
Indole-containing pharmaceuticals: targets, pharmacological activities, and SAR studies
 *a
*a
Abstract
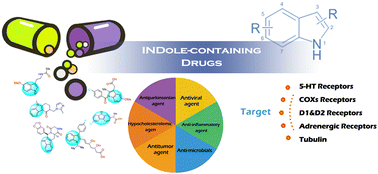
Indole is a prestigious heterocyclic skeleton widely found in both naturally-occurring and biologically-active compounds. Pharmaceutical agents containing an indole skeleton in their framework possess a wide range of pharmacological properties, including antiviral, antitumor, analgesic, and other therapeutic activities, and many indole-containing drugs have been proven to have excellent pharmacokinetic and pharmacological effects. Over the past few decades, the FDA has approved over 40 indole-containing drugs for the treatment of various clinical conditions, and the development of indole-related drugs has attracted significant attention from medicinal chemists. This review aims to provide an overview of all the approved drugs that contain an indole nucleus, focusing on their targets, pharmacological activities, and SAR studies.
 Please wait while we load your content...
Please wait while we load your content...

