 Open Access Article
Open Access ArticleTargeting ketone drugs towards transport by the intestinal peptide transporter, PepT1†
David
Foley
a,
Patrick
Bailey
*b,
Myrtani
Pieri
c and
David
Meredith
c
aSchool of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: david.foley@postgrad.manchester.ac.uk
bFaculty of Natural Sciences, Keele University, Keele, Staffordshire ST5 5BG, UK. E-mail: p.bailey@natsci.keele.ac.uk; Fax: +44 (0)1782 584593; Tel: +44 (0)1782 584583
cSchool of Life Sciences, Oxford Brookes University, Headington, Oxford OX3 0BP, UK
First published on 21st January 2009
Abstract
Thiodipeptide prodrugs of the ketone nabumetone are shown to have affinity for, and be transported by, PepT1 in vitro.
The oral bioavailability of a compound is a crucial factor in its success or failure as a therapeutic agent, particularly given the convenience of this route of administration. There are two main mechanisms of absorption from the GI tract: passive diffusion and carrier mediated transport. The physicochemical properties of molecules required to facilitate good passive diffusion have been well studied and are summarised by Lipinski's “rule of five”.1 Intestinal carrier mediated transport, be it passive or active, is usually reserved for important nutrients like glucose (e.g. SGLT-1), amino acids (e.g. ATB0) and short peptides (e.g. PepT1).2 These transporters can also sometimes accommodate compounds that structurally mimic their natural substrates. The oral bioavailability of poorly absorbed drugs can be improved either by modifying their physiochemical properties to aid passive diffusion and/or by targeting of the compounds towards carrier mediated transport.
The prodrug approach is a common tool used to overcome poor bioavailability or other properties of the parent drug which adversely affect its clinical use.3Prodrugs are inactive drug precursors that are metabolised to active drugs. The majority of prodrugs involve modifications to amine, hydroxyl or carboxylic acid functionality in the parent drug. Recently, the use of the hydroxyimine functionality as a prodrug for ketones has been described.4In vivo studies suggested that the hydroxyimines 2 and 4 of the non-steroidal anti-inflammatory drugs nabumetone 1 and ketoprofen 3, respectively, are metabolised by cytochrome P450 enzymes back to the active parent drug (Scheme 1). These authors also suggested that “the hydroxyimine is a useful intermediate prodrug structure for ketone drugs”.
 | ||
| Scheme 1 The in vivometabolism by cytochrome P450 of the hydroxyimine prodrugs of nabumetone 1 and ketoprofen 3. | ||
We initially planned to prepare prodrugs that could be hydrolysed directly to hydroxyimines, but the potential instability of (for example) hydroxyimine esters also led us to consider hydroxyimine ethers, for which oxidative release of the ketone might occur (see below). The transport mechanism we intended to exploit was via PepT1, which is a proton coupled oligopeptide transporter expressed principally in the small intestine, but also in the kidney and liver.5 It has a broad substrate specificity including most di- and tripeptides, β-lactam antibiotics and ACE inhibitors.5
There are many examples of targeting PepT1 to improve the oral bioavailability of a compound.5 This has mostly been achieved by modifying compounds so that they resemble the natural di- or tripeptide substrates. We have recently lodged a patent6 for a set of thiodipeptide substrates that we hope can act as “carriers” for drug transport by PepT1 generally. The basic premise involves attaching the desired drug to the thiodipeptide through a hydrolysable linker. The prodrug formed is then recognised as a substrate of the transporter and absorbed from the intestine. Cellular metabolism may result in cleavage of the prodrug, followed by release of the drug moiety by passive diffusion or active transport into the bloodstream. Alternatively, basolateral oligopeptide transporters5 similar to PepT1 may transport the prodrug intact into the bloodstream, where metabolism will eventually release the active drug. This general approach overcomes the limitation that the prodrug must resemble a di- or tripeptide.
The use of ester or amide bonds to the carrier thiodipeptides restricts the range of suitable drugs to those containing alcohol, amine or carboxylic acid groups. We wished to investigate if this range could be expanded to ketones by way of the hydroxyimine previously described.4 If successful this method could then be applied in efforts to improve the oral bioavailability of a wide range of ketone drugs. We describe the synthesis and in vitrotransportvia PepT1 of two prodrugs 16 and 17 (Scheme 2). Nabumetone was chosen as a representative ketone drug as the in vivo studies on the hydroxyimine prodrug had already been performed.4 A glycol spacer was chosen to improve the water solubility of the prodrug to aid in vitro biological testing and to investigate the effect of chain length on transport.
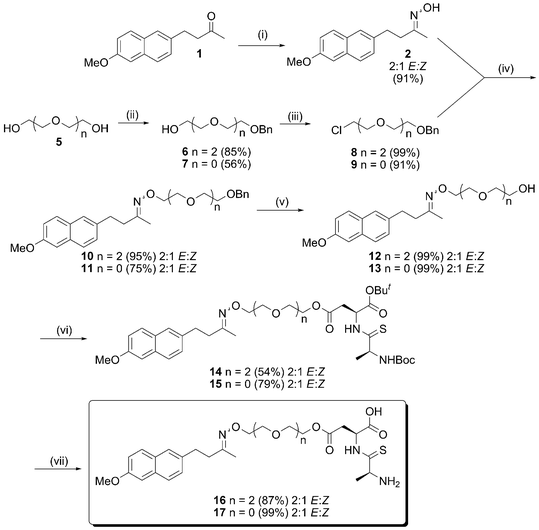 | ||
| Scheme 2 Synthesis of hydroxyimine prodrug linked to PepT1 carrier. (i) NH2OH.HCl, EtOH, rt, 72 h, then 4 M NaOH. (ii) n = 0 Ethylene glycol, NaH, KI, BnBr, 48 h. n = 2 Triethylene glycol, Ag2O, BnBr, 72 h. (iii) SOCl2, cat. pyridine, 65 °C, 4 h. (iv) KI, KOBut, DMF, rt, 24–30 h. (v) H2, 5% Pd(OH)2-C, EtOH, rt, 2 h. (vi) 23, HBTU, DIPEA, rt, 2–5 days. (vii) TFA, DCM, rt, 5 h. | ||
We initially investigated the possibility of attaching the hydroxyimine 2 directly to the aspartate thiodipeptide 23 in an effort to make the ester prodrugs. Despite evidence for the formation of the oxime ester from crude NMR and mass spectrometry, this compound proved too unstable to isolate or use. Whilst the ethers are considerably more stable, there is sufficient precedent for their chemical and metabolic degradation for us to be confident that release of free nabumetone will occur in vivo over a reasonable time-frame; this could take place through direct hydrolysis of the oxime,7 or through oxidation of the PEG spacer in the liver,8 which should liberate the oxime for further oxidative hydrolysis (as in Scheme 1). The oxime of nabumetone, 2, was prepared from nabumetone and hydroxylamine hydrochloride, using a modified method to that previously reported.4 Stirring at room temperature in ethanol for three days followed by addition of aqueous sodium hydroxide gave 2 as a white solid in high yield (91%) by simple filtration (c.f. the literature method required the use of pyridine and column purification). The imine 2 was formed as a 2:1 mix of E:Z isomers and this ratio was unchanged throughout the rest of the synthesis.
The required monobenzyl glycol ethers 6 and 7 were synthesised from the corresponding glycols in moderate to good yields (56–85%) using either standard sodium hydride based desymmetrisation chemistry or the method of Bouzide.9 These were converted to the glycol chlorides 8 and 9 in excellent yields (91–99%) using the method of Mirviss.10 When forming the chloride, it was found that increasing reaction times led to decomposition and lower yields. A reaction time of four hours was found to give the optimum balance between conversion and decomposition.
Hydroxyimine ethers 10 and 11 were formed in excellent yield by the reaction of 2 with the glycol chlorides 8 and 9 in the presence of potassium tert-butoxide. Potassium iodide was added to accelerate the rate of reaction. No reaction at the imine carbon was detected under these conditions. The benzyl ether was then removed using Pearlman's11catalyst (palladium hydroxide on carbon) to give 12 and 13 in quantitative yields.
The synthesis of the thiodipeptide carrier 23 was carried out in five steps from Fmoc-Asp(OBzl)-OH, under conditions previously optimised in our group (Scheme 3).6 Lawesson's12 reagent was used to effect the oxygen-sulfur substitution selectively at the amide carbonyl. This carrier was coupled to 12 and 13 to give 14 and 15 in moderate yields (54–79%). Finally acidolysis (50% trifluoroacetic acid in dichloromethane) was used to remove the Boc carbamate and the tert-butyl ester protecting groups. The resulting thiodipeptide conjugates 16 and 17 were lyophilised prior to in vitro testing.
 | ||
| Scheme 3 Outline of the synthesis of the thiodipeptide carrier 23. (i) tert-butyl-2,2,2-trichloroacetimidate, DCM/Et2O, 2 days. (ii) TBAF in THF, 6 h. (iii) Boc-Ala-OH, DPPA, TEA, DMF, 18 h. (iv) Lawesson's reagent, refluxing toluene, 4 h. (v) Na/NH3(l) in THF, −78 °C, 3 h. | ||
The binding affinities of 16 and 17 for PepT1 were determined by measuring the concentration at which it inhibited uptake of radiolabelled D-Phe-L-Gln in Xenopus laevisoocytes expressing rabbit PepT1 (Fig. 1). Inhibition constants were calculated from standard Michaelis-Menten kinetics.13,14 It is known that increasing the hydrophobicity of a PepT1 substrate usually increases its affinity.15 Our results are in agreement with this trend. PepT1 is a low affinity, high capacity transporter and compounds with an affinity < 1 mM are generally classed as high affinity substrates.5
![Inhibition of the transport of [3H]-d-Phe-l-Gln into PepT1-expressing oocytes by 16 and 17. Data obtained for the known PepT1 substrate, l-Phe-l-Ala is included for reference.](/image/article/2009/OB/b818606e/b818606e-f1.gif) | ||
| Fig. 1 Inhibition of the transport of [3H]-D-Phe-L-Gln into PepT1-expressing oocytes by 16 and 17. Data obtained for the known PepT1 substrate, L-Phe-L-Ala is included for reference. | ||
Both 16 and 17 were competitive inhibitors of PepT1 (data not shown), as indicated by an increase in the apparent KM of D-Phe-L-Gln in their presence using methods previously reported.16
As binding studies only show affinity for PepT1 and do not provide information as to whether the compound is a substrate or an inhibitor, further transport assays were undertaken. Trans-stimulation of radiolabelled D-Phe-L-Gln efflux from rabbit PepT1 expressing oocytes in the presence of 10 mM Gly-L-Gln (a standard PepT1 substrate) and 10 mM 16 or 17 were compared (Fig. 2).14Trans-stimulation is a consequence of having PepT1 substrates on both sides of the membrane, removing the need for the re-orientation of an empty PepT1 transporter (the rate-limiting step in the transport cycle) and therefore in this case increasing the efflux rate of the radiolabelled dipeptide injected into the cell compared to when no substrate is present extracellularly.17 Since trans-stimulation of efflux can only occur if the test compound is a substrate, this simple assay strongly indicated that both 16 and 17 are transported by PepT1 with comparable efficiency to Gly-L-Gln. Free nabumetone did not cause trans-stimulation in this assay (data not shown).
![Comparison of the efflux of 0.4 μM [3H]-d-Phe-l-Gln from PepT1-expressing oocytes in the absence (control) and presence of Gly-l-Gln, 16 or 17 (all 10 mM).](/image/article/2009/OB/b818606e/b818606e-f2.gif) | ||
| Fig. 2 Comparison of the efflux of 0.4 μM [3H]-D-Phe-L-Gln from PepT1-expressing oocytes in the absence (control) and presence of Gly-L-Gln, 16 or 17 (all 10 mM). | ||
To investigate further the extent and rate of trans-epithelial transport of these compounds, assays in Caco-2 cells were performed. Caco-2 cells were chosen as they are widely accepted as a good overall model for the small intestinal epithelium.18 This method also has the advantage of being high-throughput, although it has recently been suggested that Caco-2 cells may underestimate the in vivo trans-epithelial rate of transport.19 Apical to basolateral transport of 2 mM 16 or 17, applied to the apical side, was monitored by high performance liquid chromatography (HPLC) after one hour. This allowed us to rapidly determine if the intact compounds were crossing the membranes. We compared the extent of transport of 16 and 17 to a known hydrolysis resistant PepT1 substrate, Pheψ[CS-NH]-Ala (FSA),6 by normalisation (Fig. 3A). This normalisation allowed direct comparison between different batches of Caco-2 monolayers and gives an indication as to how well the compounds were crossing the monolayer, relative to a compound known to be transported in vivo.6 The rate of PepT1 mediated transport was determined by comparing the difference in apical to basolateral transport of 2 mM 16 or 17 with excess (20 mM) Gly-L-Gln. This level of excess Gly-L-Gln completely saturates the PepT1 transporter, so the reduction in overall transport (after one hour) of the test compounds corresponds to the PepT1 mediated component of transport. This was again compared to the PepT1 mediated rate of FSA transport by normalisation (Fig. 3B).
 | ||
| Fig. 3 A: Relative rate of mediated apical to basolateral transport of 16 and 17 in Caco-2 monolayers, normalised to the transport rate of FSA in the same culture of cells. B: Percentage of the 2 mM apical load of 16 and 17 that was transported across the Caco-2 monolayer in 1 hour by a mediated transport route (normalised to a mean value for 2 mM FSA of 1.22 ± 0.30%, n = 3 cultures). | ||
The rate of PepT1 mediated transport of 17 was 1.3 times faster than FSA [Papp (17) = 4.4 ± 0.1 × 10−6 cm sec−1versus Papp (FSA) = 3.3 ± 0.3 × 10−6 cm sec−1] with overall transport 1.7 times that of FSA. 16 gave a PepT1 mediated transport rate 6.3 times that of FSA [Papp (16) = 6.3 ± 0.7 × 10−6 cm sec−1versus Papp (FSA) = 1.0 ± 0.1 × 10−6 cm sec−1] with overall transport 3.8 times that of FSA. These Papp values are similar in magnitude to other PepT1 (pro)drug substrates previously reported.2016 was transported faster than 17. This is consistent with it having lower affinity for PepT1 and implies that the release of the substrate to give an empty transporter for re-orientation is part of the rate-limiting step in the catalytic transport cycle of PepT1.21
The pH dependence of this transport was shown by examining the PepT1 mediated rate of transport after one hour (as per method above) at pH 5.5 and 7.4 (Fig. 4). If the compounds are substrates of the proton-coupled PepT1 transporter, as previous results suggested, the transport rates should be faster at pH 5.5 than at pH 7.4.5 In each case, a significantly slower transport rate was observed at pH 7.4, approximately 50% that of the pH 5.5 rate. This pH dependence data is fully consistent with the previous data in demonstrating that the compounds are substrates of PepT1.
 | ||
| Fig. 4 pH dependence of apical to basolateral PepT1 transport rates across Caco-2 monolayers after one hour of FSA (standard), 16 and 17 (normalised to the mean rate of transport for each compound at pH 5.5, n = 3 cultures). ** p < 0.01, * p < 0.05. | ||
In conclusion, the synthesis, in vitro binding and transport of hydroxyimine prodrugs of the anti-inflammatory ketone drug nabumetone via PepT1 has been shown. This is the first example of targeting a ketone prodrug towards PepT1 absorption. Whilst the model ketone drug studied is itself orally active, we feel this approach could be used for other ketone containing drugs with low oral activity, such as the antibiotic pleuromutilin and certain cathepsin K inhibitors under development by GSK.22 Additionally, as prodrugs continue to play an ever increasing role in drug development and optimisation, expanding the range of functionality that can be converted into prodrugs will increase the number of tools available to medicinal chemists for drug development. Furthermore, active targeting of these prodrugs to membrane transporters such as PepT1 may overcome a variety of problems such as oral bioavailability, specific tissue distribution and excretion.
The authors wish to acknowledge the University of Oxford, where most of the biological testing described was carried out by DM and MP. The Wellcome Trust is thanked for generous financial support.
Notes and references
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS
.
-
E. M. Wright, B. A. Hirayama, D. D. F. Loo, E. Turk, K. Hager, in Physiology of the Gastrointestinal Tract, Raven Press, New York, 3rd edn., 1994, vol. 2, pp. 1751-1772. V. Ganapathy, M. Brandsch, F. H. Leibach, in Physiology of the Gastrointestinal Tract, Raven Press, New York, 3rd edn., 1994, vol. 2, pp. 1773-1794 Search PubMed
- B. Testa, Biochem. Pharmacol., 2004, 68, 2097–2106 CrossRef CAS
- H. Kumpulainen, N. Mahonen, M. L. Laitinen, M. Jaurakkajarvi, H. Raunio, R. O. Juvonen, J. Vepsalainen, T. Jarvinen and J. Rautio, J. Med. Chem., 2006, 49, 1207–1211 CrossRef CAS
- M. Brandsch, I. Kütter and E. Bosse-Doenecke, J. Pharm. Pharmacol., 2008, 60, 543–585 CrossRef CAS
-
P. D. Bailey, (The University of Manchester, UK). European Patent Office, Int. Application No.: WO2005067978, 2005 Search PubMed
- M. A. Brown, D. W. Gammon and J. E. Casida, J. Agric. Food Chem., 1983, 31, 1091–1096 CrossRef CAS
- M. A. Tirmenstein, Toxicology in Vitro, 1993, 7, 645–652 CrossRef CAS
- A. Bouzide and G. Sauvé, Tetrahedron Lett., 1997, 38, 5945–5948 CrossRef CAS
- S. B. Mirviss, J. Org. Chem., 1989, 54, 1948–1951 CrossRef CAS
- W. M. Pearlman, Tetrahedron Lett., 1967, 8, 1663–1664 CrossRef
- B. S. Pedersen, S. Scheibye, N. H. Nilsson and S. O. Lawesson, Bull. Soc. Chim. Belg., 1978, 87, 223–228
- C. S. Temple, J. R. Bronk, P. D. Bailey and C. A. R. Boyd, Pflügers Arch. - Eur. J. Physiol., 1995, 430, 825–829 Search PubMed
- D. Meredith, C. A. R. Boyd, J. R. Bronk, P. D. Bailey, K. M. Morgan, I. D. Collier and C. S. Temple, J. Physiol. (London), 1998, 512, 629–634 Search PubMed
- B. S. Vig, T. R. Stouch, J. K. Timoszyk, Y. Quan, D. A. Wall, R. L. Smith and T. N. Faria, J. Med. Chem., 2006, 49, 3636–3644 CrossRef CAS
- D. Meredith, C. S. Temple, N. Guha, C. J. Sword and C. A. R. Boyd, Eur. J. Biochem., 2000, 267, 3723–3728 CrossRef CAS
- C. S. Temple, A. K. Stewart, D. Meredith, N. A. Lister, K. M. Morgan, I. D. Collier, R. D. Vaughan-Jones, C. A. R. Boyd, P. D. Bailey and J. R. Bronk, J. Biol. Chem., 1998, 273, 20–22 CrossRef CAS
- I. J. Hidalgo, T. J. Raub and R. T. Borchardt, Gastroenterology, 1989, 96, 736–749 CAS
- P. V. Balimane, S. Chong, K. Patel, Y. Quan, J. Timoszyk, Y.-H. Han, B. Wang, B. Vig and T. N. Faria, Arch. Pharm. Res., 2007, 30, 507–518 Search PubMed
- R. K. Bhardwaj, D. Herrera-Ruiz, P. J. Sinko, O. S. Gudmundsson and G. Knipp, J. Pharmacol. Exper. Ther., 2005, 314, 1093–1100 CrossRef CAS
- C. S. Temple, P. D. Bailey, J. R. Bronk and C. A. R. Boyd, J. Physiol., 1996, 494, 795–808 CAS
- G. Brooks, W. Burgess, D. Colthurst, J. D. Hinks, E. Hunt, M. J. Pearson, B. Shea, A. K. Takle, J. M. Wilson and G. Woodnutt, Bioorg. Med. Chem. Lett., 2001, 9, 1221–1231 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details for the synthesis, characterisation, testing and analysis of 16, with characterisation data for 17. See DOI: 10.1039/b818606e |
| This journal is © The Royal Society of Chemistry 2009 |