Importance of the backbone conformation of (−)-ternatin in its fat-accumulation inhibitory activity against 3T3-L1 adipocytes†
Kenichiro
Shimokawa
a,
Ryoka
Miwa
a,
Kaoru
Yamada
a and
Daisuke
Uemura
*ab
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa, Nagoya 464-8602, Japan. E-mail: uemura@chem3.chem.nagoya-u.ac.jp; Fax: +81-52-789-5869; Tel: +81-52-789-5869
bInstitute for Advanced Research, Nagoya University, Furo-cho, Chikusa, Nagoya 464-8602, Japan
First published on 7th January 2009
Abstract
Key relationships between the intramolecular H-bond-derived backbone conformation and the bioactivity of the novel fat-accumulation inhibitor (−)-ternatin are examined by analyses of the NMR spectroscopic data and CD spectra of designed analogues. The results reveal that the β-turn structure of (−)-ternatin is responsible for its potent fat-accumulation inhibitory effect against 3T3-L1 murine adipocytes.
Introduction
Intramolecular H-bonds are the most important chemical interactions that form active structures/foldings of biomolecules and provide critical functions. In particular, proteins are important representatives of such molecules and show wide structural diversity. Key components that determine their structures are their secondary structures, e.g., helix, sheet, and turn structures, all of which are constructed by intramolecular H-bonds. Interestingly, these unique features are also found in naturally occurring cyclic peptides, such as cyclosporin A1 and gramicidin S.2 To date, considerable effort has been devoted to studying intramolecular H-bond-derived conformations, in peptides3,4 as well as peptidomimetics.5
Our research group recently isolated (−)-ternatin (1), a novel cyclic peptide with the primary structure cyclo [D-allo-Ile1-NMe-L-Ala2-NMe-L-Leu3-L-Leu4-NMe-L-Ala5-NMe-D-Ala6-(2R,3R)-3-hydroxy-Leu7], from the mushroom Coriolus versicolor as a novel fat-accumulation inhibitor against 3T3-L1 murine adipocytes.6 Recently, we examined the in vivo biological activity of 1 in high-fat-fed mice,6b and studied its structure–activity relationships (SARs),7 which enabled the installation of probe tags, e. g., biotin and fluorescent units, in the correct positions.
Apart from our on-going bioorganic investigation to clarify the mode of action of 1, we also focused on its unique physical and conformational properties. The X-ray crystal structure of 1 reported by Miller et al. has a type II β-turn structure in the region between the L-Leu4 and the β-OH-D-Leu7 [(2R,3R)-3-hydroxy-Leu7] moieties, which was formed by intramolecular H-bond B with the assistance of two additional H-bonds A and C in the solid state (Fig. 1).8 Similarly, 1 also shows a single conformer in solution, which can be observed in the 1H NMR spectrum of 1. Our preliminary NMR-based study of 1 supported the existence of H-bonds A and B in solution by a H–D exchange experiment.7a In addition, H-bond C may be important for a single conformation of 1 in solution as well as for its potent bioactivity. For the unique cis-amide bond region, a ROESY correlation was observed between Hα(NMe-L-Ala2) and Hα(NMe-L-Leu3). Therefore, in solution, 1 should have the same conformation as that found in an X-ray crystal structure. However, a better understanding of the importance of each H-bond to both the conformation and fat-accumulation inhibitory activity of 1 is needed. In this paper, we describe the interrelationships between the intramolecular H-bonds, backbone conformation, and fat-accumulation inhibitory activity of 1.
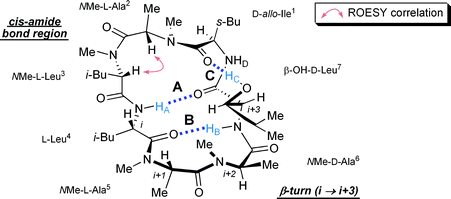 | ||
| Fig. 1 Proposed stereostructure of (−)-ternatin (1) fixed by three intramolecular H-bonds A, B, and C. | ||
Results and discussion
Design and synthesis of analogues
In order to examine the importance of each H-bond, we designed three ternatin analogues, [NMe-L-Leu4]ternatin (2) which lacks the NH proton HA, [NMe-β-OH-D-Leu7]ternatin (3) which lacks the NH protonHB, and [D-Leu7]ternatin (4) which lacks the OH protonHC (Fig. 2). Synthetic accesses to these analogues were achieved by using our synthetic route developed for the large-scale preparation of 1, which is summarized in Scheme 1. The left fragment 6, which was prepared from the unusual amino-acid ester β-OH-D-Leu-OEt (5), and the right fragment 8 which was prepared from L-Leu-OMe (7), are the common intermediates used for the solution-phase synthesis of 1 and its analogues.6b,7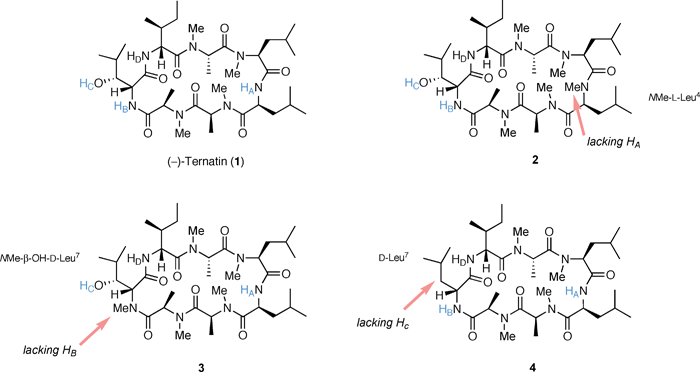 | ||
| Fig. 2 Design of H-bond-lacking ternatin analogues. | ||
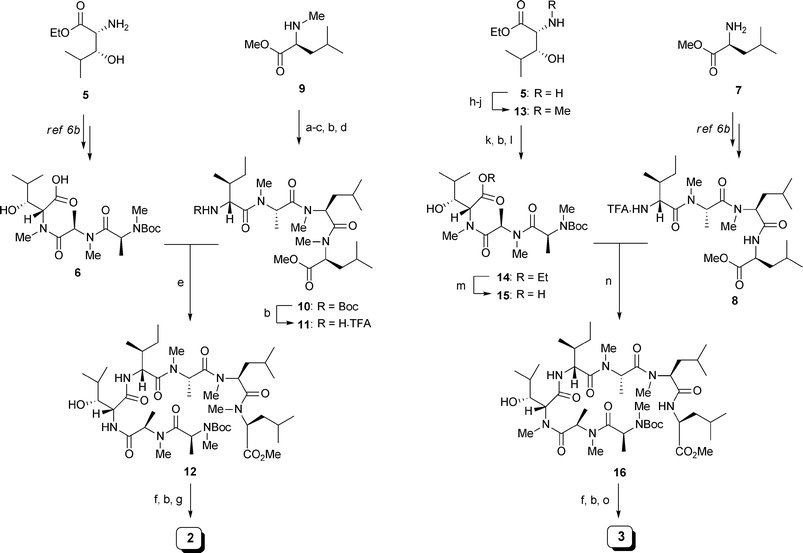 | ||
| Scheme 1 Synthesis of analogues 2 and 3. Reagents and Conditions: (a) Boc-NMe-L-Leu-OH, HATU, DIPEA, CH2Cl2, DMF, 97%; (b) 50% TFA/CH2Cl2; (c) Boc-NMe-L-Ala-OH, HATU, DIPEA, CH2Cl2, DMF, 67% in 2 steps; (d) Boc-D-allo-Ile-OH, HATU, DIPEA, CH2Cl2, DMF, 100% in 2 steps; (e) 6 (1.0 eq.), HATU, DIPEA, CH2Cl2, DMF, 79% in 2 steps; (f) LiOH, THF, t-BuOH, H2O; (g) HATU, HOAt, DIPEA, CH2Cl2 (1.5 mM), DMF, 10% in 3 steps; (h) PhCHO, MeOH; NaBH3CN; (i) (CH2O)n, MeOH; NaBH3CN; (j) H2, Pd(OH)2/C, 47% in 3 steps; (k) Boc-NMe-D-Ala-OH, HATU, DIPEA, CH2Cl2, DMF, 100%; (l) Boc-NMe-L-Ala-OH, HATU, DIPEA, CH2Cl2, DMF, 77%; (m) NaOH, 1,4-dioxane, H2O; (n) 8 (1.0 eq.), HATU, DIPEA, CH2Cl2, DMF, 70% in 2 steps; (o) HATU, HOAt, DIPEA, CH2Cl2 (1.5 mM), DMF, 33% in 3 steps. | ||
The synthesis of 2 began with preparation of the modified right fragment 11. Starting from NMe-L-Leu-OMe (9), sequential couplings with Boc-amino acids and Boc deprotections were conducted from C- to N-terminus to provide tetrapeptide 10. Removal of the Boc group of 10 with 50% TFA/CH2Cl2 gave 11. Construction of the macrocyclic structure began with fragment coupling between the left fragment 6 and 11 to give heptapeptide 12 in moderate yield. Methyl ester hydrolysis followed by Boc deprotection of 12 provided the cyclic precursor. Finally, the key HATU/HOAt-mediated macrolactamization was performed at a low concentration (1.5 mM). After HPLC purification of the crude materials, analogue 2 was obtained in 10% yield. Analogue 3 was synthesized in the same manner. First, N-methylation of amine 5 was carried out in accordance with White's protocol.9 The sequential reductive alkylation with benzaldhyde and formaldehyde was followed by the removal of the resulting benzyl group to afford NMe-β-OH-D-Leu-OEt (13). Next, 13 was coupled with Boc-amino acids to provide tripeptide 14. 14 was then subjected to alkaline hydrolysis, which gave the modified left fragment 15. Fragment coupling between 15 and the right fragment 8 provided heptapeptide 16. Methyl ester hydrolysis and Boc deprotection followed by macrolactamization under dilute conditions afforded analogue 3 in 33% yield after HPLC purification. The synthesis of 4 was reported previously.7a
Conformational analysis
The key NMR parameters needed for the current study were obtained by detailed analyses of 1D- and 2D-NMR spectra (see ESI†). The measurement of 1H NMR spectra in C6D6 revealed that 1 and 2 existed as single conformers, 3 as a mixture of conformers, and 4 as a 4:1 mixture of two conformers. The same tendency was observed in the measurements in DMSO-d6. Based on the results, the significant loss of the intramolecular H-bond B, a key interaction for the β-turn structure, might be an important cause of the multiple conformations of 3. Meanwhile, H-bond C is thought to contribute toward tightening the β-turn structure of 1, since 4, which lacks the hydroxy protonHC, existed as a mixture of conformers (see the 1H NMR spectra of 1 and 4 in the ESI†).We then evaluated the H-bonding properties of 1 and its analogues except for 3 using NMR spectroscopy. First, the H–D exchange properties of the NH and OH protons were examined in C6D6 with the addition of D2O10 (Table 1, and see also the ESI†). These experiments demonstrated a common trend in compounds 1 and 4, showing slow (or no) H–D exchanges of the two NH protons HA and HB and a rapid exchange of HD. In addition, NH protons HA and HB in 1 and 4 gave rise to sharp signals at quite a low field (the chemical shift values: >7.5 ppm), which were expected to be involved in intramolecular H-bonds. On the other hand, slow H–D exchanges of the two NH protonsHB and HD were observed in compound 2. Unfortunately, the hydroxy protons in 1 and 2 could not be examined due to their flexible nature toward H–D exchanges. Next, the temperature coefficients (−Δδ/ΔT) of NH protons were investigated in DMSO-d6. The data showed low values for NH protons HA and HB in 1 and 4, and for NH protonsHB and HD in 2, which indicates the H-bonding or solvent-shielding of each NH proton (Table 2). Since the results are consistent with the H–D exchange profiles, the existence of H-bonds A and B in compounds 1 and 4 and their β-turn structures are strongly suggested. In compound 2, however, it is possible that H-bonds may involve the NH protonsHB and HD.
| Parameter | Substrate | ||
|---|---|---|---|
| 1 | 2 | 4 | |
| a The H–D exchange experiment was conducted in C6D6 with the addition of D2O (20 µL in C6D6 solution). The tendency of H–D exchange was classified as follows; 50% H–D exchange within 1 h (F; fast) 5 h (W; weak), and 24 h (M; medium), and no exchange within 24 h (S; strong). | |||
| NHA (3J Leu4α,NH) | 7.57 (8.0 Hz) | – | 7.89 (8.0 Hz) |
| NHA H–D exchange | S | – | M |
| NHB (3Jβ-OH-Leu7α,NH) | 7.92 (8.0 Hz) | 6.80 (4.0 Hz) | 7.98 (8.5 Hz) |
| NHB H–D exchange | S | S | S |
| OHC (3Jβ-OH-Leu7β,OH) | 5.87 (s) | 3.23 (s) | – |
| OHC H–D exchange | F | F | – |
| NHD (3J Ile1α,NH) | 6.22 (7.2 Hz) | 6.64 (9.5 Hz) | 6.92–6.88 (br d) |
| NHD H–D exchange | W | M | F |
| Substrate | −Δδ/ΔT (ppb/K) | |||
|---|---|---|---|---|
| NHA | NHB | OHC | NHD | |
| a 1H NMR spectra of substrates were taken in the range of 25–60 °C. | ||||
| 1 | 2.0 | 0.1 | 2.0 | 4.6 |
| 2 | – | 3.2 | 3.0 | 0.8 |
| 4 | 1.1 | 0.8 | – | 5.0 |
The conformations of compounds 1–4 in solution were then investigated by circular dichroism (CD) spectroscopy (Fig. 3).11 Consistent with the former NMR studies, compounds 1 and 4 showed evidence of a type II β-turn structure with a maximum ellipticity at nearly 208 nm.12 Hence, 2 gave a curve with a different shape (a maximum ellipticity at 199 nm and a negative band around 228 nm), which was recognized as a β-turn structure similar to type I11a and/or another conformation derived from H-bond B with an additional intramolecular H-bond that involves NH proton HD. The low intensity and non-defined shape of the curve found for 3 may be attributed to an equilibrium of conformers.
 | ||
| Fig. 3 CD spectra of compounds 1–4 in MeOH. | ||
In summary, these spectroscopic data suggested that compounds 1 and 4 form the β-turn structure in solution, while 2 forms another type of conformation and 3 exists as multiple conformers. The key interactions for this β-turn structure are both transannular H-bonds A and B, while H-bond C can stabilize its conformation.
Bioactivity
To understand the relationships between conformation and bioactivity, the fat-accumulation inhibitory activities of the analogues were evaluated with 3T3-L1 murine adipocytes (Table 3). The bioassay consisted of the treatment of confluent 3T3-L1 preadipocytes with each sample and insulin (an inducer of adipogenesis), and further incubation for 7 days. After this period, the control cells were differentiated into mature adipocytes. Both the rate of fat accumulation and the cell viability were calculated. Among the samples tested, no cytotoxicity was observed at the concentration that gave 50% fat accumulation inhibition (IC50).| Compound | Fat accumulation-inhibitory effect: IC50 (µM) | Relative potency | Cell viabilityb: IC50 (µM) |
|---|---|---|---|
| a Values are the means of quadruplicate determinations. b Cell viability was calculated independently to exclude undesired fat-accumulation inhibition arising from the toxicity of the tested compounds. At the respective IC50 values, no cell toxicities were observed for any of the compounds. | |||
| (−)-Ternatin (1) | 0.027 ± 0.003 | 1 | 0.28 ± 0.03 |
| 2 | 22 ± 3 | 1/815 | > 130 |
| 3 | 77 ± 5 | 1/2850 | > 130 |
| 4 | 0.22 ± 0.02 | 1/8 | 3.3 ± 0.2 |
Based on the results, none of the samples showed more potent bioactivity than the natural compound 1 which showed an IC50 value for fat-accumulation inhibition of 0.027 µM. In particular, the bioactivities of analogues 2 and 3 were quite weak with IC50 values of 22 µM and 77 µM, respectively. The potencies of bioactivity were in the order 1 > 4 (1/8-fold less potent) > 2 (1/815-fold) > 3 (1/2850-fold), which were consistent with the trend in the β-turn structure observed in our conformational studies. Therefore, these results clearly indicate that the β-turn structure is absolutely responsible for potent bioactivity.
Conclusions
In conclusion, we evaluated (−)-ternatin (1) and its analogues 2–4 which were designed to understand the interrelationships between intramolecular H-bonds, conformation, and bioactivity. This study clarified that intramolecular H-bonds A and B directly contribute to the β-turn conformation of 1 as well as to its potent bioactivity (fat-accumulation inhibition against 3T3-L1 adipocytes). Meanwhile, H-bond C is also a key interaction that is responsible for tightening the bioactive conformation of 1. Further studies on this bioactive molecule are underway.Experimental section
Materials and methods
Reagents and solvents were purchased from commercial sources. All reactions were performed under a nitrogen atmosphere unless otherwise noted. Column chromatography was performed with Fuji Silysia silica gel FL-60D (Aichi, Japan). High-pressure liquid chromatography (HPLC) was performed with Develosil ODS HG-5 reversed-phase column (Nomura Chemical Co. Ltd., Aichi, Japan). Optical rotations were measured with a JASCO DIP-1000 polarimeter. IR spectra were recorded on a JASCO FT/IR-230 spectrometer. CD measurements were obtained on a JASCO J-720 spectrometer. The 1H, 13C and 2D NMR spectra were recorded on JEOL JNM-A400 and JNM-A600. The chemical shifts are referenced to the solvent peaks δH 7.26 and δC 77.0 for chloroform-d (CDCl3), δH 3.30 and δC 49.0 for methanol-d4 (CD3OD), δH 7.16 and δC 128.0 for benzene-d6 (C6D6), and δH 2.49 and δC 39.5 for dimethylsulfoxide-d6 (DMSO-d6). Mass spectra were determined on a JEOL JMS-700 spectrometer operating in the positive FAB mode (m-nitrobenzyl alcohol as a matrix).To a stirred solution of Boc-NMe-L-Leu-NMe-L-Leu-OMe (213 mg, 0.55 mmol) in dry CH2Cl2 (1 mL) cooled to 0 °C was added TFA (1 mL). After s.m. consumption was indicated by TLC, the solution was concentrated in vacuo. The resulting oil was lyophilized three times with toluene to afford amine as a colorless paste. To a stirred solution of amine in dry CH2Cl2 (0.4 mL) and DMF (0.15 mL) was added Boc-NMe-L-Ala-OH (112 mg, 0.55 mmol) and HATU (231 mg, 0.61 mmol). After the solution was cooled to 0 °C, DIPEA (290 µL, 1.66 mmol) was added dropwise. The solution was warmed to room temperature and allowed to stir for 2 h. The reaction was quenched with 0.1 M HCl aq., and extracted with EtOAc (5 mL × 2). The extracts were washed with saturated aqueous NaHCO3 (5 mL), water (5 mL), and brine (5 mL × 2), dried with Na2SO4, and then concentrated in vacuo. The resulting oil was purified by column chromatography on silica-gel (hexane/ether, 9/1→4/1→1/1→) to give Boc-NMe-L-Ala-NMe-L-Leu-NMe-L-Leu-OMe (174 mg, 67%) as a colorless oil. 1H NMR (400 MHz, CDCl3, a major rotamer) δ 5.52–5.42 (m, 1 H), 5.15 (dd, J = 10.0, 5.8 Hz, 1 H), 4.73–4.64 (m, 1 H), 3.63 (s, 3 H), 2.88 (s, 3 H), 2.75 (s, 3 H), 2.71 (s, 3 H), 1.74–1.29 (m, 6 H), 1.40 (s, 9 H), 1.21 (d, J = 6.8 Hz, 3 H), 0.92–0.81 (m, 12 H); 13C NMR (100 MHz, CDCl3) δ 171.4, 171.2, 170.3, 155.0, 79.7, 56.8, 54.3, 51.7, 50.2, 37.9, 37.7 (2 C), 37.2, 36.7, 30.8, 29.2, 27.9 (3 C), 24.5 (2 C), 24.3 (2 C), 22.8; HRMS (FAB) calcd for C24H46N3O6 (M + H)+ 472.3387, found 472.3386.
To a stirred solution of Boc-NMe-L-Ala-NMe-L-Leu-NMe-L-Leu-OMe (38 mg, 81 µmol) in dry CH2Cl2 (0.6 mL) cooled to 0 °C was added TFA (0.6 mL). After s.m. consumption was indicated by TLC, the solution was concentrated in vacuo. The resulting oil was lyophilized three times with toluene to afford amine as a colorless paste. To a stirred solution of amine in dry CH2Cl2 (0.2 mL) and DMF (0.1 mL) was added Boc-D-allo-Ile-OH·1/2H2O (20 mg, 81 µmol; after co-evaporation with toluene (3 mL × 3)) and HATU (34 mg, 89 µmol). After the solution was cooled to 0 °C, DIPEA (42 µL, 0.24 mmol) was dropwise added. The solution was warmed to room temperature and allowed to stir for 2 h. The reaction was quenched with 0.1 M HCl aq., and extracted with EtOAc (5 mL × 2). The extracts were washed with saturated aqueous NaHCO3 (5 mL), water (5 mL), and brine (5 mL × 2), dried with Na2SO4, and then concentrated in vacuo. The resulting oil was purified by column chromatography on silica-gel (hexane/ether, 8/1→4/1→1/1→) to give 10 (41.3 mg, 83%) as a colorless oil. 10: [α]24D−94.4 (c = 1.0, CHCl3); IR (CHCl3) 3424, 2959, 2363, 1735, 1702, 1630, 1499 cm−1; 1H NMR (400 MHz, CDCl3, a major rotamer) δ 5.56–5.44 (m, 2 H), 5.23–5.17 (m, 1 H), 5.15 (d, J = 9.3 Hz, 1 H) 4.50 (dd, J = 9.3, 5.8 Hz, 1 H), 3.68 (s, 3 H), 2.99 (s, 3 H), 2.93 (s, 3 H), 2.87 (s, 3 H), 1.84–1.33 (m, 9 H), 1.40 (s, 9 H), 1.27–1.21 (m, 3 H), 0.98–0.81 (m, 18 H); 13C NMR (100 MHz, CDCl3) δ 172.1, 171.9, 170.9, 169.8, 156.0, 79.6, 54.8, 53.8, 52.1, 51.5, 49.7, 37.7, 37.4, 37.1, 31.2, 30.2, 29.6, 28.3 (3 C), 26.3, 25.0, 24.8, 23.2, 23.0, 22.0, 21.3, 14.7, 14.2, 11.8; HRMS (FAB) calcd for C30H57N4O7 (M + H)+ 585.4227, found 585.4253.
To a stirred solution of Boc-NMe-D-Ala-NMe-β-OH-D-Leu-OEt (173 mg, 0.46 mmol) in dry CH2Cl2 (1 mL) cooled to 0 °C was added TFA (1 mL). After s.m. consumption was indicated by TLC, the solution was concentrated in vacuo. The resulting oil was lyophilized three times with toluene to afford amine as a colorless paste. To a stirred solution of amine in dry CH2Cl2 (1 mL) and DMF (0.1 mL) was added Boc-NMe-L-Ala-OH (94 mg, 0.46 mmol) and HATU (193 mg, 0.51 mmol). After the solution was cooled to 0 °C, DIPEA (241 µL, 1.39 mmol) was added dropwise. The solution was warmed to room temperature and allowed to stir for 2 h. The reaction was quenched with 0.1 M HCl aq., and extracted with EtOAc (5 mL × 2). The extracts were washed with saturated aqueous NaHCO3 (5 mL), water (5 mL), and brine (5 mL × 2), dried with Na2SO4, and then concentrated in vacuo. The resulting oil was purified by column chromatography on silica-gel (hexane/ether, 2/1→1/1→0/1) to give 14 (164 mg, 77%) as a colorless oil. 14: [α]24D +28.7 (c = 2.6, CHCl3); IR (CHCl3) 3496, 3011, 2972, 2350, 1649, 1473, 1395 cm−1; 1H NMR (400 MHz, CDCl3, a major rotamer) δ 5.43 (q, J = 7.2 Hz, 1 H), 5.04 (q, J = 6.8 Hz, 1 H), 4.74 (d, J = 8.4 Hz, 1 H), 4.19 (q, J = 7.2 Hz, 2 H), 3.96–3.90 (m,1 H), 3.35–3.20 (br s, 1 H), 2.96 (s, 3 H), 2.95 (s, 3 H), 2.73 (s, 3 H), 1.73–1.62 (m, 1 H), 1.43 (s, 9 H), 1.28 (d, J = 7.2 Hz, 3 H), 1.27 (d, J = 6.8 Hz, 3 H), 1.22 (t, J = 6.8 Hz, 3 H), 1.01 (d, J = 6.8 Hz, 3 H) 0.87 (d, J = 6.8 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 171.9, 171.8, 171.7, 155.5, 128.9, 80.1, 73.7, 61.5, 60.6, 50.5, 49.2, 38.6, 34.0, 29.8, 28.3 (3 C), 20.2, 15.3, 14.4, 14.3, 14.1; HRMS (FAB) calcd for C22H42N3O7 (M + H)+460.3023, found 460.3012.
To a stirred solution of 15 and 8 (0.11 mmol) in dry CH2Cl2 (1 mL) and DMF (0.1 mL) was added HATU (48 mg, 0.13 mmol). After the solution was cooled to 0 °C, DIPEA (60 µL, 0.34 mmol) was added dropwise. The solution was warmed to room temperature and allowed to stir for 2 h. The reaction was quenched with 0.1 M HCl aq. and extracted with EtOAc (5 mL × 2). The extracts were washed with saturated aqueous NaHCO3 (5 mL), water (5 mL), and brine (5 mL × 2), dried with Na2SO4, and then concentrated in vacuo. The resulting oil was purified by column chromatography on silica-gel (hexane/ether, 1/2→0/1) to give 16 (71 mg, 70%) as a colorless paste. 16: [α]23D−48.2 (c = 1.5, CHCl3); IR (CHCl3) 3365, 2959, 2881, 2344, 1682, 1644, 1512, 1466 cm−1; 1H NMR (600 MHz, CDCl3, a major rotamer) δ 6.90 (d, J = 8.4 Hz, 1 H), 6.46 (d, J = 8.5 Hz, 1 H), 5.47 (q, J = 6.6 Hz, 1 H), 5.44 (d, J = 6.5 Hz, 1 H), 5.10–4.89 (m, 4 H), 4.78 (dd, J = 8.4, 5.1 Hz, 1 H), 4.54–4.46 (m, 1 H), 3.92–3.85 (m, 1 H), 3.71(s, 3 H), 2.97, (s, 3 H), 2.95 (s, 3 H), 2.93 (s, 3 H), 2.82 (s, 3 H), 2.75 (s, 3 H), 1.76–0.83 (m, 18 H), 1.44 (s, 9 H), 1.27 (d, J = 6.6 Hz, 3 H), 1.26 (d, J = 6.6 Hz, 3 H), 0.95 (d, J = 6.6 Hz, 3 H), 0.90 (d, J = 6.2 Hz, 3 H), 0.88 (d, J = 6.6 Hz, 3 H), 0.86 (d, J = 7.7 Hz, 3 H), 0.82 (d, J = 6.2 Hz, 3 H), 0.80 (d, J = 7.0 Hz, 3 H); 13C NMR (150 MHz, CDCl3) δ 173.0, 172.4, 171.3, 170.6, 170.1, 168.9, 168.7, 154.9, 80.1, 58.0, 55.1, 52.5, 52.2, 50.6, 50.4, 50.3, 49.4, 41.1, 38.7, 36.8, 36.7, 36.1, 29.8, 29.1, 28.3 (3 C), 26.8, 26.7, 25.0, 24.7, 23.1, 22.8, 21.8, 21.7, 21.5, 20.1, 15.1, 14.7, 14.5, 14.4, 14.2, 11.7, 11.6; HRMS (FAB) calcd for C44H81N7O11Na (M + Na)+ 906.5892, found 906.5885.
cyclo[D-allo-Ile-NMe-L-Ala-NMe-L-Leu-L-Leu-NMe-L-Ala-NMe-D-Ala-NMe-β-OH-D-Leu] (3)
To a stirred solution of 16 (53 mg, 60 µmol) in t-BuOH (1 mL), THF (0.25 mL), and H2O (0.25 mL) was added LiOH·H2O (13 mg, 0.30 mmol). The solution was stirred at r.t. until TLC indicated s.m. consumption. The reaction was quenched with 0.1 M HCl aq. and extracted with EtOAc (5 mL × 2). The extracts were dried with Na2SO4 and concentrated in vacuo. The resulting residue was dissolved in dry CH2Cl2 (0.5 mL) and then cooled to 0 °C. To this mixture was added TFA (0.5 mL). After s.m. consumption was indicated by TLC, the solution was concentrated in vacuo. The resulting oil was lyophilized three times with toluene to afford cyclic precursor. To a stirred solution of this compound in dry CH2Cl2 (40 mL) and DMF (0.5 mL) was added a solution of HATU (46 mg, 0.12 mmol) and HOAt (16 mg, 0.12 mmol). Afer the solution was cooled to 0 °C, DIPEA (47 µL, 0.27 mmol) was added. The reaction mixture was stirred for 2 d at room temperature. The reaction mixture was quenched with 0.1 M HCl aq. (10 mL) and diluted with EtOAc (100 mL). The extracts were washed with saturated aqueous NaHCO3 (30 mL) and brine (30 mL × 2), dried with Na2SO4, and then concentrated in vacuo. The resulting paste was purified by column chromatography on silica-gel (CHCl3/MeOH, 1/0→39/1→) to give crude product. Further purification was conducted by HPLC using Develosil ODS HG-5 column (ϕ 20 × 250 mm) eluting with 55% CH3CN aq., at a flow rate of 5 mL/min monitoring at 215 nm, to give pure 3 (16 mg, 33%, tR = 49.9 min) as an amorphous solid. 3 exists as the mixture of conformers in the 1H NMR spectrum as shown in the ESI†; IR (CHCl3) 3292, 2959, 2363, 1617, 1466 cm−1; HRMS (FAB) calcd for C38H70N7O8 (M + H)+ 752.5286, found 752.5302.Bioassay
The murine preadipocyte cell line 3T3-L1 was purchased from the Human Science Research Resources Bank, Japan Health Sciences Foundation (Osaka, Japan). Fetal calf serum (FCS) was purchased from ICN Biomedicals Inc. (Aurora, Ohio). Penicillin and streptomycin were purchased from Nacalai Tesque Inc. (Kyoto, Japan). Insulin was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). All other reagents were purchased from Sigma-Aldrich Fine Chemicals (St. Louis, MO) unless otherwise stated.The preadipocyte cell line 3T3-L1 was cultured in Dulbecco's modified Eagle medium (DMEM) with 10% FCS in two 96-well plates at 37 °C, 5% CO2 for 4–7 days. After the cells reached 100% confluence, the culture buffer was changed to a differentiation buffer (150 µL per well) and samples that were dissolved in MeOH or water (7.5 µL) were added. The differentiation buffer was composed of DMEM containing 10% FCS, 1 µM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine (IBMX), 90 U/mL penicillin, 90 µg/mL streptomycin, and 10 µg/mL insulin. As a control, MeOH or water (7.5 µL) was added in place of samples. After 7 days, the differentiated 3T3-L1 adipocytes in the 96-well plate were treated with 2% Triton-X 100 (10 µL/well) for 30 min at room temperature followed by sonication for 1 min. Fat accumulation was determined by measuring the liberated triglyceride using a Triglyceride E-test Kit (Wako), and the absorbance at 630/690 nm (Microplate Reader EL-800, BIO-TEK Instruments, Inc) was measured according to manufacture's instructions. The fat-accumulation rate was calculated as a percentage of the control. To determine the cell viability of the differentiated 3T3-L1 adipocytes, another 96-well plate was treated with a Cell Counting Kit-8 Test (Wako), and the absorbance at 450 nm was measured according to manufacture's instructions. The viability was calculated as a percentage of the control.
Acknowledgements
This study was supported in part by Grants-in-Aid for Scientific Research for Creative Scientific Research (Grant No. 16GS0206) and the Global COE program in Chemistry at Nagoya University (Grant No. B-021), from the Ministry of Education, Culture, Sports, Science and Technology, Japan. We are indebted to Banyu Pharmaceutical Co., Ltd. for their financial support.References
- A. Rüegger, M. Kuhn, H. Lichti, H. R. Loosli, R. Huguenin, C. Quiquerez and A. von Wartburg, Helv. Chim. Acta, 1976, 59, 1075 CrossRef CAS.
- G. F. Gauze and M. G. Brazhnikova, Am. Rev. Soviet Med., 1944, 2, 134 Search PubMed.
- (a) M. Köck, H. Kessler, D. Seebach and A. Thaler, J. Am. Chem. Soc., 1992, 114, 2676 CrossRef; (b) M. Ohnishi and D. W. Urry, Biochem. Biophys. Res. Commun., 1964, 36, 194; (c) H. Kessler, J. W. Bats, C. Griesinger, S. Koll, M. Will and K. Wagner, J. Am. Chem. Soc., 1988, 110, 1033 CrossRef CAS; (d) S. Nagpal, V. Gupta, K. J. Kaur and D. M. Salunke, J. Biol. Chem., 1999, 274, 23296 CrossRef CAS; (e) S.-T. Yang, S. Y. Shin, C. W. Lee, Y.-C. Kim, K.-S. Hahm and J. I. Kim, FEBS Lett., 2003, 540, 229 CrossRef CAS; (f) for a review, see: E. Vass, M. Hollósi, F. Besson and R. Buchet, Chem. Rev., 2003, 103, 1917 Search PubMed.
- For conformational studies on peptides with β-turn structure, see also references 11 and 12.
- (a) C. R. Jones, M. K. N. Qureshi, F. R. Truscott, S.-T. D. Hsu, A. J. Morrison and M. D. Smith, Angew. Chem., Int. Ed., 2008, 47, 7099 CrossRef CAS; (b) A. S. M. Ressurreicão, A. Bordessa, M. Civera, L. Belvisi, C. Gennari and U. Piarulli, J. Org. Chem., 2008, 73, 652 CrossRef CAS; (c) J. S. Nowick, K. S. Lam, T. V. Khasanova, W. E. Kemnitzer, S. Maitra, H. T. Mee and R. Liu, J. Am. Chem. Soc., 2002, 124, 4972 CrossRef CAS; (d) for reviews, see: A. Giannis and T. Kolter, Angew. Chem., Int. Ed. Engl., 1993, 32, 1244 Search PubMed; (e) J. Gante, Angew. Chem. Int. Ed. Engl., 1994, 33, 1699 CrossRef; (f) K. Burgess, Acc. Chem. Res., 2001, 34, 826 CrossRef CAS.
- (a) K. Shimokawa, I. Mashima, A. Asai, K. Yamada, M. Kita and D. Uemura, Tetrahedron Lett., 2006, 47, 4445 CrossRef CAS; (b) K. Shimokawa, K. Yamada, M. Kita and D. Uemura, Bioorg. Med. Chem. Lett., 2007, 17, 4447 CrossRef CAS; (c) K. Shimokawa, I. Mashima, A. Asai, T. Ohno, K. Yamada, M. Kita and D. Uemura, Chem. Asian. J., 2008, 3, 438 CrossRef CAS.
- (a) K. Shimokawa, Y. Iwase, K. Yamada and D. Uemura, Org. Biomol. Chem., 2008, 6, 58 RSC; (b) K. Shimokawa, Y. Iwase, R. Miwa, K. Yamada and D. Uemura, J. Med. Chem., 2008, 51, 5912 CrossRef CAS.
- R. Miller, N. M. Galitsky, W. L. Duax, D. A. Langs, V. Z. Pletnev and V. T. Ivanov, Int. J. Pept. Protein Res., 1993, 42, 539 CAS.
- K. N. White and J. P. Konopelski, Org. Lett., 2005, 7, 4111 CrossRef CAS.
- (a) The heterogeneous solvent system C6D6/D2O worked sufficiently in the H–D exchange experiment of 1 (ref 7a). The combination of CDCl3/D2O for H–D exchange experiment is also reported, see: T. Rezai, B. Yu, G. L. Millhauser, M. P. Jacobson and R. S. Lokey, J. Am. Chem. Soc., 2006, 128, 2510 Search PubMed; (b) R. Kishore, A. Kumar and P. Balaram, J. Am. Chem. Soc., 1985, 107, 8019 CrossRef CAS.
- (a) A. Perczel and G. D. Fasman, Protein Sci., 1992, 1, 378 CAS; (b) A. Perczel, M. Hollósi, in Circular Dichroism and the Conformational Analysis of Biomolecules, ed. G. D. Fasman, Plenum Press, New York and London, 1996, ch. 9, pp 285-380 Search PubMed.
- (a) C. A. Bush, S. K. Sarkar and K. D. Kopple, Biochemistry, 1978, 17, 4951 CrossRef CAS; (b) S. Prasad, R. B. Rao and P. Balaram, Biopolymers, 1995, 35, 11 CrossRef CAS; (c) A. Dutt, A. Dutta, R. Mondal, E. C. Spencer, J. A. K. Howard and A. Pramanik, Tetrahedron, 2007, 63, 10282 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1D- and 2D-NMR spectra of analogues 1-4; H/D exchange experiment; measurement of temperature coefficients of amide protons. See DOI: 10.1039/b818903j |
| This journal is © The Royal Society of Chemistry 2009 |