DOI:
10.1039/B811497H
(Paper)
Soft Matter, 2009,
5, 790-796
Capacity-controllable nanocarriers for metal ions
Received
7th July 2008
, Accepted 31st October 2008
First published on 8th December 2008
Abstract
We report on capacity-controllable nanocarrier system for metal ions based on a novel kind of polymeric microemulsion. These microemulsions are formed in mixed systems of negatively charged metal-bisligand coordination polymers (cp), positively charged homopolyelectrolyte (hp), and positive-neutral diblock copolymers (dp), and are called Complex Coacervate Core microemulsions (C3-µE). The stoichiometric mixtures of cp and dp mixed system forms complex coacervate core micelles (C3M), also called polyion complex (PIC) micelles, or block ionomer complex micelles (BIC), with metal ions residing in the core as a constituent of the coordination polymers. In the presence of an additional stoichiomeric mixture of hp and cp, the micellar core swells and C3-µE are formed. In this way the amount of metal ions in a core can be increased up to 10 times, whereas by increasing the length of the dp one can only increase the amount of metal ions up to 2.5 times. The radius of the particles (Rh ≈ 140 nm) can be up to 6 times larger than the original micelles, but they still have core-shell structures. The large C3-µE particles, having densities larger than that of water, gradually sediment with time, but they do not agglomerate: they can be easily redispersed by simple shaking. C3-µEs are stable in the temperature range of 10–80 °C.
1. Introduction
Recently, nanoparticles (NPs) with controllable sizes have received considerable attention in chemistry, biology, and applied fields.1 Changing the size of NPs can result in modulation of their physical and chemical properties. For instance, drug delivery vehicles of different sizes feature different biodistribution, efficacy, and safety.2 Complex coacervate core micelles (C3Ms)3–5 are promising vehicles for charged molecules. These micelles are also known as polyion complex micelles (PICs)6–8 or block ionomer complex micelles (BICs).9,10 Recently, we have opened a new vista to this field: by combining metal ions, bisligands, and neutral-charged diblock copolymers, novel C3Ms that contains metal ions in the core were prepared.11–14 In these novel micelles, metal ions and bisligand molecules form an anionic coordination polymer.15,16 Together with the cationic block of the diblock copolymer they form the complex coacervate cores of the C3Ms.11 These cores are stabilized against unbounded growth by the neutral blocks of the diblock copolymers which form the micellar corona. This micellar system can be used as nanocarrier for metal ions, which might be useful as contrast agent, for example, for nuclear magnetic resonance imaging (MRI), magnetic targeting, or for therapeutic applications. For applications, it is very important that the amount of the metal ions in the particle core can be controlled; there may be several ways of doing this. The first idea one may come up with is to vary the cationic block length of the diblock copolymer. However, this is feasible only within certain limits, since synthesis of rather long polyelectrolytes is not practical. In this work, we explore another option, namely, to control the loading amount of metal ions through formation of complex coacervate core microemulsions (C3-µEs). This would seem to be a very general method.
C3-µEs, in which an additional amount of complex coacervate is solubilized in the cores of the particles, are actually C3Ms swollen by a certain amount of extra complex coacervate, in analogy with droplet microemulsions formed from oil and surfactant.18 The principle to make C3-µEs is to use a mixture of charged-neutral diblock copolymers (dp) and like-charged homopolymers (hp) instead of only dp, in combination with oppositely charged hp, which is illustrated in Scheme 1. Optimal formation of particles occurs for isoelectric mixtures.
 |
| | Scheme 1 Schematic representation of formation of a complex coacervate core microemulsion (C3-µE) droplet. The core comprises of anionic homopolymer (green, hp−), the cationic homopolymer (purple, hp+), and the cationic block (red, dp+) of the diblock copolymer. The corona consists of the electroneutral hydrophilic blocks (blue) of the diblock copolymer. | |
The formation of such a kind of C3-µE from covalent polymers has been demonstrated recently by our group.17 Here we propose to use an anionic metal-bisligand coordination polymer (cp) instead of a covalent anionic hp. The amount of metal ions in the C3-µEs made in this way can be expressed in terms of the mixing parameter β, which is defined as β = 1 + N+hp/N+dp, where N+hp and N+dp represent the number of positive charges carried by the hp and dp, respectively. Static and dynamic light scattering, transmission electron microscopy, and electrophoresis were employed for characterization.
2. Results and discussion
The dp used in this work is poly(N-methyl-2-vinylpyridinium iodide)-b-poly(ethylene oxide), P2MVP41-b-PEO205, the same as used in our previous work.11–14 The hp is poly(N-methyl-4-vinylpyridinium iodide), i.e., P4MVP48. The cp self-assembles from zinc ions and bisligand compound pyridine-2,6-dicarboxylic acid groups connected at the 4-position of the pyridine ring by four ethylene oxide spacers (L2EO4) at a 1:1 molar ratio, the same as in our previous work.11–14 The structures of the dp, hp, bisligand molecule L2EO4, and formation of cp between zinc ions and L2EO4 are illustrated in Scheme 2.
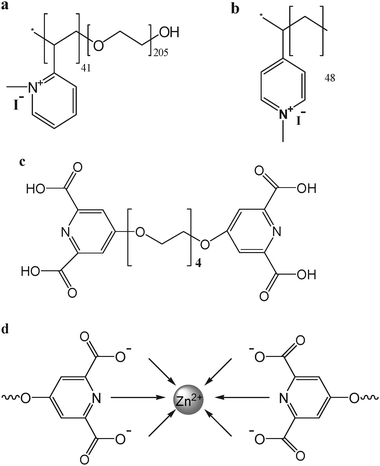 |
| | Scheme 2 Structure of (a) dp: P2MVP41-b-PEO205; (b) hp: P4MVP48; (c) bisligand L2EO4 and (d) schematic representation of coordination formation between zinc ions and L2EO4 (formation of cp). | |
The C3-µEs were prepared through two procedures: one is titration of the negatively charged cp to the positively charged mixture of hp and dp; the other one is direct mixing of a cp solution and a hp/dp mixed solution. These two methods result in C3-µEs of different polydispersities and sizes, indicating that history effects play a role. In the following, we present the corresponding results.
2.1 C3-µEs prepared by titration
With the first protocol, formation of particles was directly followed by light scattering. The variation of scattered light intensity with changing the charge fraction f− in the system is shown in Fig. 1a. The charge fraction f− is defined as
where N− and N+ are the overall amount of negative and positive charges carried by the polyelectrolytes in the system, respectively, with N+ = N+hp + N+dp.
![a) Variation of the scattered light intensity with f− upon addition of cp to the mixture of hp/dp for several values of β. The intensities have been normalized against the original total positive charge concentration ([+] = 0.87, 0.89, 0.91, 0.93 mM for β =1, 1.5, 2, 3, respectively. These concentrations correspond to 0.41, 0.42, 0.43, 0.44 g/L, respectively.). b) variation of the CUMULANT hydrodynamic radius with f− during titration of hp/cp mixtures of different β; c) variation of the hydrodynamic radius (solid squares) and polydispersity index (PDI, open triangles) with mixing ratio of hp/cp at f− =0.5. d) CONTIN size distribution of the C3-µEs at f− = 0.5 obtained by titration.](/image/article/2009/SM/b811497h/b811497h-f1.gif) |
| | Fig. 1 a) Variation of the scattered light intensity with f− upon addition of cp to the mixture of hp/dp for several values of β. The intensities have been normalized against the original total positive charge concentration ([+] = 0.87, 0.89, 0.91, 0.93 mM for β =1, 1.5, 2, 3, respectively. These concentrations correspond to 0.41, 0.42, 0.43, 0.44 g/L, respectively.). b) variation of the CUMULANT hydrodynamic radius with f− during titration of hp/cp mixtures of different β; c) variation of the hydrodynamic radius (solid squares) and polydispersity index (PDI, open triangles) with mixing ratio of hp/cp at f− =0.5. d) CONTIN size distribution of the C3-µEs at f− = 0.5 obtained by titration. | |
A sharp increase of the scattered light intensity was observed for all the systems (Fig. 1a), which indicates that C3-µEs were indeed formed when cp was added into the mixture of hp/dp. The scattered intensities for all the hp/dp mixtures of different β rise sharply around f− = 0.5 and level off at larger f−, indicating that optimal formation of C3-µEs occurs at the stoichiometric ratio and above. Apparently, the structure of these C3-µEs is not affected by excess coordination polymer in contrast to C3-µEs formed from conventional polyelectrolyte systems.17 Similar to the formation of normal C3Ms by using coordination polymers, this is attributed to the effect that coordination polymers only form in the core due to the presence of the oppositely charged block copolymers.11
Another striking feature accompanied with the formation of the C3-µEs is the significant increase of the scattered light intensity as β was increased. When no hp was added (β =1), the plateau scattered intensity is 76 kHz mM−1 under the experimental conditions; while the intensities are 570, 1130, 1760 kHz mM−1 for β = 1.5, 2, and 3, respectively. Actually, the scattered intensity was so strong for even larger β that it exceeded the limit of the detector under the same condition. Therefore, the laser power has to be lowered in order to measure systems with β ≥ 4. The greatly increased scattering intensity with increasing β indicates that the size of the C3-µEs increases when more dp are substituted by hp; this is clearly seen in Fig. 1b where the CUMULANT hydrodynamic radii of the above C3-µEs are shown. The plateau Rh values were taken as the size of the particles. Before the sizes reach constant, the particles are under the way of formation. Note that in Fig. 1b for the β = 1 system the particle size exhibits a maximum prior to level off, which is due to the formation of some worm-like micelles at f− around 0.3.12 These worm-like micelles coexist with spherical micelles and are actually a very small fraction in the whole system, which therefore contributes limit to the scattered light intensity. That is why we don't see a corresponding increase in the scattered light intensity in Fig. 1a. This phenomenon has been reported in a great detail in our previous work.12,18 From β = 1 to β = 3, the size of the C3-µEs increases from 25 nm to 130 nm. The variation of the particle size with β is shown in Fig. 1c. It is clear that the increase of the particle size is very steep as β changes from 1 to 3, whilst the increase levels off at β > 3, and Rh becomes constant beyond β = 5. Beyond β = 10 precipitation occurs, which is probably due to loss of colloidal stability, because there are no longer enough water soluble PEO blocks to stabilize the C3-µEs. This may be analogous to the phenomenon ‘emulsification failure’ in ordinary microemulsions at which the amount of oil exceeds the maximum emulsification capacity of the surfactant.19
The particle size distribution at charge neutral mixing ratio, i.e., f− = 0.5 was further analyzed with the CONTIN method. As is revealed in Fig. 1d, except in the micellar system, i.e.β = 1, two groups of particles are observed, where the small portion of larger particles are almost negligible, other systems with β > 1 show one broad peak, indicating that the particles in these systems are rather polydisperse (PDI ≥ 0.17, Fig. 1c).
2.2 C3-µEs obtained by direct mixing
2.2.1 Kinetic effects on particle size.
As we know from our previous work, kinetic effects do play a role in the formation of complex coacervate core particles.12 With the titration method some large objects are often formed. This does not occur with the direct mixing method. For instance, in titrations, two populations of micelles are seen for the β = 1 case (Fig. 1 d). The small peak at 150 nm does not appear upon direct mixing. When cp and dp were mixed directly, only small micelles were observed.11,12 To check whether effects of preparation history occur with the formation of C3-µEs, we also prepared them by direct mixing cp and dp/hp solutions at f− = 0.5.
It is obvious in Fig. 2a that the sizes of the C3-µEs freshly prepared by direct mixing are much smaller than those formed by titration. However, these sizes change with time. It takes about 2 days for the directly mixed systems to grow to the same size as the particles formed by titration. The maximum sizes were achieved within 4 days. Further prolongation of the waiting time resulted in a concentration gradient in the sample cuvette: the bluish hue increases in the vertical direction (from the upper to the lower) and ‘precipitates’ were observed in the bottom of the sample cuvette, indicating sediment occurs in the system. Correspondingly, a smaller particle size was detected by DLS. This is because the larger particles have sunk to the bottom of the cuvette, so that only smaller ones were left at the level where the laser beam traverses. This sediment can be easily redispersed simply by turning the cuvette up-side-down several times, and the redispersed particles have almost the same size as those before precipitation. As shown in Fig. 2b, the maximum size for the C3-µEs particles in the β = 8 system is about 142 nm; 1 week later, it becomes 120 nm; after shaking, the size comes back to 140 nm, and the size distribution did not change significantly. This means that the sediment does not consist of agglomerated material but consists of stable C3-µEs particles. Due to the presence of metal ions, the density of the cores is larger than water, so that the particles tend to sediment with time. The sedimentation follows Stokes' law:
| |  | (1) |
where
s is the distance the particles traveled during sedimentation,
ρp and
ρs are the densities of the particles and
solvent, respectively;
g, the acceleration due to gravity;
t, time to travel a distance
s;
η, viscosity of the
solvent; and
R, the radius of the particles. If we assume the density of the C3-
µE particles to be the same as that of the micelles formed in
cp/
dp mixed system, namely, 1.83 g/cm
3 (measured by densitometer
18), the time for 140 nm particles to sediment to the bottom of the cuvette is about 2 days. Since smaller particles sediment more slowly, one will observe a decrease of the number of the large particles in the beam, so that
DLS detects smaller particles upon standing for longer time. Comparison of the peak widths in
Fig. 1d and
2b indicates that the size distributions of the C3-
µE prepared by direct
mixing cp with the mixture of
hp/
dp are much narrower than those prepared by
titration, as can be clearly evidenced by comparison of the peak widths in
Fig. 1d and
2b. The distribution obtained by
titration at
β = 8 ranges from 40 to 800 nm, whereas the one obtained by direct
mixing is only between 70 and 300 nm!
 |
| | Fig. 2 a) Variation of the hydrodynamic radius with β for the direct mixing samples. The different symbols indicate results measured at different time intervals. The inset is the variation of radii with time for the β = 8 system. Solid circle: obtained by titration; square: standing for 10 minutes after direct mixing (same for the follows); open circle: standing for 1 days; upward triangle: standing for 2 days; diamond: standing for 7 days; downward triangle: standing for 4 days. b) CONTIN size distribution for β = 8 system after 3 days, 3 weeks of rest, and the latter where the precipitate was redispersed by gently shaking. | |
The fact that particles do not agglomerate indicates that they are stabilized by a PEO corona similarly as with normal unswollen C3Ms.20–22 Actually, in the temperature range of 10–80 °C, the size of the particles does not change, suggesting that the coacervates in the core are very stable and are well-stabilized by a neutral PEO corona. This is confirmed by the electrophoresis measurements. As summarized in Table 1, the ζ potentials of the particles have very small absolute values (−2 to +2 mV) regardless of the charge mixing ratio f−. These values are far smaller than the potentials required for stabilizing colloidal particles, indicating that the coacervate cores are sterically stabilized by the neutral PEO corona.
Table 1 ζ potential for the β = 8 cp/hp/dp mixed systems at different f−
|
f
−
|
0.2 |
0.3 |
0.5 |
0.80 |
| ζ (mV) |
1.2 |
−0.9 |
−1.2 |
−1.5 |
2.2.2 Morphology of the particles.
To further characterize the structure of the C3-µEs, we observed them by TEM. Owing to the presence of zinc ions in the cores of the particles, the electronic contrast between the particles and background is very strong, so that clear images were obtained. As shown in Fig. 3a–d, C3-µEs are spherical and rather polydisperse. Increase of size with increasing β is clearly indicated. The average radii obtained by TEM are 15–25 nm smaller than those from DLS measurements carried out on the same samples. This difference is simply the thickness of the PEO corona that stabilizes the particles, because TEM reveals just the cores of the particles. Due to the low electronic contrast of the PEO block, the PEO coronas cannot be directly inferred from the TEM pictures. The PEO corona thickness can also be deduced from half of the distance between two closely neighboring particles in Fig. 3d, which is about 18–24 nm. This agrees well with the results deduced from the comparison between DLS and TEM.
![TEM images for the particles in Zn-L2EO4/P2MVP41-PEO205/P4MVP48 mixed systems with various β, at f− = 0.5. [+] = [−] = 0.84 mM (corresponds to 0.40 g/L). From a to d: β = 1 (micelles), 1.5(C3-µEs), 3(C3-µEs), 7(C3-µEs), respectively.](/image/article/2009/SM/b811497h/b811497h-f3.gif) |
| | Fig. 3 TEM images for the particles in Zn-L2EO4/P2MVP41-PEO205/P4MVP48 mixed systems with various β, at f− = 0.5. [+] = [−] = 0.84 mM (corresponds to 0.40 g/L). From a to d: β = 1 (micelles), 1.5(C3-µEs), 3(C3-µEs), 7(C3-µEs), respectively. | |
2.3 Comparison with micelles formed by longer dp
The successful enlargement of the capacity for loading metal ions by preparation of C3-µEs is very significant for the potential application of the systems. Such a large increase of the loading capacity cannot be achieved by simply increasing the length of P2MVP block of the diblock copolymer. It has been demonstrated by many groups that only when the degree of the polymerization of the neutral block is 2–5 times that of the charged block, micelles can be formed.4,23 In case of using PEO205, it means that the micelles are formed only if the polymerisation degree of the P2MVP block does not exceed 100. This corresponds to an upper limit of a factor of 100/41 ≈ 2.5. Actually, for P2MVP41-PEO71 with a block ratio of 1:1.7, precipitates were indeed formed immediately. In this case, the system is a whitish suspension upon shaking, indicating agglomeration of particles. In contrast, all our C3-µEs systems are bluish, no matter fresh or old, shaken or not, which is the typical appearance for a colloidal solution.
The size of C3-µEs is much larger than we have expected. From geometrical considerations, the particle size is expected to be
| | | Rtotal = Rcorona + R0coreVrelcore (ref. 17), | (2) |
where
R0core is the core radius without added additional homopolyelectrolyte, which can be read from the cryo-TEM images directly, and equals about 9 nm.
11Vrelcore is the relative volume of the coacervate cores, which is the same as
β (in the case of
hp +
dp) or equal to
β (in the case of long
dp, where the definition for
β is not applicable). In combination with the DLS results, the corona thickness,
Rcorona can be derived as 15.5 nm, which is constant for the systems in
Table 2. According to
eqn (2), the predicted particle size with a complex coacervate volume of 1.7 should be 30.8 nm. When such a volume increase was achieved by increasing the charge block length, for instance, substitution of P2MVP
42-
b-PEO
446 by P2MVP
71-
b-PEO
452 in the mixed system,
i.e., by increasing the volume of the complex coacervates by a factor of 70%, the micellar size increases from 24.5 to 31 nm, which agrees perfectly with the experimental result, as shown in
Table 2. However, when such a complex coacervate volume was accomplished by
mixing a
homopolymer with a
diblock copolymer, the particle size becomes 80 nm, which is obviously much larger than the predicted value. The larger particle size in the latter case is probably a result of particle polydispersity. As can be seen from the TEM results, the C3-
µEs are rather polydisperse, whereas the C3Ms are more monodisperse. Actually, the size dependence of the C3-
µEs on
β is rather complicated, which may also be related to the type of background electrolytes.
17,24
Table 2 Particle size (Rh) enlarged by increasing the coacervate volume. The relative volume of coacervate (Vrelcore) formed between P2MVP42-b-PEO446 (dp) and cp was set as V = 1. V = 1.7 was achieved either by using P2MVP71-b-PEO446 (long dp) or mixing P2MVP42-b-PEO446 and a homopolymer P4MVP48 (hp + dp). The predicted size by using geometrical rule is listed for comparison
|
V
rel
core
|
Cationic polymers |
Denotation |
R
h/nm |
| 1 |
P2MVP42-b-PEO446 |
dp
|
24.5 |
| 1.7 |
P2MVP42-b-PEO446 + P4MVP48 |
hp + dp |
80 |
| 1.7 |
P2MVP71-b-PEO446 |
Long dp |
31 |
| 1.7 |
— |
Prediction |
30.8 |
The swelling of the complex coacervate core micelles contributes a new approach for controlling supramolecular assembly and lures it to application. In a similar way, by simply varying the mixture composition, we can reduce the amount of metal ions in the system by substituting part of the coordination polymer by metal-free covalent polyanions. The adjustability of the amount of metal ions per particle greatly improves their potential as nanocarriers for metal ions. When proper metal ions (or mixture thereof) are used, these C3-µEs can be used for target delivery and cell imaging, including magnetic resonance imaging (e.g. Fe2+, Fe3, Ga3+, etc.), fluorescence imaging (e.g. Ho3+ etc), and therapeutic applications. Our present report helps to relate fundamental research on the complex coacervate micelles and their possible application as functional materials.
3. Experimental
3.1 Materials
The diblock copolymer (dp) poly(N-methyl-2-vinyl pyridinium iodide)-b-poly(ethylene oxide) (dp = P2MVP41-b-PEO205) (Scheme 2a), was quaternized as previously described11–13 from poly(2-vinylpyridine)-b-poly(ethylene oxide) (PVP41-b-PEO205, obtained from Polymer Source, Mw/Mn = 1.03, Mw = 13.3 kg/mol) up to 90% quaternization degree. Other diblock copolymers, PVP42-b-PEO446, PVP71-b-PEO452, and PVP41-b-PEO71 were prepared in the same procedure. The homopolymer (hp) poly (N-methyl-4-vinyl pyridinium iodide) (hp = P4MVP48, Scheme 2b.) was purchased from Polymer Source at a quaternization degree of 98% and used as received.
The coordination polymer was prepared according to Vermonden et al15,16 by mixing equimolar amounts of zinc nitrate and a bisligand compound based on pyridine-2,6-dicarboxylic acid groups connected at the 4-position of the pyridine ring by a spacer of four ethylene oxide units. The structure of the bisligand and its coordination with zinc ions is shown in Scheme 2c and 2d, respectively. The coordination polymers formed between zinc ions and the bisligand are mainly small rings at concentration lower than 20 mM,15,16 but they exist as long chains in the core of complex coacervate core micelles due to the high local concentration.11 In this study, coordination polymer solutions of < 1 mM were used, that means we started from small rings of the coordination polymer for all of the experiments. All the stock solutions were prepared in 20 mM PIPES-NaOH buffer of pH 5.4 before mixing.
3.2 Methods
3.2.1 Light scattering titration.
Light scattering (LS) measurements were performed with an ALV laser light scattering apparatus, equipped with a 400 mW argon ion laser operating at a wavelength of 514.5 nm. A refractive index matching bath of filtered cis-decalin surrounded the cylindrical scattering cell, and the temperature was controlled within ±0.5 °C at 25 °C using a Haake C35-F8 thermostat. Titrations were carried out using a Schott-Geräte computer-controlled titration setup to control both sequential addition of titrant and stirring of the cell. The concentrations of both the titrant and titrated solutions are expressed in terms of ionic concentrations. The charge concentration of the titrant is always about 5 times higher than that of the titrated solution. After every dosage, the solution is automatically stirred with a stirring bar for 60 seconds, during which no LS data are taken. Then, after a waiting time of 30 seconds, the LS measurement is started. After each titration step, the laser light scattering intensity (I) at 90°, and the intensity auto correlation function were recorded. The CUMULANT method was used to analyze data to obtain both the mean apparent hydrodynamic radius (Rh) and the polydispersity index (PDI). Rh is calculated from the inverse decay time Γ of the 2nd order fitting and the width of the distribution as well as the PDI is calculated from the second moment µ2, according to the following formulas:| | ![[D with combining macron]](https://www.rsc.org/images/entities/i_char_0044_0304.gif) = = ![[capital Gamma, Greek, macron]](https://www.rsc.org/images/entities/char_e0ba.gif) /q2 /q2 | (3) |
| | q = 4πn![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) sin(θ/2)/λ sin(θ/2)/λ | (4) |
| | | Rh = kT/6πη | (5) |
where q is the scattering vector, k the Boltzmann constant, T the absolute temperature, η the viscosity of the solvent, and the mean diffusion coefficient of the particles. Variations in the mean Rh and I are studied as a function of the mole fraction of negative charge ratios f− for several values of β. For all experiments, the scattering intensity I is corrected for volume changes and normalized with the initial concentration of the titrated species in the following way:where Im is the measured intensity, Vd and Vt are the volumes of the dosed agent and of the titrated species, respectively, and C is the original molar concentration of the positive charges (titrated species) before titration. The CONTIN method is used to analyze the distribution of particle radii. This method extracts a discrete distribution G(Γ) from the measured field correlation function, g1(t):| |  | (8) |
Analogously to eqn (1) and (3), each relaxation rate Γn can be related to a diffusion coefficient:
each with an associated hydrodynamic radius.
3.2.2 Transmission electronic microscopy (TEM).
A few microliters of diluted emulsion were placed on a formvar-coated copper TEM grid (Plano, 300 mesh), and the excess liquid was removed with filter paper. This sample was observed directly with JEOL100 (CX) operated at 80 kV.
3.2.3 ζ-Potential measurement.
The electrophoretic mobility of the particles in 20 mM pH 5.4 PIPES buffer was determined with a Zetasizer 2000 (Malvern Instruments) with an attached PC running the accompanying software (PCS v1.51). This instrument was operating at 25 °C with a 15 mW laser operating at a wavelength of 635 nm. The ζ-potential of the particles was calculated from the measured electrophoretic mobility according to the Smoluchowski equation as follows:Where η is the viscosity of the solution, u is the electrophoretic mobility, ε0 is the dielectric constant in vacuum, and εr is the relative dielectric constant. The product ε0εr is the absolute dielectric constant or dielectric permittivity.
4. Conclusion
We have successfully prepared novel, capacity-controllable nanocarriers for metal ions, through formation of complex coacervate core microemulsions. The C3-µEs particles are sterically stabilized by the PEO coronas surrounding the complex coacervate cores. By using combinations of diblock copolymer and homopolymer of like charge, the amount of metal ions per particle can be increased by as much as 10 times. By using an oppositely charged homopolyelectrolyte, we expect to decrease the amount of metal ions in these C3-µEs particles. Hence, the particle size and internal composition can be simply set by choosing the compositions of the mixture from which they are formed.
Acknowledgements
The authors thank Dr. A. T. M. Marcelis (Laboratory of Organic Chemistry, Wageningen University, Dreijenplein 8, 6703 HB Wageningen, The Netherlands) for the help with synthesis of L2EO4 bisligands; Dr. I. K. Voets for providing P2MVP42-b-PEO446, P2MVP71-b-PEO446, and P2MVP41-b-PEO71. Financial support is from the EU POLYAMPHI/Marie Curie program (RT6-2002, proposal 505027) and SONS Eurocores program (Project JA016-SONS-AMPHI).
References
- A. C. Balazs, T. Emrick and T. P. Russell, Science, 2006, 314, 1107 CrossRef CAS.
- L. E. Euliss, J. A. DuPont, S. Gratton and J. De Simone, Chemical Society Reviews, 2006, 35, 1095 Search PubMed.
- M. A. Cohen Stuart, N. A. M. Besseling and R. G. Fokkink, Langmuir, 1998, 14, 6846 CrossRef.
- S. Van der Burgh, A. de Keizer, Stuart and M. A. Cohen Stuart, Langmuir, 2004, 20, 1073 CrossRef CAS.
- M. A. Cohen Stuart, B. Hofs, I. K. Voets and A. de Keizer, Curr. Opin. Colloid Interface Sci., 2005, 10, 30 CrossRef.
- A. Harada and K. Kataoka, Macromolecules, 1995, 28, 5294 CrossRef CAS.
- A. Harada and K. Kataoka, Macromolecules, 1998, 31, 288 CrossRef CAS.
- Y. Lee, S. Fukushima, Y. Bae, S. Hiki, T. Ishii and K. Kataoka, J. Am. Chem. Soc., 2007, 129, 5362 CrossRef CAS.
- A. V. Kabanov, T. K. Bronich, V. A. Kabanov, K. Yu and A. Eisenberg, Macromolecules, 1996, 29, 6797 CrossRef CAS.
- A. V. Kabanov, T. K. Bronich, V. A. Kabanov, K. Yu and A. Eisenberg, J. Am. Chem. Soc., 1998, 120, 9941 CrossRef CAS.
- Y. Yan, N. A. M. Besseling, A. de Keizer, A. T. M. Marcelis, M. Drechsler and M. A. Cohen Stuart, Angew. Chem. Int. Ed, 2007, 46, 1807 CrossRef CAS.
- Y. Yan, N. A. M. Besseling, A. de Keizer, R. Fokkink, M. Drechsler and M. A. Cohen Stuart, J. Phys. Chem. B, 2007, 111, 11662 CrossRef CAS.
- Y. Yan, N. A. M. Besseling, A. de Keizer, R. Fokkink and M. A. Cohen Stuart, J. Phys. Chem. B, 2007, 111, 5811 CrossRef CAS.
- Y. Yan, A. de Keizer, M. A. Cohen Stuart, M. Drechsler and N. A. M. Besseling, J. Phys. Chem. B, 2008, 112, 10908 CrossRef CAS.
- T. Vermonden, W. M. De Vos, A. T. M. Marcelis and E. R. Sudhölter, Eur. J. Inorg. Chem, 2004, 2847 CrossRef CAS.
- T. Vermonden, J. Van der Gucht, P. De Waard, A. T. M. Marcelis, N. A. M. Besseling, E. J. R. Sudhölter, G. J. Fleer and M. A. Cohen Stuart, Macromolecules, 2003, 36, 7035 CrossRef CAS.
- B. Hofs, A. de Keizer, S. van der Burgh, F. A. M. Leermakers and M. A. Cohen Stuart, Soft matter, 2008, 4, 1473 RSC.
- Y. Yan, L. Harnau, N. A. M. Besseling, A. de Keizer, M. Ballauff, S. Rosenfeldt and M. A. Cohen Stuart, Soft matter., 2008, 4, 2207 RSC.
-
V. Degiorgio and M. Corti, Physics of Amphiphiles: Micelles, Vesicles and Microemulsions, North-Holland, Amsterdam, 1985 Search PubMed.
- M. Oishi, K. Kataoka and Y. Nagasaki, Bioconjugate Chem., 2006, 17, 677 CrossRef CAS.
- D. Wakebayashi, N. Nishiyama, K. Itaka, K. Miyata, Y. Yamasaki, A. Harada, H. Koyama, Y. Nagasaki and K. Kataoka, Biomacromolecules, 2004, 5, 2128 CrossRef CAS.
- D. Wakebayashi, N. Nishiyama, Y. Yamasaki, K. Itaka, N. Kanayama, A. Harada, Y. Nagasaki and K. Kataoka, J. Controlled Release, 2004, 95, 653 CrossRef CAS.
- J. F. Berret, A. Sehgal, O. Sandre, A. Vacher and M. Airian, J. Colloid. Interface Sci., 2006, 303, 315 CrossRef CAS.
- A. Harada and K. Kataoka, Macromolecules, 2003, 36, 4995 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here. 