[18F]- and [11C]-Labeled N-benzyl-isatin sulfonamide analogues as PET tracers for Apoptosis: synthesis, radiolabeling mechanism, and in vivo imaging study of apoptosis in Fas-treated mice using [11C]WC-98†
Dong
Zhou
a,
Wenhua
Chu
a,
Delphine L.
Chen
a,
Qi
Wang
a,
David E.
Reichert
a,
Justin
Rothfuss
a,
Andre
D'Avignon
b,
Michael J.
Welch
a and
Robert H.
Mach
*a
aDivision of Radiological Sciences, Washington University School of Medicine, Campus Box 8225 510 S. Kingshighway Blvd. St. Louis, Missouri 63110, USA. E-mail: rhmach@mir.wustl.edu
bDepartment of Chemistry, Washington University, One Brookings Dr., St. Louis, Missouri 63130, USA
First published on 16th February 2009
Abstract
The radiolabeled isatin sulfonamide caspase-3 inhibitor, [18F]2 (WC-II-89), is a potential PET radiotracer for noninvasive imaging of apoptosis. The radiolabeling mechanism was studied by 13C NMR, ESI/MS, and computational calculations. It was found that the high electrophilicity of the C3 carbonyl group in the isatin ring, which served as a trap for [18F]fluoride, was responsible for the failure of the radiolabeling via nucleophilic substitution of the mesylate group in 7a by [18F]fluoride. Once treated with a strong base, 7a opened the isatin ring completely to form an isatinate intermediate 16, which lost the ability to trap [18F]fluoride, thereby allowing the displacement of the mesylate group to afford the 18F-labeled isatinate 17. [18F]17 can be converted to isatin [18F]2 efficiently under acidic conditions. The ring-opening and re-closure of the isatin ring under basic and acidic conditions were confirmed by reversed phase HPLC analysis, ESI/MS and 13C NMR studies. Computational studies of model compounds also support the above proposed mechanism. Similarly, the ring-opening and re-closure method was used successfully in the synthesis of the 11C labeled isatin sulfonamide analogue [11C]4 (WC-98). A microPET imaging study using [11C]4 in the Fas liver apoptosis model demonstrated retained activity in the target organ (liver) of the treated mice. Increased caspase-3 activation in the liver was verified by the fluorometric caspase-3 enzyme assay. Therefore, this study provides a useful method for radio-synthesis of isatin derivative radiotracers for PET and SPECT studies, and [11C]4 is a potential PET radiotracer for noninvasive imaging of apoptosis.
Introduction
“Molecular imaging is the visualization, characterization and measurement of biological processes at the molecular and cellular levels in humans and other living systems.”1Positron emission tomography (PET), one of the in vivo imaging techniques used in molecular imaging, is being used more frequently in clinical and research fields because of its high sensitivity, minimal physiological effect from PET tracers, good spatial resolution and ease of accurate quantification. One important application of molecular imaging is to study programmed cell death (apoptosis) at the molecular level. Apoptosis is critical for the normal development and function of multicellular organisms as a common and universal mechanism of cell death.2 The abnormal regulation of cellular deathviaapoptosis is believed to play a key role in a variety of human diseases.3 In addition, the beneficial effect of chemotherapy, radiotherapy, and other antitumor therapies can be attributed to their activation of the apoptotic process.4 Therefore, the development of a noninvasive imaging procedure that can study the process of apoptosis in a variety of disease states and monitor the ability of a drug or other treatment either to induce or to halt apoptosis would be of tremendous value to the research and clinical community.One of the most commonly used agents so far for imaging apoptosisin vivo is based on Annexin V, which is a 36 kDa protein that binds selectively with high affinity to phosphatidylserine, a protein that is externalized in the early stages of apoptosis after the activation of caspase-3. Annexin V has been labeled with different radioisotopes for PET and single photon emission computed tomography (SPECT) studies.599mTc-labeled Annexin V using SPECT showed promising results and is undergoing evaluation in clinical trials.6 However, since the externalization of phosphatidylserine also occurs in necrosis, radiolabeled Annexin V is not specific for imaging apoptosisin vivo. An alternative strategy for visualizing cells and tissues undergoing apoptosis is the use of peptides or small molecules directed toward enzymes that are intrinsic to the biochemical pathways of apoptosis. The enzymes responsible for the regulation and execution of apoptosis are the caspases, which exist as inactive zymogens (pro-caspases) in the cytosol and become activated when cells receive an apoptotic signal. Caspases activated early in the process of apoptosis are known as initiator caspases, such as caspase-2, -8, -9, and -10. The function of the initiator caspases is to activate the executioner caspases, caspase-3, -6, and -7, which are responsible for the proteolytic cleavage of proteins that are necessary for cellular function. Cellular apoptosis can be initiated via two different pathways, extrinsic and intrinsic.7 Since both pathways converge at the level of caspase-3 activation, immunohistochemical staining for activated caspase-3 provides an unambiguous marker for apoptosis.

A number of isatin (1) sulfonamide analogues have been reported to have high inhibition potency for the executioner caspases, caspase-3 and -7, and high selectivity against other caspases.8 One of these isatin sulfonamide analogues, 1-[4-(2-fluoroethoxy)-benzyl]-5-(2-phenoxymethyl-pyrrolidine-1-sulfonyl)-1H-indole-2,3-dione, 2 (WC-II-89), has been labeled with 18F and evaluated in biodistribution studies with the cycloheximide liver injury model in rats, a well-established murine model of chemically-induced apoptosis. The biodistribution studies of [18F]2 revealed higher uptake in the liver and spleen in cycloheximide-treated rats when compared with untreated control rats. We also reported the first microPET imaging study directly measuring caspase-3 activation in tissues undergoing apoptosis using [18F]2 as a PET radiotracer.9 Given the central importance of apoptosis in a variety of disease states as well as in new drug development, the development of [18F]2 as a promising radiotracer for PET imaging of apoptosis is significant in studying apoptosisin vivo.
The requisite radiolabeling reactions usually take place via the nucleophilic substitution of a good leaving group such as mesylate, tosylate or triflate under basic conditions for 18F labeling10 or methylation using [11C]CH3I in the presence of a base for 11C labeling.11 These labeling reactions are very different from the normal chemical synthesis: the half lives of 18F (t½ = 110 min) and 11C (t½ = 20.4 min) are short, limiting the reaction time; therefore, fast reaction times and simple procedures are required. Secondly, only tracer amounts of radioactive isotopes, such as [18F]fluoride and [11C]CH3I, are present to react with a large excess of substrate. The labeling reaction can benefit from the large amount of substrate to increase the rate of the reaction, but at the same time, any amount of reactive impurities or other reactive functional groups in the substrate might react with [18F]fluoride or [11C]CH3I, generating side-products slowing down or even stopping the desired radiolabeling reaction.
A variety of reactions can take place on the isatin ring in the isatin analogues.12 One such reaction is the nucleophilic attack on the C3 carbonyl carbon of the isatin ring, the “warhead group” that is crucial to the inhibition of caspase-3 by isatin sulfonamide analogues.13 Although isatin sulfonamide analogues exhibit very good potency for inhibiting caspase-3, the preparation of radiolabeled isatin sulfonamide analogues with 18F or 11C for PET imaging is limited because of the difficulty of radiolabeling with 18F or 11C using the standard methods.14 In order to improve the synthesis of such compounds, we sought a better understanding of the radiolabeling reaction, and examined the mechanism through both experimental and modeling studies. Furthermore, related isatin derivatives have been reported as drug candidates for their inhibition of SARS Coronavirus 3C-like Protease,15GSK-3 protease,16 carboxylesterases17 and HIV-1 reverse transcriptase.18 They have also exhibited cytotoxicity to cancer cellsin vitro.19 This study could provide a useful method to synthesize isatin derivative radiotracers for both PET and SPECT studies. Here we report the radiolabeling reactions of N-benzyl-isatin sulfonamide analogues with 18F and 11C, the mechanism and modeling studies of the labeling reactions and evaluation of the 11C compound in an in vivo microPET imaging study with the Fas liver injury model in mice.
Results and discussion
Synthesis and Radiolabeling
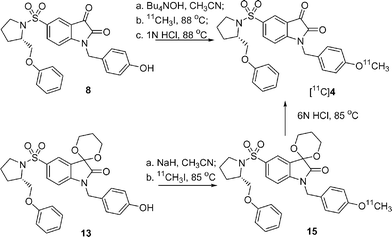
The syntheses of standard compounds 2, 4 and precursors 7a, 7b and 12 for 18F labeling of 2 and 8, and 13 for 11C labeling of 4, are shown in Scheme 1. The isatin nitrogen of 5-(2-phenoxymethyl-pyrrolidine-sulfonyl)-1H-2,3-dione 3 was alkylated by treatment of 3 with sodium hydride in DMF at 0 °C for 20 min and followed by addition of various alkyl halides to give compounds 2, 4, 5, and 6 respectively. Compound 5 was then heated to reflux with silver methanesulfonate or silver p-toluenesulfonate in CH3CN to generate precursors 7a or 7b. Precursor 8 was prepared by hydrolysis of 6 with sodium hydroxide in aqueous methanol. For the synthesis of precursors 12 and 13, the C3 carbonyl group in 3 was first protected as 1,3-dioxane by refluxing 3 and 1,3-propanediol in benzene using p-toluenesulfonic acid as a catalyst to give 9, which was then alkylated with 4-bromoethyloxybenzyl bromide or 4-acetoxybenzyl chloride to give compounds 10 or 11 respectively. Then precursors 12 and 13 were prepared with the same procedure as for 7a and 8. | ||
| Scheme 1 Synthesis of standards and their precursors. | ||
The methods for synthesis of [18F]2 are summarized in Scheme 2. First, precursor 7a or 7b was converted completely to the isatinate salt by treatment with strong base such as Bu4NOH or K2CO3/Kryptofix 222 (K222) in order to avoid the trapping effect due to the C3 carbonyl group in isatin; then the nucleophilic substitution reaction took place under standard labeling conditions to replace the mesylate or tosylate groups affording the 18F labeled isatinate salt of 2 in high yield, which was then converted quickly to [18F]2 in high yield with 1 N HCl under microwave irradiation. [18F]2 was confirmed by reversed phase HPLC co-eluting with non-radioactive standard 2. The synthesis time is 90 min, the labeling yield is high, the chemical and radiochemical purities are good and the specific activity is high (2600 ± 1276 mCi/mmol, n = 15). Alternatively, [18F]2 was synthesized from the C3 carbonyl-protected mesylate precursor 12, followed by hydrolysis of 14 using 1 N HCl. In a similar manner, [11C]4 was synthesized as shown in Scheme 3. All of these reactions provide [18F]2 and [11C]4 with high yield and high specific activity, suitable for use in a molecular imaging study of apoptosis.

Mechanism of the Radiolabeling Reaction
The typical procedure for 18F labeling of organic compounds via nucleophilic substitution of good leaving groups by [18F]fluoride is described as following: aqueous [18F]fluoride, produced by the nuclear reaction 18O(p,n)18F, is dried in the presence of Kryptofix 222 (K222) and K2CO3viaazeotropic distillation with successive additions of CH3CN to give free [18F]fluoride, which is a good nucleophile. Fluorination is then carried out in polar aprotic solvents, such as DMSO, DMF or CH3CN under conventional heating or microwave irradiation.20 However, the labeling reaction of mesylate precursor 7a using the standard method afforded either no labeling products or a more polar product in low yield according to TLC or HPLC analysis. Interestingly, if the precursor 7a was first treated with 2 equivalent of dried Bu4NOH, then the labeling reaction took place efficiently to form a more polar product in high yields, which could then be converted to the desired product [18F]2 gradually at room temperature or quickly at elevated temperature under acidic conditions. These results suggest that the [18F]fluoride cannot displace the mesylate group directly in 7a, but it does displace the mesylate group from an intermediate formed under basic conditions. A proposed mechanism for this observation in the 18F labeling reaction of 2 is shown in Scheme 4.![Labeling mechanism of [18F]2](/image/article/2009/OB/b819024k/b819024k-s4.gif) | ||
| Scheme 4 Labeling mechanism of [18F]2 | ||
In terms of nucleophilic addition on the isatin ring, nucleophiles such as H2O, OH− or F− can add to the C3 carbonyl carbon easily to form the corresponding species in a reversible reaction. However, the OH−group can also attack the C2 carbonyl carbon resulting in the opening of the isatin ring. This reaction is irreversible under basic conditions. Therefore, the isatin might exist in different forms in solution, such as the reversible hydrated form, 18, and the irreversible ring-opened form, 16, depending on the pH of the solution and the solvent used. Isatin-5-sulfonate itself was reported to exist as an isatin and a hydrated form in a 3–4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by NMR.21 In aqueous solution, the hydrate form is suggested by HPLC peak fronting observed in reversed phase C18 HPLC chromatography using CH3CN and water (pH = 7 or 4) as solvents. The peak fronting should be due to the equilibrium of isatin and its more polar hydrated form, resulting from the interaction with water in the HPLC solvents. This peak fronting can be suppressed by addition of methanol to the HPLC solvent system. The formation of the hydrated form corroborates the high electrophilicity of the isatin ring at the C3 carbon. In the case of 18F labeling reactions of isatin analogues, free [18F]fluoride, as a good nucleophile, can add to the C3 carbonyl carbon to form an intermediate 20. Although the intermediate 20 is in equilibrium with 7a, the limiting amount of [18F]fluoride (<10−7 mol for 100 mCi [18F]fluoride with specific activity of >1000 mCi/μmol) used in the labeling reaction is only one twentieth or even one hundredth of the amount of the precursor substrate used (1 mg, 2 × 10−6 mol) in the reaction. Therefore, the C3 carbonyl group in isatin analogues serves as a trap for [18F]fluoride and results in the failure of the substitution of mesylate or tosylate groups by [18F]fluoride. When the precursor 7a was treated with strong base (OH−), either the ring opening form 16 or the hydrated intermediate 18 was formed completely so that the chance for [18F]fluoride to be trapped by the isatinC-3 carbonyl group was eliminated. Under these conditions, [18F]fluoride will substitute the mesylate group to give the radiolabeled product 17 or 19, which will be converted to [18F]2 under acidic conditions.
:1 ratio by NMR.21 In aqueous solution, the hydrate form is suggested by HPLC peak fronting observed in reversed phase C18 HPLC chromatography using CH3CN and water (pH = 7 or 4) as solvents. The peak fronting should be due to the equilibrium of isatin and its more polar hydrated form, resulting from the interaction with water in the HPLC solvents. This peak fronting can be suppressed by addition of methanol to the HPLC solvent system. The formation of the hydrated form corroborates the high electrophilicity of the isatin ring at the C3 carbon. In the case of 18F labeling reactions of isatin analogues, free [18F]fluoride, as a good nucleophile, can add to the C3 carbonyl carbon to form an intermediate 20. Although the intermediate 20 is in equilibrium with 7a, the limiting amount of [18F]fluoride (<10−7 mol for 100 mCi [18F]fluoride with specific activity of >1000 mCi/μmol) used in the labeling reaction is only one twentieth or even one hundredth of the amount of the precursor substrate used (1 mg, 2 × 10−6 mol) in the reaction. Therefore, the C3 carbonyl group in isatin analogues serves as a trap for [18F]fluoride and results in the failure of the substitution of mesylate or tosylate groups by [18F]fluoride. When the precursor 7a was treated with strong base (OH−), either the ring opening form 16 or the hydrated intermediate 18 was formed completely so that the chance for [18F]fluoride to be trapped by the isatinC-3 carbonyl group was eliminated. Under these conditions, [18F]fluoride will substitute the mesylate group to give the radiolabeled product 17 or 19, which will be converted to [18F]2 under acidic conditions.
13C-NMR Studies
In order to distinguish which intermediate, 16 or 18, was involved in the following labeling reaction, a 13C NMR study was carried out on samples of 7a, under the conditions simulating the labeling reaction. The 13C chemical shift of the C3 carbonyl carbon should be around 90 ppm for the hydrated form 18 and 200 ppm for the ring-opening form 16.21 In DMSO, once precursor 7a was treated with 1 or 2 equivalent of 1 M Bu4NOH in water, a clean formation of the ring-opened product isatinate 16 was observed based on the observation that a new set of 13C NMR peaks formed with the complete disappearance of those of 7a and that the chemical shift for C3 carbonyl carbon is shifted from 181.5 ppm for 7a to 200.6 ppm, which is the chemical shift expected for the ring-opened isatinate form 16. The 13C NMR peaks (around 90 ppm) of the C3 carbonyl carbon for the hydrated form 18 were not observed. (For example, the C3 of 12 is at 93.3 ppm). Similarly, the C3 of N-benzyl isatin 21, which was used for the modeling study, shifts from 183.1 ppm to 201.0 ppm in its ring-opened isatinate form 22 (ESI Figure S7–S14†). Therefore, intermediate 18 is not likely to be involved in the labeling reaction and the ring-opened isatinate form 16 should be the species that is being labeled. We also attempted to study the formation of the fluoride adduct with 21 by 13C and 19F NMR. The 13C NMR study of a reaction mixture of 21 and Bu4NF in DMSO indicated the gradual formation of isatinate which should be due to the unavoidable OH−/H2O in Bu4NF solution. By comparison with a control reaction, in which no 21 was added, the signal-to-noise ratio in 19F NMR of Bu4NF was reduced after addition of 21. This observation indicated that there is a fast exchange of fluoride with isatin and the observation supports our assumption that [18F]fluoride is trapped by the isatin C3 carbonyl group instead of substituting the mesylate group during the 18F labeling of 2 under standard conditions (ESI Figure S5–S6†).Molecular Modeling Studies
In order to evaluate the electrophilicity of isatin and their isatinates, DFT calculations were performed using G9822 in order to compare the reactivity of the C3 carbon towards nucleophilic attack. Calculations were carried out on the simplified isatin precursor 23 and its analogues to obtain the nucleophilic reactivity Fukui function (FF).23 The frontier electron theory or Fukui function has been widely used as a local density functional descriptors to model chemical reactivity and site selectivity. The atom with the highest FF is highly reactive compared to the other atoms in the molecule. As confirmed by the theoretical calculations, the C3 carbon of the simplified precursor 23 has the highest electrophilicity (FF= 0.24) in the molecule before the isatin ring opens; while its electrophilicity is lowered tremendously once it was converted into its isatinate form 24 (FF= 0.04). Its reactivity also decreases considerably in the hydrate form 25 (FF= 0.12), while the reactivity of the carbon attached to the mesylate group (C-OMs) undergoes no change. This is in good agreement with the proposed radiolabeling mechanism.
Optimization of Radiolabeling Conditions
Once the labeling mechanism was elucidated, the amount of K2CO3 used in the labeling reaction and the effect of water on the reaction yield were studied. When only K2CO3 was used in the labeling reaction, it was found that about 2 mg of K2CO3 was needed in order to obtain a high yield of the labeling product (Table 2, entry 4). When less than 1 mg of K2CO3 was used, there was no incorporation after initial microwave irradiation, and only after more K2CO3/K222 in DMSO was added, the yield increased to a comparable value as when 2 mg K2CO3 used (Table 1, entry 3). When [18F]fluoride in the presence of K222 and K2CO3 was dried very carefully, the yield was low. This is understandable because K2CO3/K222 is a weaker base than Bu4NOH and it doesn't provide OH− in the absence of water to open the isatin ring. Therefore, if K2CO3 was used as a base in the labeling reaction, it needs a longer reaction time and a trace amount of H2O to open the isatin ring completely in order to avoid the trapping effect from the C3 carbonyl group. Normally, water should be excluded in the labeling reaction because water can solvate [18F]fluoride, reducing its nucleophilicity, lowering its reactivity and resulting in the failure of the nucleophilic substitution. As expected when the isatin precursor was treated with wet Bu4NOH, a very low yield of labeling product was obtained (Table 1, entry 2) in comparison to treatment with dry Bu4NOH (Table 1, entry 1). Therefore, the labeling reaction conditions were optimized by using a large amount of K2CO3 to open the isatin ring completely and to consume trace amounts of water left during the drying of [18F]fluoride, affording [18F]2 in very high yields.| Reaction Entry | Substrate/mg | K2CO3/K222/mg | Solvent | Reaction Conditionb | % Incorporation Yield (product)d |
|---|---|---|---|---|---|
| a Bu4NOH pre-treatment method: with 2 equivalents of Bu4NOH; b MW: Microwave (60 W) with an interval of 1 min between each irradiation; c The reaction mixture was treated further with K2CO3 (3mg) and K222 (15mg) in DMSO; d Incorporation yields were determined by radioTLC (silica gel, 20% MeOH, 80% CH2Cl2). | |||||
| 1 | 7a (1.1)a | 1.4/5.5 | DMSO | 30 + 25 s (MW) | 90 (17) |
| 2 | 7a (1.2) awet | 1.6/5.5 | DMSO | 30 + 25 s (MW) | 19 (17) |
| 3 | 7a (1.0) | 0.8/4 | DMSO | 30 (MW) 30 c(MW) | 0 (17) 86 (17) |
| 4 | 7a (1.1) | 1.8/8.6 | DMSO | 30 + 30 Sec (MW) | 89 (17) |
| 5 | 12 (1.2) | 1.4/5.0 | DMSO | 30 + 25 s (MW) | 80 (14) |
| 6 | 7b (1.0) | 2.1/12 | DMSO | 30 (MW) | 65 (17) |
An alternative route to avoid the trapping of [18F]fluoride by the C3 carbonyl group in the isatin ring is to protect the C3 carbonyl group. Once protected, it loses the ability for nucleophilic addition by [18F]fluoride. The mesylate precursor 12 for [18F]2 was protected at the C3 position with 1,3-dioxane was stable to the basic labeling condition. Under standard labeling conditions, the mesylate group was replaced with [18F]fluoridevia nucleophilic substitution to form the 18F labeled intermediate 14 (Table 2, entry 5). Then 14 was deprotected with 1 N HCl under microwave irradiation to afford [18F]2. The successful labeling of the C3 carbonyl-protected precursor 12 again indicates the involvement of this carbonyl in the 18F labeling of 2 from the unprotected precursor 7a.
Hydrolysis of isatin and N-methyl isatin and recyclization of isatinates have been studied,24 but the study of other N-substituted isatins is quite limited. The stability of N-benzylisatins towards base hydrolysis is expected to be better than that of isatin or N-methyl isatin because of the steric protection from the benzyl substitute on the nitrogen atom in the isatin ring. This stability enables N-benzyl-isatinsulfonamide analogues to function as potential tracers for imaging caspases-3/7 activity in vivo. The hydrolysis of N-benzyl-isatinsulfonamides was studied by reversed phase HPLC and ESI/MS. According to HPLC analysis of the reaction mixture, Bu4NOH can hydrolyze isatin analogues to form isatinates exclusively and quickly; the hydrolysis by K2CO3/K222 was quick, but the hydrolysis by NaHCO3/H2O was slow and in neutral solution the analogues were very stable. Once the isatinates were treated with 1 N HCl, they were converted back to isatin analogues gradually at room temperature or quickly under heating or microwave irradiation. The base hydrolyzed isatin analogues and acid recyclized isatinates were purified by reversed phase HPLC and the HPLC purified products were further analyzed by ESI/MS. Molecular ion plus H2O was observed as the major peak for the hydrolyzed product in ESI/MS, together with the 13C NMR study, indicating the formation of isatinate; The formation of isatin analogues from acid recyclization of isatinates was confirmed by comparison of the retention time in HPLC and MS spectrum with the corresponding isatin analogues (ESI Figure S3, S4†).
For the classic alkylation of hydroxyl or nitrogen groups, NaH is commonly used as a base to deprotonate, and it was used in the N-alkylation of 3 during the synthesis of standard and precursor N-benzylisatin sulfonamides. However, in the 11C methylation of 8 by [11C]CH3I, where excess of NaH was used, the 11C labeling of 8 failed to afford the expected radiolabeled product [11C]4, but led to the formation of numerous polar radioactive peaks as observed by reversed phase HPLC. To circumvent this problem, the ketal precursor 13 was prepared, and in the presence of NaH, 13 was labeled with [11C]CH3I in DMF or CH3CN under standard conditions to afford 15. However, it was difficult to remove the dioxolane protecting group of 15 with 1 N HCl even under microwave irradiation, the condition which was used to remove the protecting group in 14 for the 18F labeling of 2. A 6 N HCl solution was needed to remove the dioxolane group in 15, and this harsh hydrolysis condition caused the labeling results to be inconsistent in terms of radiochemical yield and specific activity (Table 3).
| Method | Substrate/mg | Base/Solvent | Hydrolysis (HCl) | Specific activity | Yield (%) |
|---|---|---|---|---|---|
| 1 | 8 (0.2) | Bu4NOH/CH3CN | 1 N | 5600 ± 110 | 62 ± 12 (n = 4) |
| 2 | 13 (1.0) | NaH/CH3CN | 6 N | 3920 ± 4400 | < 10 (n = 4) |
The ring-opening and reclosure method used in the 18F labeling of 2 was also used in the 11C labeling of 4. Two equivalents of Bu4OH in H2O was used to treat as low as 0.2 mg of substrate 8, opening the isatin ring exclusively and quickly. Then the O-methylation took place under the standard condition to afford the 11C labeled isatinate intermediate, which was converted to [11C]4 by the treatment with 1 N HCl. This method is simple, the yield is very high, and the chemical and radiochemical purities are very good (Table 3). Therefore, the ring-opening/reclosure method can also be used in the 11C methylation labeling reaction of isatin analogues with very good labeling results.
In vivo microPET study of Fas-treated liver injury apoptosis model
Once the radiolabeling conditions were established, [11C]4 (WC-98) was evaluated in a well-characterized mouse model of liver apoptosis induced by administration of anti-Fas (Jo2) antibody, which results in massive caspase-3 activation in the liver. Both microPET imaging and biodistribution studies were performed.25 The microPET imaging results are shown in Fig. 1. There is clearly retained activity in the liver in the Fas-treated mice compared to the control, with retention of the activity in the liver demonstrated on time-activity curves (Fig. 1). The ex vivo analysis of the liver is illustrated in Fig. 2. At 5 min, there is no difference in liver uptake between treated and control mice; however, at 30 min, there is clearly increased tracer activity in the liver samples taken from the treated mice compared with controls. Liver samples from the biodistribution study were taken and analyzed for caspase-3 activation by fluorometric enzyme analysis using the caspase-3 fluorogenic substrate Ac-DEVD-AMC (Biomol) (Fig. 3). Enzyme analysis verified increased caspase-3 enzymatic activity in the treated animals and correlated with the findings from the microPET imaging and biodistribution studies. Since [11C]4 (WC-98) is a competitive inhibitor of caspase-3 and binds to the activated form of caspase-3 in tissues undergoing apoptosis, we see expected retention of the tracer in the liver of the treated mice. The results obtained with [18F]4 suggest again that radiolabeled N-benzyl-isatin sulfonamide analogues are potential radiotracers for imaging caspase-3 activity in vivo.![MicroPET imaging with [11C]4 (WC-98). Images were acquired dynamically over 60 min and summed. Single slice coronal summed images demonstrate increased retention of [11C]4 (WC-98) in the treated animal, which is confirmed by time-activity curves generated from a volume of interest (in green) placed over the liver. Gallbladder (GB) activity is marked by yellow arrows.](/image/article/2009/OB/b819024k/b819024k-f1.gif) | ||
| Fig. 1 MicroPET imaging with [11C]4 (WC-98). Images were acquired dynamically over 60 min and summed. Single slice coronal summed images demonstrate increased retention of [11C]4 (WC-98) in the treated animal, which is confirmed by time-activity curves generated from a volume of interest (in green) placed over the liver. Gallbladder (GB) activity is marked by yellow arrows. | ||
![Ex
vivo evaluation of [11C]4 (WC-98) uptake in liver comparing mice treated with the Fas antibody to untreated controls. Error bars indicate the standard deviation. There is increased uptake of [11C]4 (WC-98) at 30 min in the treated group. N= 3 in each group at 5 min, N= 5 in each group at 30 min.](/image/article/2009/OB/b819024k/b819024k-f2.gif) | ||
| Fig. 2 Ex vivo evaluation of [11C]4 (WC-98) uptake in liver comparing mice treated with the Fas antibody to untreated controls. Error bars indicate the standard deviation. There is increased uptake of [11C]4 (WC-98) at 30 min in the treated group. N= 3 in each group at 5 min, N= 5 in each group at 30 min. | ||
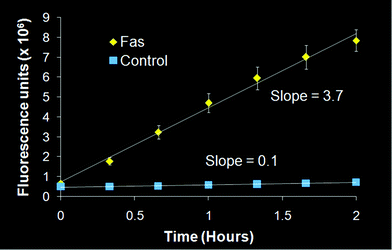 | ||
| Fig. 3 Fluorometric caspase-3 enzymatic assay. Liver protein samples (200 μg) were loaded into each well of a 96-well plate in triplicate. Error bars indicate the standard deviation; error bars that are not visible are within the data point. | ||
Conclusion
We have demonstrated that 11C- and 18F-labeled isatin analogues can be synthesized by using the base/acid ring-opening/recyclization procedure or by protecting the C-3 carbonyl group as a 1,3-dioxane. It was found that the high electrophilicity of the C-3 carbonyl group in the isatin ring served as a trap for [18F]fluoride, resulting in the failure of 18F radiolabeling under typical conditions. Therefore, the protection of the C-3 carbonyl carbon in isatin analogues is critical to avoid unwanted nucleophilic attacks during the synthesis. The base/acid ring-opening/recyclization procedure proved to be a highly efficient method for this purpose. Utilizing this method, we have synthesized 18F and 11C labeled isatin analogues with high chemical and radiochemical purities. In vivo evaluation of [11C]4 (WC-98) in the Fas liver injury apoptosis model demonstrated increased uptake of the tracer in treated animals, with verification of caspase-3 activation by fluorometric caspase-3 enzymatic assay. The correlation of retaining tracer and caspase-3 enzyme activity in the liver of treated mice indicates that [11C]4 (WC-98), a benzyl-isatin sulfonamide analogue caspase-3 inhibitor, is a potential radiotracer for imaging caspases3/7 activation in apoptosis. The methods described in this paper should be applicable to the radiolabeling of other isatin analogues.Experimental
General methods and materials
All chemicals were obtained from standard commercial sources and used without further purification. All reactions were carried out by standard air-free and moisture-free techniques under an inert argon atmosphere with dry solvents unless otherwise stated. Flash column chromatography was conducted using Scientific Adsorbents, Inc. silica gel, 60A, “40 Micron Flash” (32–63 μm). Melting points were determined using MEL-TEMP 3.0 apparatus and are uncorrected. Routine 1H NMR spectra were recorded on a Varian Unity-300 (300 MHz) NMR spectrometer. All chemical shifts were reported as a part per million (ppm) downfield from tetramethylsilane (TMS). All coupling constants (J) are given in Hertz (Hz). Splitting patterns are typically described as follows: s, singlet; d, doublet; t, triplet; m, multiplet. 19F NMR spectra were recorded at 282.2 MHz, and chemical shifts are reported as Hz upfield from an external CFCl3 standard. High resolution 13C NMR was recorded on a Varian Unity Inova-600 at 150.9 MHz. ESI/MS was performed on a Waters ZQ 4000 single quadrupole mass spectrometer equipped with an electrospray ionization (ESI) LC-MS interface. Elemental analyses (C, H, N) were determined by Atlantic Microlab, Inc. High performance liquid chromatography (HPLC) was performed with an ultraviolet detector operating at 251 nm and a well-scintillation NaI (Tl) detector and associated electronics for radioactivity detection. Alltech Econosil C18 250 × 10 mm semi-preparative column and Altech Altima C18 250 × 4.6 mm analytical column were used for preparation and analysis respectively.H218O was purchased from Rotem Industries (Israel). [18F]Fluoride was produced at Washington University by the 18O(p,n)18F reaction through proton irradiation of enriched (95%) 18O water using RDS111 cyclotron. Materials were heated using a custom-designed microwave cavity, model 420BX (Micro-Now Instruments, Skokie, IL). Screw-cap test tubes used for microwave heating were purchased from Fisher Scientific (Pyrex No. 9825). Classic C-18 Sep-Pak cartridges were purchased from Waters Corporation. For the TLC analyses, EM Science Silica Gel 60 F254TLC plates were purchased from Fisher Scientific (Pittsburgh, PA). Radio-TLC was accomplished using a Bioscan 200 imaging scanner (Bioscan, Inc., Washington, DC). Radioactivity was counted with a Beckman Gamma 6000 counter containing a NaI crystal (Beckman Instruments, Inc., Irvine, CA). [11C]CH3I was produced from [11C]CO2 using a GE PETtrace MeI Microlab. [11C]CO2 was produced at the Cyclotron Facility of Washington University School of Medicine using a JSW BC-16/8 cyclotron by irradiating a gas target of 0.2% O2 in N2 for 15–30 min with a 40 μA beam of 16 MeV protons. [11C]CO2 was converted to [11C]CH3I by the GE PETtrace MeI Microlab using a nickel catalyst [Shimalite-Ni (reduced), Shimadzu, Japan P.N.221–27719] in the presence of H2 at 360 °C and followed by reaction with iodine in the gas phase at 690 °C. [11C]CH3I was delivered in the gas phase with helium approximately 12 min following the end of bombardment.
:3) to afford 4 (87 mg, 69%) as a yellow solid, mp 125.9–127.4 °C (from diethyl ether) (Found: C, 64.05; H, 5.17; N, 5.50. C27H26N2O6S requires C, 64.02; H, 5.17; N, 5.53%); δH(300 MHz, CDCl3) 8.00 (d, J 1.8, 1H, isatin C4-H), 7.95 (dd, J 8.25 and 1.8, 1H, isatin C6-H), 7.25 (m, 2H, Ar-H), 7.21 (d, J 7.5, 2H, Ar-H), 6.92-6.85 (m, 3H, Ar-H, 1H, isatin C7-H), 6.81 (d, J 7.5, 2H, Ar-H), 4.85 (s, 2H, ArCH2), 4.15 (m, 1H, pyrrole C2-H), 3.95-3.89 (m, 2H, ArOCH2), 3.80 (s, 3H, OCH3), 3.49 (m, 1H, pyrrole C5-H), 3.22 (m, 1H, pyrrole C5-H), 2.02 (m, 2H, pyrrole C3-H), 1.78 (m, 2H, pyrrole C4-H); δC(75 MHz, CDCl3) 182.00, 159.98, 158.42, 157.98, 153.57, 142.30, 137.30, 134.33, 129.73, 129.30, 125.88, 124.62, 124.50, 121.27, 117.69, 114.83, 114.57, 111.41, 69.35, 58.84, 55.56, 49.73, 44.17, 29.24, 24.36.
:2) to afford 5 (445 mg, 74%) as a yellow solid, mp 163.6-164.2 °C (from diethyl ether) (Found: C, 55.94; H, 4.50; N, 4.59. C28H27BrN2O6S requires C, 56.10; H, 4.54; N, 4.67%); δH(300 MHz, CDCl3) 8.01 (s, 1H, isatin C4-H), 7.96 (dd, J 8.25 and 1.8, 1H, isatin C6-H), 7.28-7.20 (m, 4H, Ar-H), 6.96-6.80 (m, 5H, Ar-H, 1H, isatin C7-H), 4.86 (s, 2H, ArCH2), 4.28 (t, J 6.0, 2H, BrCH2), 4.15 (m, 1H, pyrrole C2-H), 3.93 (m, 2H, ArOCH2), 3.63 (t, J 6.0, 2H, BrCH2CH2), 3.50 (m, 1H, pyrrole C5-H), 3.21 (m, 1H, Pyrrole C5-H), 2.02 (m 2H, pyrrole C3-H), 1.78 (m, 2H, pyrrole C4-H); δC(75 MHz, CDCl3) 181.99, 158.53, 158.44, 158.01, 153.52, 147.59, 137.37, 134.37, 129.78, 129.45, 126.75, 124.59, 121.32, 117.72, 115.66, 114.59, 111.43, 69.35, 68.19, 58.87, 49.79, 44.15, 29.29, 29.18, 24.40.
:2) to afford 6 (167mg, 60%) as a yellow solid, mp 192.4–194.2 °C (from diethyl ether) (Found: C, 62.82; H, 4.89; N, 5.19. C28H26N2O7S requires C, 62.91; H, 4.90; N, 5.24%); δH(300 MHz, CDCl3) 8.03 (s, 1H, isatin C4-H), 7.98 (d, J 8.4, 1H, isatin C6-H), 7.35 (d, J 8.4, 2H, Ar-H), 7.24 (t, J 7.8, 2H, Ar-H), 7.10 (d, J 8.7, 2H, Ar-H), 6.96-6.81 (m, 3H, Ar-H, 1H, isatin C7-H), 4.91 (s, 2H, ArCH2), 4.16 (m, 1H, pyrrole C2-H), 3.93 (m, 2H, ArOCH2), 3.50 (m, 1H, pyrrole C5-H), 3.21 (m, 1H, pyrrole C5-H), 2.30 (s, 3H, CH3CO), 2.03 (m, 2H, pyrrole C3-H), 1.79 (m, 2H, pyrrole C4-H); δH(75 MHz, CDCl3) 181.75, 169.60, 158.45, 158.00, 153.32, 151.10, 137.47, 134.50, 131.61, 129.78, 129.11, 124.70, 122.73, 121.30, 117.71, 114.60, 111.33, 69.37, 58.89, 49.79, 44.10, 29.28, 24.37, 21.36.
:2) to afford 7b (122 mg, 71%) as a yellow solid, mp 71.7–72.9 °C (from diethyl ether) (Found: C, 60.68; H, 4.99; N, 3.59. C35H34N2O9S2 requires C, 60.85; H, 4.96; N, 4.06%); δH(300 MHz, CDCl3) 8.00 (s, 1H, isatin C4-H), 7.95 (d, J 8.4, 1H, isatin C6-H), 7.80 (d, J 8.4, 2H, Ar-H), 7.34 (d, J 8.1, 2H, Ar-H), 7.25-7.20 (m, 4H, Ar-H), 6.95-6.78 (m, 5H, Ar-H, 1H, isatin C7-H), 4.84 (s, 2H, ArCH2), 4.34 (t, J 3.9, 2H, TsOCH2), 4.15 (m, 2H, TsOCH2CH2, 1H, pyrrole C2-H), 3.92 (m, 2H, ArOCH2), 3.49 (m, 1H, pyrrole C5-H), 3.20 (m, 1H, pyrrole C5-H), 2.44 (s, 3H, Ts-CH3), 2.01 (m, 2H, pyrrole C3-H), 1.78 (m, 2H, pyrrole C4-H); δC(75 MHz, CDCl3) 181.99, 158.44, 158.02, 153.51, 147.63, 145.34, 137.39, 134.37, 133.03, 130.16, 129.78, 129.35, 128.85, 128.27, 126.74, 124.59, 121.30, 117.72, 115.52, 114.91, 114.59, 111.44, 69.35, 68.19, 65.86, 58.88, 49.79, 44.13, 29.28, 24.39, 21.93.
:2) to afford 10 (254 mg, 77%) as a white solid, mp 84.3–86.1 °C (from diethyl ether) (Found: C, 56.70; H, 5.11; N, 4.21. C31H33BrN2O7S requires C, 56.62; H, 5.06; N, 4.26%); δH(300 MHz, CDCl3) 7.90 (s, 1H, isatin C4-H), 7.73 (d, J 8.1, 1H, isatin C6-H), 7.29 (d, J 8.4, 2H, Ar-H), 7.21 (d, J 9.00, 2H, Ar-H), 6.98-6.86 (m, 5H, Ar-H), 6.70 (d, J 8.4, 1H, isatin C7-H), 5.00 (t, J 12.3, 2H, OCH2), 4.78 (s, 2H, ArCH2), 4.30 (m, 1H, pyrrole C2-H), 4.27 (t, J 6.0, 2H, BrCH2), 4.03-3.85 (m, 2H, OCH2, 2H, ArOCH2), 3.63 (t, J 6.00, 2H, BrOCH2CH2), 3.49 (m, 1H, pyrrol C5-H), 3.08 (m, 1H, pyrrole C5-H), 2.42 (m, 1H, OCH2CH2), 2.00 (m, 2H, pyrrole C3-H), 1.69 (m, 2H, pyrrole C4-H, 1H, OCH2CH2); δC(75 MHz, CDCl3) 158.65, 131.38, 129.78, 128.99, 124.15, 121.20, 115.50, 114.75, 109.54, 68.16, 61.63, 58.55, 49.61, 43.05, 29.21, 29.10, 25.48, 24.14.
:2) to afford 11 (189 mg, 64%) as a white solid, mp 67.5–69.1 °C (from diethyl ether) (Found: C, 63.10; H, 5.43; N, 4.66. C31H32N2O8S requires C, 62.82; H, 5.44; N, 4.73%); δH(300 MHz, CDCl3) 7.91 (d, J 2.1, 1H, isatin C4-H), 7.75 (dd, J 8.1 and 1.8, 1H, isatin C6-H), 7.31-7.26 (m, 4H, Ar-H), 7.07 (d, J 8.7, 2H, Ar-H), 6.98-6.91 (m, 3H, Ar-H), 6.71 (d, J 8.4, 1H, isatin C7-H), 4.99 (t, J 12.3, 2H, OCH2), 4.83 (s, 2H, ArCH2), 4.29 (dd, J 8.7 and 2.7, 1H, pyrrole C2-H), 4.03-3.83 (m, 2H, ArOCH2, 2H, OCH2), 3.50 (m, 1H, pyrrole C5-H), 3.07 (m, 1H, pyrrole C5-H), 2.41 (m, 1H, OCH2CH2), 2.29 (s, 3H, CH3CO), 1.97 (m, 2H, pyrrole C3-H), 1.71 (m, 2H, pyrrole C4-H, 1H, OCH2CH2); δC(75 MHz, CDCl3) 158.67, 131.44, 129.78, 128.65, 124.24, 122.47, 121.19, 114.76, 109.46, 69.95, 61.65, 58.60, 49.62, 43.03, 29.10, 25.46, 24.15, 21.38.
:2) to afford 12 (122 mg, 73%) as a white solid, mp 80.5–82.1 °C (from diethyl ether) (Found: C, 57.28; H, 5.47; N, 4.19. C32H36N2O10S2 reqires C, 57.13; H, 5.39; N, 4.16%); δH(300 MHz, CDCl3) 7.90 (s, 1H, isatin C4-H), 7.73 (d, J 8.1, 1H, isatin C6-H), 7.32-7.20 (m, 4H, Ar-H), 6.98-6.85 (m, 5H, Ar-H), 6.69 (d, J 8.1, 1H, isatin C7-H), 4.99(t, J 9.9, 2H, OCH2), 4.78 (s, 2H, ArCH2), 4.54 (t, J 4.8, 2H, MsOCH2), 4.29 (m, 1H, pyrrole C2-H), 4.21 (t, J 4.8, 2H, MsOCH2CH2), 4.03-3.82 (m, 2H, OCH2, 2H, ArOCH2), 3.49 (m, 1H, pyrrole C5-H), 3.09 (m, 1H, pyrrole C5-H), 3.07 (s, 3H, Ms-CH3), 2.41 (m, 1H, OCH2CH2), 1.97 (m, 2H, pyrrole C3-H), 1.68 (m, 2H, pyrrole C4-H, 1H, OCH2CH2); δC(75 MHz, CDCl3) 171.84, 158.64, 157.99, 146.26, 132.30, 131.33, 129.74, 129.01, 128.50, 127.89, 124.11, 121.17, 115.34, 114.74, 109.48, 93.32, 69.92, 68.02, 66.13, 61.55, 58.55, 49.58, 42.98, 38.01, 29.08, 25.43, 24.12.
:2) to afford 13 (116 mg, 85%) as a yellow solid, mp 92.8–94.5 °C (from diethyl ether) (Found: C, 63.10; H, 5.67; N, 4.90. C29H30N2O7S requires C, 63.26; H, 5.49; N, 5.09%); δH(300 MHz, CDCl3) 7.90 (s, 1H, isatin C4-H), 7.73 (d, J 8.7, 1H, isatin C6-H), 7.28 (d, J 8.7, 2H, Ar-H), 7.14 (d, J 8.4, 2H, Ar-H), 6.98-6.90 (m, 3H, Ar-H), 6.78 (d, J 8.7, 2H, Ar-H), 6.71 (d, J 8.1, 1H, isatin C7-H), 5.07 (s, 1H, ArOH), 5.00 (t, J 12.0, 2H, OCH2), 4.76 (s, 2H, ArCH2), 4.29 (dd, J 8.4 and 2.7, 1H, pyrrole C2-H), 4.03-3.85 (m, 2H, ArOCH2), 3.51 (m, 1H, pyrrole C5-H), 3.06 (m, 1H, pyrrole C5-H), 2.42 (m, 1H, OCH2CH2), 1.97 (m, 2H, pyrrole C3-H), 1.68 (m, 2H, pyrrole C4-H, 1H, OCH2CH2); δC(75 MHz, CDCl3) 171.90, 158.65, 155.68, 147.60, 146.37, 132.18, 131.38, 129.78, 129.09, 128.48, 126.91, 124.13, 121.21, 116.13, 114.76, 116.13, 114.76, 109.62, 93.37, 69.93, 61.65, 58.58, 49.62, 43.12, 29.10, 25.47, 24.15.
General procedure for 18F radiolabeling of isatin sulfonamides
Pretreatment. Into a 10 mL Pyrex tube were added anhydrous CH3CN (1 mL) and 1 M Bu4NOH in H2O (3.6 μL, 3.6 μmol). After the water was removed by azeotropic distillation at 80 °C under a gentle stream of nitrogen, a solution of 7a (1.1 mg, 1.8 μmol) in DMSO (300 μL) was added inside and the solution was vortexed and heated under microwave irradiation on medium power (60 W) for 30 s. The solution was allowed to cool down to ambient temperature for next step.
18F labeling . [18F]fluoride (100–200 mCi) in H218O (100–300 μL) was transferred to a glass tube containing K222 (5 mg) and K2CO3 (1.1 mg). Water was removed by azeotropic distillation at 110 °C under a gentle stream of nitrogen with successive addition of three aliquots (1.0 mL) of CH3CN. During the final evaporation, the container was removed from the oil bath when the final volume reached about 100 μL. Evaporation to dryness of the residue was conducted at room temperature. [18F]fluoride was resolubilized in anhydrous DMSO (300 μL) and the solution was transferred to the Pyrex tube containing the intermediate prepared in the first step. A 3 mm glass bead was also added to the tube for a more homogeneous heat distribution during the microwave irradiation and the tube was capped firmly. The mixture was vortexed and irradiated by microwave (60 W) for 30 and 25 s with 1 min between each irradiation. RadioTLC indicated that up to 85% [18F] fluoride was incorporated.
Hydrolysis . After the reaction solution was cooled down to ambient temperature, 1 N HCl (500 μL) was added and the mixture was heated under microwave irradiation on medium power (60 W) for 25 s. RadioTLC indicated that almost all [18F] intermediates were converted to [18F]2. The crude reaction mixture was diluted with water (20 mL) and the solid-phase extraction was performed using a Waters Oasis HLB cartridge previously rinsed with methanol (6 mL) and MilliQ water (2 × 6 mL). After the radioactive sample had been applied, the cartridge was rinsed with additional water (2 × 10 mL) to eliminate unreacted fluoride and salts. Then the radiolabeled product was eluted with CH3CN (10 mL). Upon the completion of removal of CH3CN under reduced pressure, the residue was taken up in CH3CN (1.25 mL) and methanol (1.25 ml), followed by the addition of water (2 ml) for HPLC purification. The purification was carried out by semipreparative HPLC (Alltech Econosil C18 250 × 10 mm 10 μm) eluting with 25% CH3CN, 45% MeOH and 30% 0.1 M ammonium formate buffer (pH = 4.5) at a flow rate of 5 mL min−1 and UV at 251 nm. The radioactive peak corresponding to [18F]2 was detected at 13 to 16 min according to the radioactivity detector and was collected for further purification.
The HPLC collection of [18F]2 was diluted in water (200 mL), then the activity was collected in a Waters C18 Classic Sep-Pak by passing the above solution through it under pressure. After the Sep-Pak was washed with water (10 mL) to remove any trace of CH3CN and dried by nitrogen, the activity was eluted with ethanol (2 mL). The ethanol solution was used for the preparation of final solution with saline for animal study. The total synthesis time is 100 min.
General procedure for 11C radiolabeling of isatin sulfonamides [11C]4
:1 CH3CN–water (1.5 mL). [11C]4 was purified by a reversed phase HPLC (Alltech Econosil C18 250 × 10 mm 10 μm) eluting with 25% CH3CN, 25% 0.1 M ammonium formate buffer (pH = 4.5) and 50% MeOH with UV at 237 nm and a flow rate at 4 mL min−1. The corresponding radioactivity for [11C]4 at 11–13 min of HPLC elution was diluted in water (150 mL), and extracted in a Water classic C18 Sep-Pak by passing the diluted solution through it under pressure. After the Sep-Pak was washed with water (10 mL), [11C]4 was eluted from the Sep-Pak with ethanol (1.5 mL). The final doses for further studies were prepared in 10% ethanol/90% saline. Total preparation time was ca 60 min.
Fukui function (FF) calculations
23, 24, and 25 were built by hand and optimized at the B3LYP/6-31G* level in Gaussian98. Following the method of Thanikaivelan,23 the CHELPG method was utilized to assign the electron populations used for the Fukui analysis.Evaluation of Fas Induced Liver Apoptosis Model Using [11C]4 (WC-98)
Female Balb/c mice (19.3 ± 1 g) were injected intravenously with anti-Fas (Jo2) antibody (10 μg) (BD Biosciences, San Jose, CA). Then injected with [11C]4 90 min later. For the microPET imaging study, 60 min of imaging were acquired dynamically after obtaining a transmission scan for attenuation correction. Regions of interest were drawn over the left lobe of the liver to avoid spillover activity from the gallbladder. For the biodistribution study, animals were sacrificed at 5 min and 30 min after tracer injection, the liver was removed, weighed, and counted in a Beckman Gamma 6000 counter. The percent injected dose per gram of tissue (%ID/g) was presented as the mean ± standard deviation.Fluorometric enzyme analysis of caspase-3 activity
Protein was extracted from liver samples obtained from the biodistribution study according to a previously published protocol.26Protein (200 μg) was loaded in triplicate into a 96-well plate with Ac-DEVD-AMC substrate (20 μM) (Biomol International, LP, Plymouth Meeting, PA), with or without the caspase-3 inhibitor Ac-DEVD-CHO (1 μM) (Sigma-Aldrich, St. Louis, MO) in the assay buffer. The plate was read every 15 min for 4 h on a Victor3 V microplate reader (Model 1420, Perkin-Elmer, Fremont, CA). Background fluorescence was subtracted from each time point, and readings were plotted over time. Linear regression was performed on the linear portion of the curve (within 2 h of reading) to determine the slope, which is the rate of caspase-3 enzyme activity.All animal experiments were conducted in compliance with the Guidelines for the Care and Use of Research Animals published by the Animal Studies Committee of Washington University in St. Louis, School of Medicine.
Acknowledgements
This work was supported by grants P01 HL013851–40, R21 CA121952, and K08 EB006702.References
- Definition according to Society of Nuclear Medicine, 54th annual meeting, June 2nd–6th, 2007, Washington, D.C.
- M. D. Jacobson, M. Weil and M. C. Raff, Cell, 1997, 88, 347–354 CrossRef CAS.
- J. C. Reed, Nat. Rev. Drug Discov., 2002, 1, 111–121 CrossRef CAS; I. Rodriguez, K. Matsuura, C. Ody, S. Nagata and P. Vassalli, J. Exp. Med., 1996, 184, 2067–2072 CrossRef CAS.
- E. White, Gene Dev., 1996, 10, 1–15 Search PubMed; A. Ashkenazi and V. M. Dixit, Science, 1998, 281, 1305–1308 CrossRef CAS; G. Evan and T. Littlewood, Science, 1998, 281, 1317–1322 CrossRef CAS; D. R. Green and J. C. Reed, Science, 1998, 281, 1309–1312 CrossRef CAS; N. A. Thornberry and Y. Lazebnik, Science, 1998, 281, 1312–1316 CrossRef CAS; C. Dive and J. A. Hickman, Br. J. Cancer, 1991, 64, 192–196 CAS.
- Review see: C. M. M. Lahorte, J. Vanderheyden, N. Steinmetz, C. Van, De Wiele, R. A. Dierckx and G. Slegers, Eur. J. Nucl. Med. Mol. Imaging, 2004, 31, 887–919 Search PubMed.
- G. J. Kemerink, X. Liu, D. Kieffer, S. Ceyssens, L. Mortelmans, A. M. Verbruggen, N. D. Steinmetz, J. Vanderheyden, A. M. Green and K. Verbeke, J. Nucl. Med., 2003, 44, 947–952 CAS.
- M. Neuss, M. T. Crow, A. Chesley and E. G. Lakatta, Cardiovasc. Drugs Ther., 2001, 15, 507–523 CrossRef CAS.
- W. Chu, J. Zhang, C. Zeng, J. Rothfuss, Z. Tu, Y. Chu, D. E. Reichert, M. J. Welch and R. H. Mach, J. Med. Chem., 2005, 48, 7637–7647 CrossRef CAS; K. Kopka, A. Faust, P. Keul, S. Wagner, H. J. Breyholz, C. Höltke, O. Schober, M. Schäfers and B. Levkau, J. Med. Chem., 2006, 49, 6704–6715 CrossRef CAS.
- D. Zhou, W. Chu, J. Rothfuss, C. Zeng, J. Xu, L. Jones, M. J. Welch and R. H. Mach, Bioorg. Med. Chem. Lett., 2006, 16, 5041–5046 CrossRef CAS.
- R. Bolton, J. Labelled Compd. Radiopharm., 2002, 45, 485–528 CAS; M.-C. Lasne, C. Perrio, J. Rouden, L. Barré, D. Roeda, F. Dolle and C. Crouzel, Top. Curr. Chem., 2002, 222, 201–258 CAS.
- R. Bolton, J. Labelled Compd. Radiopharm., 2001, 44, 701–736 CrossRef CAS.
- J. F. M. Da Silva, S. J. Garden and A. C. Pinto, J. Braz. Chem. Soc., 2001, 12, 273–324 CAS.
- D. Lee, S. A. Long, J. L. Adams, G. Chan, K. S. Vaidya, T. A. Francis, K. Kikly, J. D. Winkler, C. M. Sung, C. Debouck, S. Richardson, M. A. Levy, W. E. Jr. DeWolf, P. M. Keller, T. Tomaszek, M. S. Head, M. D. Ryan, R. C. Haltiwanger, P. H. Liang, C. A. Janson, P. J. McDevitt, K. Johanson, N. O. Concha, W. Chan, S. S. Abdel-Meguid, A. M. Badger, M. W. Lark, D. P. Nadeau, L. J. Suva, M. Gowen and M. E. Nuttall, J. Biol. Chem., 2000, 275, 16007–16014 CrossRef CAS.
- Under the reported condition, only the ring-opened product was observed by us. A. Faust, S. Wagner, M. P. Law, S. Hermann, U. Schnöckel, P. Keul, O. Schober, M. Schäfers, B. Levkau and K. Kopka, Q. J. Nucl. Med. Mol. Imaging, 2007, 51, 67–73 Search PubMed.
- Y. Liu, T.-F. Zheng, F. Jin, L. Zhou, Z.-M. Liu, P. Wei and L.-H. Lai, Huaxue Xuebao, 2007, 65, 1707–1712 CAS; L. Zhou, Y. Liu, W. Zhang, P. Wei, C. Huang, J. Pei, Y. Yuan and L. Lai, J. Med. Chem., 2006, 49, 3440–3443 CrossRef CAS; P.-H. Liang, Curr. Top. Med. Chem., 2006, 6, 361–376 CrossRef CAS; L.-R. Chen, Y.-C. Wang, Y. W. Lin, S.-Y. Chou, S.-F. Chen, L. T. Liu, Y.-T. Wu, C.-J. Kuo, T. S.-S. Chen and S.-H. Juang, Bioorg. Med. Chem. Lett., 2005, 15, 3058–3062 CrossRef CAS.
- L. Meijer, A.-L. Skaltsounis, P. Magiatis, P. Polychronopoulos, M. Knockaert, M. Leost, X. P. Ryan, C. A. Vonica, A. Brivanlou, R. Dajani, C. Crovace, C. Tarricone, A. Musacchio, S. M. Roe, L. Pearl and P. Greengard, Chem. Biol., 2003, 10, 1255–1266 CrossRef CAS.
- J. L. Hyatt, T. Moak, M. J. Hatfield, L. Tsurkan, C. C. Edwards, M. Wierdl, M. K. Danks, R. M. Wadkins and P. M. Potter, J. Med. Chem., 2007, 50, 1876–1885 CrossRef CAS.
- D. Sriram, P. Yogeeswari and K. Meena, Pharmazie, 2006, 61, 274–277 CAS; D. Sriram, T. R. Bal and P. Yogeeswari, Bioorg. Med. Chem., 2004, 12, 5865–5873 CrossRef CAS.
- K. L. Vine, J. M. Locke, M. Ranson, S. G. Pyne and J. B. Bremner, Bioorg. Med. Chem., 2007, 15, 931–938 CrossRef CAS; K. L. Vine, J. M. Locke, M. Ranson, S. G. Pyne and J. B. Bremner, J. Med. Chem., 2007, 50, 5109–5117 CrossRef CAS; I. Chiyanzu, C. Clarkson, P. J. Smith, J. Lehman, J. Gut, P. J. Rosenthal and K. Chibale, Bioorg. Med. Chem., 2005, 13, 3249–3261 CrossRef CAS.
- N. Elander, J. R. Jones, S.-Y. Lu and S. Stone-Elander, Chem. Soc. Rev., 2000, 29, 239–249 RSC; A. de la Hoz, A. Díaz-Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164–178 RSC.
- H. Stünzi, Aust. J. Chem., 1981, 34, 365–371.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, N. Rega, P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle, and J. A. Pople, Gaussian 98, Revision A.11.4; Gaussian, Inc., Pittsburgh, PA, 2002 Search PubMed.
- R. G. Parr and W. Yang, J. Am. Chem. Soc., 1984, 106, 4049–4050 CrossRef CAS; P. Thanikaivelan, J. Padmanabhan, V. Subramanian and T. Ramasami, Theo. Chem. Accnts., 2002, 107, 326–335 Search PubMed.
- L. A. Casey, R. Galt and M. I. Page, J. Chem. Soc. Perkin Trans. 2, 1993, 23–28 RSC; A. M. Ismai and A. A. Zaghloul, Int. J. Chem. Kinet., 1998, 30, 463–469 CrossRef.
- J. Ogasawara, R. Watanabe-Fukunaga, M. Adachi, A. Matsuzawa, T. Kasugai, Y. Kitamura, N. Itoh, T. Suda and S. Nagata, Nature, 1993, 364, 806–809 CrossRef CAS.
- S. Kumar, Methods Mol. Biol., 2004, 282, 19–30 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/b819024k |
| This journal is © The Royal Society of Chemistry 2009 |