Tian, Z.-T. He, C.-Y. Sun, N.-Y. Wu and G.-Q. Lin, J. Am. Chem. Soc., 2013, 135, 11700–11703 CrossRef CAS PubMed
- S.-H. Kim-Lee, I. Alonso, P. Mauleón, R. G. Arrayás and J. C. Carretero, ACS Catal., 2018, 8, 8993–9005 CrossRef CAS
- For reviews:
(a) V. V. Voronin, M. S. Ledovskaya, A. S. Bogachenkov, K. S. Rodygin and V. P. Ananikov, Molecules, 2018, 23, 2442–2525 CrossRef PubMed
- For examples of catalytic reactions of acetylene:
(a)
Y. Fu, S. Yu, B. Zhang and T. Gong, Preparation method of alkenyl alkynyl boric acid ester, China Patent, CN108503662(A), 2018, China, Utility patent Search PubMed
- For reviews:
(a) M. S. Ledovskaya, V. V. Voronin and K. S. Rodygin, Russ. Chem. Rev., 2018, 87, 167–191 CrossRef CAS
- E. Y. Shmidt, N. I. Protsuk, A. M. Vasiĺtsov, A. V. Ivanov, A. I. Mikhaleva and B. A. Trofimov, Heterocycl. Compd., 2013, 49, 404–407 CrossRef CAS
- J. Li, H. Zhang, L. Li, M. Cai, Y. Li, D. Xie and J. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 10173–10184 CrossRef CAS
-
(a) H. Xie, T. Lin, L. Shi and X. Meng, RSC Adv., 2016, 6, 97285–97292 RSC
- When the steric hindrance of the aryl group in vinyl ketones increases, tandem Michael additions forming byproduct C were suppressed and the yields of 2 increased correspondingly
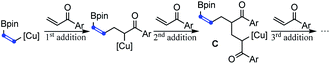 .
. - D. P. Ojha and K. R. Prabhu, Org. Lett., 2016, 18, 432–435 CrossRef CAS PubMed
- Synthetic applications were performed according to the following references:
(a) K. Matos and J. A. Soderquist, J. Org. Chem., 1998, 63, 461–470 CrossRef CAS PubMed
- No new ligands, new additives or new reagents were developed to expand the reaction scope of substituted alkynes in this work. However, by exploring acetylene's unique properties, traditionally unachievable reactions of substituted alkynes can be achieved with acetylene.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all compounds, and copies of 1H and 13C NMR spectra. CCDC 2165502 (2t). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02306g |
| This journal is © The Royal Society of Chemistry 2022 |