Sequential crosslinking to control cellular spreading in 3-dimensional hydrogels
Sudhir
Khetan
,
Joshua S.
Katz
and
Jason A.
Burdick
*
Department of Bioengineering, University of Pennsylvania, 240 Skirkanich Hall, 210 South 33rd Street, Philadelphia, PA 19104, USA. E-mail: burdick2@seas.upenn.edu
First published on 11th February 2009
Abstract
With advanced understanding of how manipulations in material chemistry and structure influence cellular interactions, material control over cellular behavior (e.g., spreading) is becoming increasingly possible. In this example, we developed a novel process that utilizes different crosslinking mechanisms to provide gel environments that are either permissive or inhibitory to cellular spreading. To accomplish this, a multi-acrylated macromer (i.e., acrylated hyaluronic acid) was first crosslinked with an addition reaction using a matrix metalloprotease (MMP) cleavable peptide containing thiol groups. When an adhesive peptide was also coupled to the network, this environment permitted the spreading of encapsulated human mesenchymal stem cells (hMSCs), whereas control systems did not. If all acrylates were not consumed during the initial crosslinking step, a photoinitiated radical polymerization could be used to crosslink the remaining acrylates and inhibit cellular spreading with the production of covalent barriers. Variations in the ratio of the two crosslink types in individual constructs controlled the degradation and mechanical properties of the hydrogels, as well as the degree of spreading of encapsulated cells. Cell spreading was further controlled spatially with the use of photomasks. Overall, this new technology is an exciting and potentially valuable tool, both to provide new insights into the relationships between gel structure and cell behavior, and for eventual tissue-engineering applications where spatial control over cells is desired.
Introduction
Cellular spreading is important in that it allows cells to interact with their environment, including receiving cues towards proliferation and even differentiation.1 Until recently, the scaffolding component in tissue engineering has been employed as a relatively inert component to the approach, providing mainly structural support and potential adhesion interactions through decoration with peptides and proteins.2–5 However, it is now clear that the dynamic interplay that occurs between cells and the extracellular matrix (ECM) is also important in the design and functionality of new biomaterials for use as synthetic cellular environments. The ECM is a dynamic and biologically active matrix with critical structural and functional roles, and ECM remodeling is necessary for cell migration and tissue morphogenesis.Cellular spreading, which varies in vivo according to cell type and biochemical and mechanical properties of different tissues, may influence cellular functions such as stem cell differentiation.6–8 Past work indicates that hMSCs seeded onto substrates coated with adhesive elements such as fibronectin,9collagen,10 and gelatin11 differentiate depending on adhesion, morphology, and spreading. Curran et al. demonstrated that differences in morphology of hMSCs adhered to glass substrates with modified surface chemistries led to differences in differentiation.12,13 The importance of cell shape in terminal differentiation has also been demonstrated for other progenitor cell types ranging from bone marrow stromal cells7 to human epidermal keratinocytes.14
Recent studies have incorporated ECM-mimetic features into hydrogels in a 3-dimensional (3-D) fashion to control encapsulated cellular behavior. For example, it has been shown that the viability and proliferation of hMSCs encapsulated in synthetic PEG-based hydrogel networks depends on the adhesiveness of the surrounding matrix.3 Beyond adhesion, Lutolf et al.15 demonstrated that spreading and random migration of fibroblasts encapsulated in PEG-based hydrogels was possible when both cell-adhesivity and MMP-degradability were incorporated into the networks. They have since explored this system for cardiac tissue-engineering applications,16 showing that multipotent cardioprogenitors encapsulated in the networks differentiated along the cardiac lineage when the stiffness of the scaffold mimicked that of native cardiac tissue. In a similar manner, Kim et al.17 incorporated cell-adhesivity and proteolytic degradability into hyaluronic acid (HA)-based scaffolds and demonstrated spreading of encapsulated hMSCs, something that was not possible in gels lacking either bioactive feature. Others have also utilized hydrogels containing these cues for tissue-engineering approaches.18–20
Despite these approaches, very few studies have investigated the spatial control that may be possible in these environments. In one example, investigators micropatterned cell-adhesive oligopeptides into precisely defined channels and demonstrated guided neurite outgrowth.21,22 However, the spatial control of encapsulated cell behavior using a cytocompatible process has not yet been achieved; the described 3-D studies all employed a single mode of crosslinking (e.g., addition reactions between peptide thiol groups and vinyl double bonds) homogeneously throughout the matrix volume, whereas the spatially controlled hydrogels did not use processes that were compatible with cells. A technique that affords such spatial control would be useful in numerous applications, from fundamental investigations of the influence of gel structure on cellular behavior to tools for advanced tissue-engineering applications.
In this work, we present a novel material-based process that utilizes multiple modes of crosslinking in a sequential manner to spatially control the behavior of cells encapsulated within 3-D hydrogels (this process is shown schematically in Fig. 1). During the primary step, hydrogels that contain both adhesive sites and MMP-cleavable dithiol crosslinkers are formed from multi-acrylate macromers (i.e., acrylated hyaluronic acid) via an addition mechanism, leaving a network that is “permissive” to remodeling and cellular spreading. Importantly, only a portion of the total number of acrylate groups is consumed during this first step, which occurs in the presence of a photoinitiator (at this point, non-reactive). During the secondary step, the gels are exposed to light to initiate radical polymerization of all the remaining acrylates, creating a network that is “inhibitory” to cell spreading based on the covalent crosslinks formed through kinetic chains. The premise is that the covalent crosslinks block cellular remodeling and prevent cellular spreading in the hydrogels since the mesh sizes are significantly smaller than typical cell diameters.17,23 Since the second step is initiated by light, which can be spatially controlled, it is anticipated that this approach may be useful to spatially control cellular spreading within the hydrogels. This report describes the process and its utility in controlling stem cell behavior in 3-D hydrogel environments.
 | ||
| Fig. 1 Schematic of sequential crosslinking of AHA using a primary addition reaction and a secondary radical polymerization. | ||
Experimental
AHA synthesis
Acrylated hyaluronic acid (AHA) was synthesized via a 3-step protocol. All 1H NMR spectra were recorded on a Bruker Avance 360MHz spectrometer.The final structure and 1H NMR spectrum of AHA are shown in Fig. 2. NMR (D2O): δ (ppm) 6.4, 0.4H, d; 6.15, 0.4H, dd; 5.9, 0.4H, d; 4.2–4.6, 2H; 3.15–4.0, 10H; 2.7, 1.2H, broad; 1.9, 3H, s.
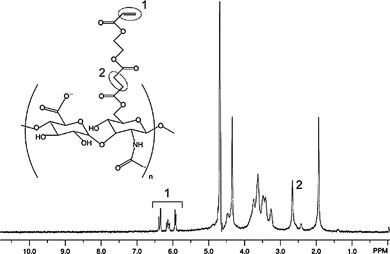 | ||
| Fig. 2 1H NMR Spectrum (D2O) of acrylated hyaluronic acid (AHA). | ||
Cells
Human mesenchymal stem cells (hMSCs) were obtained from Lonza Corporation (Wakersville, MD, USA). For encapsulation studies, hMSCs were expanded in growth media (α-MEM, 10% FBS, 1% L-glutamine & penicillin streptomycin) and encapsulated at low passage numbers (between 2 and 4) in AHA hydrogels at a density of 5 × 106cells per mL. The constructs were maintained in 1.5 mL of growth media in a 24-well plate and refreshed every 3 days until the end of day 5, at which point live/dead analysis was performed.Peptides
The cell adhesive oligopeptide GCGYGRGDSPG (MW: 1025.1 Da) and MMP-degradable oligopeptide GCRDGPQG↓IWGQDRCG (MW: 1754.0 Da), both with >95% purity (per manufacturer HPLC analysis), were obtained from GenScript Corporation (Piscataway, NJ, USA) for all studies.Crosslinking
AHA was dissolved in a triethanolamine-buffered saline (TEOA buffer: 0.2 M TEOA, 0.3 M total osmolarity, pH 8.0) containing Irgacure2959 (Ciba) photoinitiator (final concentration of 0.05 wt%). I2959 was chosen due to its aqueous solubility and good cytocompatibility.24 The cell adhesive peptide dissolved in TEOA buffer was added to the AHA solution at a final peptide concentration of 1 mM (corresponding to ∼1/20 of available acrylate groups with 3 wt% AHA), and allowed to react for 1 h at 37 °C. Following re-suspension of cells in this solution, MMP peptide dissolved TEOA buffer was added to the pre-polymer solution corresponding to the desired percent acrylate consumption, and 50 µL of this mixture was immediately pipetted into sterile molds (5 mm diameter, 2 mm height). The gels were allowed to react (primary crosslinking) for 15 min at room temperature inside the laminar flow hood. For sequential crosslinking studies, gels were then exposed to 10 mW cm−2 365 nm ultraviolet light (Omnicure S1000 UV Spot Cure System, Exfo Life Sciences Division, Mississauga, Ontario, Canada) for 4 minutes (secondary crosslinking). Gelation times were chosen based on earlier acellular experiments that measured the addition and radical polymerization durations (15 and 4 min, respectively) for which further reaction did not change the mechanical properties.Hydrogel characterization
Acellular samples were fabricated as described above. Following crosslinking and swelling to equilibrium in PBS for 24 h, the Young's modulus of each hydrogel disk was determined by unconfined submersion compression testing on a dynamic mechanical analyzer (Q800 Series; TA Instruments, New Castle, DE, USA) with an oscillating plate compression clamp attachment. Briefly, the diameter of each swelled hydrogel disk (∼5 mm) was determined using a digital caliper, and the sample was immersed in a PBS bath between unconfined parallel compression plates to prevent dehydration. An equilibrium preload force was applied by the descending plate, followed by application of a ramped strain of 10% min−1 to 60%. The Young's modulus was then determined using the slope of the stress–strain curve at low strain (<25% strain). To obtain the volumetric swelling ratio (QV), equilibrium swelled constructs were patted dry to remove surface liquid and weighed (wet weight), lyophilized, and re-weighed (dry weight). QV is reported as the ratio of the wet weight to dry weight, assuming a density of 1.23 g cm−3 for the AHA macromers.25 For degradation studies, hydrogels that crosslinked completely (i.e., 100% acrylate consumption) through addition or radical polymerization were incubated in separate wells of a 24-well plate containing 1 mL PBS with 40 nM human MMP-1 (Sigma) on an orbital shaker at 37 °C. The solutions were refreshed every 24 h for 1 week, and the supernatant samples (frozen and stored at −20 °C after collection) were analyzed in triplicate via a modified uronic acid assay.26 Briefly, 100 µL of each sample was added to a concentrated solution of sulfuric acid and sodium tetraborate decahydrate (Sigma) and heated to 100 °C for 10 min. 100 µL of 0.125% carbazole solution in ethanol was then added, and the samples were vortexed and heated to 100 °C for 15 min. The sample absorbances were then read at 530 nm and compared to a standard curve of known concentrations of HA (a range from 0.1 to 2.0 mg mL−1). All studies were performed in triplicate unless otherwise noted.Live/dead staining
Encapsulated cells were visualized for viability using a fluorescent live/dead staining kit (Molecular Probes) and imaging on an inverted microscope (Axiovert 200, Carl Zeiss Inc.) equipped with an epifluorescent lamp. For assessment of viability, three random images of each gel at 5× magnification were taken through both the live (FITC) and the dead (TRITC) filters. Cell viability was then assessed by counting the total number of live and dead cells in each image and calculating the ratio of live cells to total cells. For all cellular images, each construct was first viewed from the top to the bottom surface to ensure uniform cellular morphology throughout the construct volume and that images were obtained from the interior of the gel.Cellular aspect ratio measurements
For cellular aspect ratio measurements, three random light microscopy images at 5× magnification were taken from each gel using an inverted microscope. To quantify cellular spreading, the maximum orthogonal length and width of each cell was measured using NIH ImageJ, and the aspect ratio calculated as the longer length divided by the shorter length. Each image produced ≥ 15 measurements from randomly selected cells, or n ≥ 45 for each sample. The measured aspect ratios were then sorted into bins to form histograms of spreading for each formulation.Statistical analysis
All values are reported as the mean ± standard error of the mean. ANOVA in conjunction with Tukey's post hoc test was used to determine statistically significant differences between groups, with p ≤ 0.05.Results and discussion
Acellular hydrogel synthesis and characterization
AHA with 38% of hydroxy groups acrylate-modified was synthesized and the acrylation conversion was measured via1H NMR as described. The purified yield was ∼65% based on moles of HA present in the HA–TBA reactant and AHA product. HA, a linear glycosaminoglycan made of alternating D-glucuronic acid and N-acetyl-D-glucosamine, was used as the primary structural component due to its biocompatibility, hydrophilicity, importance in vivo including in the turnover of ECM following tissue injury, interactions with cellsvia surface receptors,27,28 and past use in tissue-engineering applications.29–33 Although HA is commonly modified with methacrylate groups, acrylate groups were used since they react much faster during the primary addition step,34,35 which allows for uniform cell suspensions. While HA was used in the current work, the sequential crosslinking technique may potentially be applied to other macromers functionalized with reactive groups that can undergo multiple modes of crosslinking, highlighting its versatility.AHA was crosslinked into 3 wt% hydrogels either with a photoinitiated polymerization alone, with an addition polymerization alone, or sequentially using both (in order) an addition and radical polymerization. For the addition polymerizations, theoretically 50, 75, or 100% of the acrylates were consumed. 1H NMR was used to confirm a decrease in the acrylate peaks upon addition crosslinking and elimination of the acrylate peaks with photopolymerization. Additionally, peak reduction corresponded to the theoretical amount of acrylate consumption (data not shown).
Both the mechanics and the swelling of the hydrogels were dependent on the type of crosslinking (and for sequentially crosslinked gels, the ratio of addition to radical crosslinking) that was used (Fig. 3A and 3B). Hydrogels crosslinked only through radical polymerization exhibited a ∼2–4-fold higher compressive modulus (18.62 ± 1.96 kPa) and swelled significantly less (QV = 27.75 ± 1.20) than either addition alone (e.g., modulus = 4.60 ± 0.71 kPa, QV = 45.42 ± 1.70 for 50% formulation) or sequentially crosslinked (e.g., modulus = 9.45 ± 2.90 kPa, QV = 38.26 ± 2.68 for 50% + photo formulation) hydrogels. 100% addition samples polymerized too quickly to obtain uniform samples suitable for mechanical testing. The kinetic chains in the radically crosslinked only hydrogels concentrate the acrylate side chains and create a more dense network than those that are reacted with a dithiol oligopeptide crosslinker molecule, as in the addition reaction. This could explain the higher modulus and decreased swelling in radical only hydrogels. Sequentially crosslinked hydrogels with identical peptide compositions but secondarily crosslinked through photopolymerization exhibited increased moduli and decreased swelling relative to their addition-only counterparts, indicative of the secondary radical polymerization. These changes were greater for the 50% case, since a higher percentage of acrylate groups was available for the radical crosslinking step.
 | ||
| Fig. 3 Characterization of sequentially crosslinked AHA hydrogels. (A) Compressive modulus and (B) swelling ratio of photopolymerized and sequentially crosslinked AHA hydrogels. The sequential crosslinking was performed with a theoretical consumption of either 50% or 75% of acrylates on the AHA during the primary crosslinking. * denotes statistically significant difference from all other groups. (C) Degradation kinetics of AHA hydrogels crosslinked using only an addition or radical mechanism (100% theoretical consumption of acrylates in both cases) in the presence of 40 nM MMP-1. | ||
AHA hydrogels synthesized completely (i.e., 100% acrylate consumption) through the addition or radical crosslinking mechanisms also differed predictably in degradation kinetics when incubated in PBS containing 40 nM MMP-1 (Fig. 3C). Hydrogels crosslinked with MMP-degradable oligopeptides underwent complete degradationvia the action of exogenous proteases by day 7, while radically crosslinked gels showed little mass loss (∼10%, with kinetics that mimicked incubation in PBS alone) potentially from hydrolysis of ester linkages in the crosslinks or a soluble fraction after crosslinking. These results support the underlying premise that the covalent kinetic chains do not allow for proteolytic degradation, whereas the MMP cleavable crosslinks degrade rapidly in the presence of the enzyme. These trends also illustrate the potential tunability of the sequential crosslinking system, as both the concentration of MMP-degradable domains (i.e., the degradation kinetics of the hydrogel in the presence of MMP) and bulk mechanical properties can be matched to the tissue-engineering application of interest.
Controlled encapsulated cell spreading in bulk polymerized gels
To determine if these results translate into cellular instructive hydrogels, hMSCs were suspended in the initial macromer solution and encapsulated using either the photoinitiated polymerization alone or the sequential crosslinking procedure.The addition alone hydrogels with 100% acrylate consumption polymerized too quickly to obtain evenly distributed cells and were not further investigated. Control hydrogels (shown in Fig. 4) were produced that contained only the MMP-degradable peptide crosslinker, but no RGD peptide (−RGD, MMP); the RGD peptide, and alternate non-degradable dithiol crosslinker dithiothreitol (+RGD, DTT); and containing RGD peptide but crosslinked using a photoinitated radical polymerization alone (+RGD, photo). As expected, hMSCs in these constructs remained rounded (Fig. 4a), with 100% of the cells exhibiting an aspect ratio (i.e., the ratio of the longest to shortest dimension of encapsulated cells) between 1 and 2 (Fig. 4b). In contrast, when adhesivity and degradability were incorporated into single gels (+RGD, MMP), robust cell spreading was observed and found to be dependant of the relative amount of each crosslinking mode (Fig. 5). Cells encapsulated in “permissive” hydrogels synthesized only through addition crosslinking using MMP-degradable peptides corresponding to 50% and 75% acrylate consumption exhibited relatively high degrees of spreading (i.e., a distribution towards much higher aspect ratios in Fig. 5b). However, cells encapsulated in “inhibitory” hydrogels formed with the sequential crosslinking procedure were similar to the radical polymerization alone and remained rounded. This inhibition was more pronounced with a lower fraction of acrylates consumed during the addition step (50% versus 75%), potentially due to the greater amount of crosslinking during the secondary radical polymerization to inhibit spreading. As others have reported,16,18 these results indicate that both adhesion and degradation sites are necessary for cellular remodeling of synthetic hydrogels.
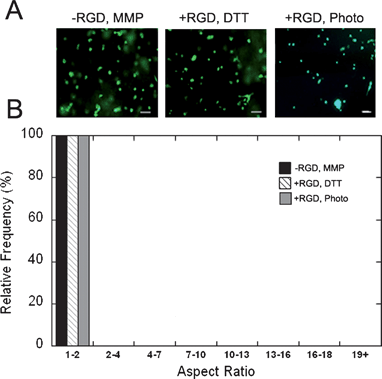 | ||
| Fig. 4 hMSC encapsulation in control AHA hydrogels. (A) Images of encapsulated hMSCs in hydrogels formed using (respectively) MMP-degradable peptide crosslinker in the absence of RGD peptide, with the non-degradable dithiol crosslinker (DTT), and containing RGD peptide but crosslinked using radical polymerization alone. Scale bar represents 100 µm. (B) Histogram of the cellular aspect ratio (the ratio of the longest to shortest dimension of encapsulated cells) for these same groups. All cultures were run for 5 days. There were no statistically significant differences between these groups. | ||
 | ||
| Fig. 5 hMSC encapsulation in sequentially crosslinked AHA hydrogels. (A) Images of encapsulated hMSCs in hydrogels formed by an addition polymerization alone or using the sequential polymerization procedure. The sequential crosslinking was performed with a theoretical consumption of either 50 or 75% of acrylates on the AHA during the primary crosslinking. Scale bar represents 100 µm. (B) Histogram of the cellular aspect ratio (the ratio of the longest to shortest dimension of encapsulated cells) for these same groups. All cultures were run for 5 days. There were statistically significant differences for the aspect ratios between the addition polymerization alone and the sequential polymerization for each formulation (i.e., 50 or 75% acrylate consumption). | ||
Cells in all conditions exhibited high viability (88–94%) as quantified from live/dead staining (dead stain overlayed on Fig. 4 and Fig. 5 live images), with no statistical differences between any of the hydrogel compositions (data not shown).
Spatially controlled encapsulated cell spreading
Although these results illustrate our ability to form gels that either permit or inhibit cell spreading, there are many instances where this would be beneficial to achieve with spatial control. As discussed, it is clear that cues such as spreading lead to changes in cell signaling and potentially differentiation; thus, spatial control over spreading could lead to control over cell lineage towards the development of advanced tissue-engineering approaches with differentiation down multiple cell lineages. In this sequential crosslinking approach, the creation of spatially controlled spreading of AHA hydrogels can be achieved by applying a photomask between the two crosslinking steps (Fig. 6A). As illustrated, regions of the hydrogel that are unmasked are exposed to light and undergo a secondary radical polymerization, while masked regions are not exposed to the light and only undergo the primary crosslinking. | ||
| Fig. 6 Spatially patterned outgrowth of hMSCs. (A) Schematic of process to spatially control cell spreading in AHA hydrogels. (B) Calcein-stained hMSCs encapsulated in a sequentially crosslinked AHA hydrogel where one half of the construct was covered with a mask during light exposure. Scale bar represents 100 µm. (C) Histogram of cell spreading in regions exposed to light (addition + radical) or covered with a mask (addition only) during the secondary crosslinking. All cultures were run for 5 days. There was a statistically significant difference between aspect ratios in regions exposed to and masked from light. | ||
To illustrate the feasibility of this approach, AHA hydrogels synthesized with 50% consumption during the primary crosslinking were exposed to light through a mask that blocked half of the sample in entirety. A live image of cells at the interface in this gel is shown in Fig. 6B and indicates spherical morphology with light exposure and spindle-like morphology in areas that were covered with the mask. The extent of outgrowth in these regions, both qualitatively from light microscopy and as quantified through aspect ratio measurements (Fig. 6C), were similar to the corresponding bulk gels assessed above. Although this is a simple example of the approach, more complex patterns could be achieved with different masks or through the use of lasers for the secondary polymerization.36,37
Conclusions
The sequential polymerization described here is a robust, novel approach towards dictating the cellular behavior in 3-dimensions. While a single AHA weight percentage was used in the current study, the versatility of the sequential crosslinking technology arises from the ability to vary this and other design parameters (e.g., HA acrylation efficiency, macromer and peptide concentrations, encapsulated cell density) to tune the remodeling kinetics to different applications. For instance, differences in cellular morphology in patterned AHA hydrogels could be useful as a signaling mechanism for spatially controlled differentiation of encapsulated stem cells. Such an approach has potential in the regeneration of tissues with anisotropic properties (e.g., vasculature or nervous tissues) or where spatially controlled organization of cells is desired. In the current work, the cells were cultured in standard growth medium to illustrate the technique of controlled spreading, and no specific cell type or tissue was targeted. Collectively, this approach may become a valuable tool in biomaterials development and regenerative medicine.Acknowledgements
The authors gratefully acknowledge the Penn Institute for Regenerative Medicine, and funding from the David and Lucile Packard Foundation (JAB) and the National Science Foundation Graduate Research Fellowship Program (SK, JSK).References
- B. Gumbiner, Cell, 1996, 84, 345–357 CrossRef CAS.
- J. Moon, M. Hahn, I. Kim, B. Nsiah and J. West, Tissue Eng., Part A, 2008 Search PubMed.
- C. Salinas and K. Anseth, J. Tissue Eng. Regen. Med., 2008, 2, 296–304 Search PubMed.
- L. Santiago, R. Nowak, J. Peter Rubin and K. Marra, Biomaterials, 2006, 27, 2962–2969 CrossRef CAS.
- Z. Zhang, S. Chen and S. Jiang, Biomacromolecules, 2006, 7, 3311–3315 CrossRef CAS.
- H. Senechal, J. Wahrmann, D. Delain and M.-C. A, In Vitro, 1984, 20, 692–698 CrossRef CAS.
- F. Scintu, C. Reali, R. Pillai, M. Badiali, M. Sanna, F. Argiolu, M. Ristaldi and V. Sogos, BMC Neurosci., 2006, 7, 14 CrossRef.
- F. Watt, J. Cell Sci. Suppl., 1987, 8, 313–326 Search PubMed.
- N. Ogura, M. Kawada, W. Chang, Q. Zhang, S. Lee, T. Kondoh and Y. Abiko, J. Oral Sci., 2004, 46, 207–213 CrossRef CAS.
- R. Salasznyk, W. Williams, A. Boskey, A. Batorsky and G. Plopper, J. Biomed. Biotechnol., 2004, 2004, 24–34 Search PubMed.
- Y. M. Shin, K.-S. Kim, Y. M. Lim, Y. C. Nho and H. Shin, Biomacromolecules, 2008, 9, 1772–1781 CrossRef CAS.
- J. Curran, R. Chen and J. Hunt, Biomaterials, 2005, 26, 7057–7067 CrossRef CAS.
- J. Curran, R. Chen and J. Hunt, Biomaterials, 2006, 27, 4783–4793 CrossRef CAS.
- F. Watt, P. Jordan and C. O'Neill, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 5576–5580 CrossRef CAS.
- M. Lutolf, G. Raeber, A. Zisch, N. Tirelli and J. Hubbell, Adv. Mater., 2003, 15, 888–892 CrossRef CAS.
- T. Kraehenbuehl, P. Zammaretti, A. Van der Vlies, R. Schoenmakers, M. Lutolf, M. Jaconi and J. Hubbell, Biomaterials, 2008, 29, 2757–2766 CrossRef CAS.
- J. Kim, Y. Park, G. Tae, K. Lee, S. Hwang, I. Kim, I. Noh and K. Sun, J. Mater. Sci., Mater. Med., 2008, 19, 3311–3318 CrossRef CAS.
- A. Gobin and J. West, FASEB J., 2002, 16, 751–753 CAS.
- B. Mann, A. Gobin, A. Tsai, R. Schmedlen and J. West, Biomaterials, 2001, 22, 3045–3051 CrossRef CAS.
- K. Nguyen and J. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS.
- Y. Luo and M. Shoichet, Nat. Mater., 2004, 3, 249–253 CrossRef CAS.
- P. Musoke-Zawedde and M. Shoichet, Biomed. Mater., 2006, 1, 162–169 Search PubMed.
- C. Chung, J. Mesa, M. Randolph, M. Yaremchuk and J. Burdick, J. Biomed. Mater. Res. A, 2006, 77, 518–525 CrossRef.
- C. Williams, A. Malik, T. Kim, P. Manson and J. Elisseeff, Biomaterials, 2005, 26, 1211–1218 CrossRef CAS.
- J. Leach, K. Bivens, C. Collins and C. Schmidt, J. Biomed. Mater. Res. A, 2004, 70, 74–82.
- T. Bitter and H. Muir, Anal. Biochem., 1962, 4, 330–334 CrossRef CAS.
- D. Jiang, J. Liang and P. Noble, Annu. Rev. Cell Dev. Biol., 2007, 23, 435–461 CrossRef CAS.
- S. Yung, G. Thomas and M. Davies, Kidney Int., 2000, 58, 1953–1962 CrossRef CAS.
- J. Baier Leach, K. Bivens, C. J. Patrick and C. Schmidt, Biotechnol. Bioeng., 2003, 82, 578–589 CrossRef CAS.
- C. Chung, J. Mesa, G. Miller, M. Randolph, T. Gill and J. Burdick, Tissue Eng., 2006, 12, 2665–2773 CrossRef CAS.
- Y. Sakai, Y. Matsuyama, K. Takahashi, T. Sato, T. Hattori, S. Nakashima and N. Ishiguro, Biomed. Mater. Eng., 2007, 17, 191–197 Search PubMed.
- S. Sahoo, C. Chung, S. Khetan and J. Burdick, Biomacromolecules, 2008, 9, 1088–1092 CrossRef CAS.
- Y. Ji, K. Ghosh, X. Shu, B. Li, J. Sokolov, G. Prestwich, R. Clark and M. Rafailovich, Biomaterials, 2006, 27, 3782–3792 CrossRef CAS.
- M. Carroll, J. Cheeseman, R. Osman and H. Weinstein, J. Phys. Chem., 1989, 93 Search PubMed.
- M. Gibas and A. Korytkowska-Walach, Polym. Bull., 2003, 51, 17–22 CrossRef CAS.
- M. Hahn, L. Taite, J. Moon, M. Rowland, K. Ruffino and J. West, Biomaterials, 2006, 27, 2519–2524 CrossRef CAS.
- S. Lee, J. Moon and J. West, Biomaterials, 2008, 29, 2962–2968 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |