Synthesis of (−)-pericosine B, the antipode of the cytotoxic marine natural product†
Yoshihide
Usami
*,
Kentaro
Suzuki
,
Koji
Mizuki
,
Hayato
Ichikawa
and
Masao
Arimoto
Osaka University of Pharmaceutical Sciences, 4-20-1 Nasahara, Takatsuki, Osaka 569-1094, Japan. E-mail: usami@gly.oups.ac.jp; Fax: +81 72 690 1005; Tel: +81 72 690 1083
First published on 13th November 2008
Abstract
The stereoselective synthesis of (−)-pericosine B, which is the antipode of the cytotoxic metabolite of the fungus Periconia byssoides OUPS-N133 separated from the sea hare, was accomplished in 9 steps in 12% total yield from (−)-quinic acid, together with the synthesis of its epimer. Every crucial step of this total synthesis, including ring opening of a β-epoxide and NaBH4reduction of an unstable β,γ-unsaturated enone, proceeded with excellent stereoselectivity.
Introduction
The isolation and structure determination of highly functionalized C-7 cyclohexenoid natural products pericosines A–E 1–5 (Fig. 1), which are cytotoxic metabolites of the fungus Periconia byssoides OUPS-N133 originally separated from the sea hare Aplysia kurodai, were reported in 1997 and 2007 by Numata and co-workers.1,2 The absolute configuration of pericosines A–D 1–4 was elucidated by total syntheses.3–9 Compound 1 was reported to exhibit significant inhibitory activity against protein kinase EGFR and human topoisomerase II, but similar biological tests on 2–4 were not reported. We were therefore interested in the biological activity of 2 against human cancer cell lines. In addition to their significant bioactivity, it is noteworthy that pericosines C 3 and E 5 exist as a mixture of enantiomers. X-ray analysis by Numata and co-workers2 established the relative stereochemistry of pericosine E 5 to originate from two monomeric pericosines 1 and 2 having different chirality. As we pointed out previously, their finding meant that the presence of the antipode of monomeric 1 or 2 in nature is possible.10 Therefore, synthesis of the antipode of 1–3 is significant. We have already reported the synthesis of the antipode of 16,7 and 33 but not that of 2.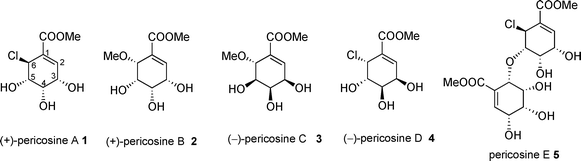 | ||
| Fig. 1 Structures of naturally occurring pericosines. | ||
The total synthesis of (+)-2, a naturally occurring enantiomer, was reported only once in 1998 by Donohoe and co-workers9 in spite of extensive effort by other groups including ours to date.11–13 However, the only successful synthesis had problems in terms of the use of expensive starting materials and a stoichiometric amount of toxic osmium tetroxide. Recently, we reported the determination of the absolute configuration of pericosine D by a synthetic approach.8 Unfortunately, the total yield of desired product 4 was quite low when relatively inexpensive (−)-quinic acid was used. Nevertheless, that work gave us a hint for a short synthesis of 2. Following several years of failed attempts at synthesizing 2,11 we describe herein a short synthesis of the antipode of 2 and its epimer.
Results and discussion
From the results of our previous work that dealt with the determination of pericosine D 4, we suspected that the introduction of a 6α-methoxy group into the pericosine core 6-membered ring is possible when MeOH is used as solvent in the stereoselective ring opening of intermediate β-epoxide 8.8The synthesis of (−)-pericosine B is summarized in Scheme 1 followed by Scheme 2. Methyl quinate derivative 6 was prepared from commercially available (−)-quinic acid in 78% according to the literature.14 Compound 6 was converted into unstable diene 7 in 2 steps. Then, without purification, crude 7 was oxidized with mCPBA at 40 °C to afford an inseparable mixture of epoxides 8 and 9 in 40% yield in 3 steps.
 | ||
| Scheme 1 Stereoselective synthesis of 6α-methoxyalchohol 10. | ||
 | ||
| Scheme 2 Completion of total synthesis of (−)-pericosine B 2. | ||
Ring opening of a mixture of 8 and 9 with a catalytic amount of HCl in MeOH gave the desired 6α-methoxypericosine derivative 10 in 54% yield, with small amount of 118 (1%) and 1-methoxylated alcohol 12 (1%) (whose configuration at C-1 could not be determined), with the recovery of 9 (32%). The relative stereochemistry of major product 10 was confirmed by NOESY analysis as shown in Scheme 1. Cross-peaks of H-4/H-6 and H-5/6-methoxy group were observed.
In next step, it was difficult to promote the SN2-type Walden inversion by Mitsunobu reaction at C-5 in 10. Close inspection of the 1H-NMR spectra of 10, which had relatively large coupling constants of J4,5 = 7.3 Hz and J5,6 = 6.6 Hz, suggested a half-chair conformation, as illustrated in Fig. 2, that inhibits SN2-type attack of the nucleophile from inside the pericosine core 6-membered ring, which is in a fixed conformation due to the cyclohexylidene bridge.
 | ||
| Fig. 2 Plausible conformation of 10. | ||
Then, the inversion of stereochemistry of C-5 in 10 was attempted by means of Dess–Martin oxidation followed by stereoselective reduction with NaBH4. Methoxy alcohol 10 was oxidized with Dess–Martin periodinane, albeit very slowly, to give crude β,γ-unsaturated enone 13. Without purification, 1315 was reduced with NaBH4 at 0 °C to give the desired diastereomer 14 as the sole product in 86% yield in 2 steps.
This total synthesis was completed by deprotection of the cyclohexylidene moiety in 14 with TFA in MeOH to afford (−)-pericosine B 2 in 82% yield. All spectral data except specific rotation agreed with those of reported natural pericosine B. The specific rotation ([α]D25−32.6) of synthesized compound showed almost the same value as previously synthesized (+)-2 ([α]D21 +30.6 (c 0.8 in EtOH)) but with the opposite sign.
 | ||
| Fig. 3 Structures of undesired compounds.15 | ||
The overall yield of this total synthesis of 2 was 12% in 9 steps starting from (−)-quinic acid. Similarly, epimer 15 was prepared in 57% yield from 10.
Conclusions
We have accomplished the stereoselective total synthesis of (−)-pericosine B 2, which has opposite chirality to the natural product, in 9 steps in 12% total yield. Its epimer 15 was also prepared. The second synthesis of pericosine B described herein is a toxic-reagent-free method and is also applicable to the synthesis of (+)-pericosine B, which was obtained as a minor component in nature and has significant biological activity, since either enantiomer of unstable diene 716 could be prepared from (−)-quinic acid. This synthetic route toward 2 is also a divergent one, as the common intermediate yielded pericosine D 4.8Experimental section
General information
IR spectra were obtained with a JEOL FT/IR-680 Plus spectrometer. HRMS was determined with a JEOL JMS-700 (2) mass spectrometer. NMR spectra were recorded at 27 °C on Varian UNITY INOVA-500 and Mercury-300 spectrometers in CDCl3 with tetramethylsilane (TMS) as internal standard. Melting points were determined on a Yanagimoto micromelting point apparatus and are uncorrected. Specific rotations were measured on a JASCO DIP-1000 polarimeter and [α]D values are given in 10−1 deg cm2 g−1. Liquid column chromatography was conducted over silica gel (Nacalai, silica gel 60, mesh 70–230 or 230–400). Analytical TLC was performed on precoated Merck glass plates (silica gel 60 F254), and compounds were detected by dipping an ethanol solution of phosphomolybdic acid, followed by heating. Dry THF was distilled over sodium benzophenone ketyl under argon atmosphere.Synthesis of a mixture of epoxides 8 and 9 from 6
To a solution of diol 6 (552 mg, 1.93 mmol) in CH2Cl2 (25 mL) were added pyridine (780.5 μL, 9.65 mmol) and catalytic DMAP (30.0 mg). A solution of Tf2O (729 μL, 4.25 mmol) in CH2Cl2 (25 mL) was added dropwise to the mixture at 0 °C with stirring. After stirring overnight at rt, the reaction mixture was treated with aqueous NaHCO3 and then extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered, and evaporated to give a crude residue containing triflates and diene 7. The mixture was dissolved in DMF (3 mL), and CsOAc (1.99 mmol) was added to the solution with stirring at rt. After 3 hr, the reaction mixture was extracted with t-butylmethylether and H2O. The organic layer was washed with brine twice, dried over MgSO4, filtered, and evaporated to afford crude diene 7. To a solution of crude diene 7 in CH2Cl2 (10 mL), mCPBA (518.8 mg, max 77%, calcd ca. 2.3 mmol) was added and the reaction mixture was kept at 40 °C overnight. The reaction mixture was treated with aqueous NaHCO3 and then extracted with CH2Cl2. The organic layer was separated, dried over MgSO4, filtered, and evaporated to afford a crude residue that was purified by column chromatography (eluent: hexane:EtOAc = 4:1) to give a mixture of 8 and 9 (204.9 mg, 40% from 6, ratio: 8:9 = 3:2 from 1H-NMR spectrum).Methyl (3R,4R,5S,6S)-3,4-O-cyclohexylidene-3,4,5-trihydroxy-6-methoxy-1-cyclohexene-1-carboxylate 10
Methyl (4S,5R,6R)-4,5-O-cyclohexylidene-4,5,6-trihydroxy-1-methoxy-2-cyclohexene-1-carboxylate 12
To a mixture of 8 and 9 (ca. 3:2) (73.9 mg, combined amount) in MeOH (5 mL) was added 1 drop of 1.0 M HCl in Et2O with a microsyringe. After stirring overnight at rt, the reaction mixture was condensed under reduced pressure to afford a crude residue that was purified by column chromatography (eluent: hexane:EtOAc = 5:3) to give 10 (44.9 mg, 54%), 11 (0.8 mg, 1%), and 12 (0.9 mg, 1%) with recovery of 9 (23.6 mg, 32%). 10: Colorless crystals (CH2Cl2); mp 132–135 °C; [α]D25−55.2 (c 0.165 in CHCl3); IRνmax (KBr)/cm−1 3395 (OH), 1719 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1660 (CC); 1H-NMR (500 MHz; CDCl3; Me4Si) δ 1.25–1.70 (10H, m), 3.60 (3H, s, 6-OMe), 3.80 (3H, s, COOMe), 3.94 (1H, dd, J = 7.3, 6.6 Hz, H-5), 4.00 (1H, dt, J = 6.6, 1.4 Hz, H-6), 4.17 (1H, dd, J = 7.3, 6.4 Hz, H-4), 4.66 (1H, ddd, J = 6.4, 3.9, 1.4 Hz, H-3), 6.68 (1H, dd, J = 3.9, 1.4 Hz, H-2); 13C-NMR (125.6 MHz; CDCl3; Me4Si) δ 23.6 (t), 24.0 (t), 25.0 (t), 35.3 (t), 37.8 (t), 52.0 (q), 60.5 (q), 70.7 (d), 72.6 (d), 75.9 (d), 78.0 (d), 111.7 (s), 132.3 (d), 134.7 (s), 166.4 (s); EIMS m/z 298 (M+, 76%); HREIMS m/z calcd for C15H22O6 (M)+ 298.1416, found 298.1415.
O), 1660 (CC); 1H-NMR (500 MHz; CDCl3; Me4Si) δ 1.25–1.70 (10H, m), 3.60 (3H, s, 6-OMe), 3.80 (3H, s, COOMe), 3.94 (1H, dd, J = 7.3, 6.6 Hz, H-5), 4.00 (1H, dt, J = 6.6, 1.4 Hz, H-6), 4.17 (1H, dd, J = 7.3, 6.4 Hz, H-4), 4.66 (1H, ddd, J = 6.4, 3.9, 1.4 Hz, H-3), 6.68 (1H, dd, J = 3.9, 1.4 Hz, H-2); 13C-NMR (125.6 MHz; CDCl3; Me4Si) δ 23.6 (t), 24.0 (t), 25.0 (t), 35.3 (t), 37.8 (t), 52.0 (q), 60.5 (q), 70.7 (d), 72.6 (d), 75.9 (d), 78.0 (d), 111.7 (s), 132.3 (d), 134.7 (s), 166.4 (s); EIMS m/z 298 (M+, 76%); HREIMS m/z calcd for C15H22O6 (M)+ 298.1416, found 298.1415.
12
: Colorless oil; [α]D25 +146.8 (c 0.5 in CHCl3); IRνmax (liquid film)/cm−1 3553 (OH), 1724 (CO), 1660 (CC); 1H-NMR (500 MHz; CDCl3; Me4Si) δ 1.35–1.77 (10H, m), 2.63 (1H, d, J = 3.4 Hz, 6-OH), 3.45 (3H, s, −OMe), 3.78 (3H, s, COOMe), 3.91 (1H, dd, J = 8.4, 3.4 Hz, H-4), 4.45 (1H, dd, J = 8.4, 7.1 Hz, H-5), 4.78 (1H, ddd, J = 7.1, 3.4, 1.1 Hz, H-6), 5.86 (1H, dd, J = 10.1, 1.1 Hz, H-1), 6.17 (1H, dd, J = 10.1, 3.4 Hz, H-2); 13C-NMR (125.6 MHz; CDCl3; Me4Si) δ 23.5 (t), 24.0 (t), 25.1 (t), 34.5 (t), 37.6 (t), 52.2 (q), 52.8 (q), 71.9 (d), 73.1 (d), 76.5 (d), 83.3 (s), 111.2 (s), 127.7 (d), 129.9 (d), 170.0 (s); EIMS m/z 298 (M+, 85%); HREIMS m/z calcd for C15H22O6 (M)+ 298.1416, found 298.1416.
Methyl (3R,4R,5R,6S)-3,4-O-cyclohexylidene-3,4,5-trihydroxy-6-methoxy-1-cyclohexene-1-carboxylate 14
To a solution of 10 (13.8 mg, 0.046 mmol) in CH2Cl2 (2 mL) was added Dess–Martin periodinane (99.1 mg, 0.23 mmol) at rt with stirring. After 24 hr, the reaction mixture was diluted with tert-butylmethylether and treated with aq. Na2S2O4 and aq. NaHCO3. The organic layer was separated, dried over MgSO4, and filtered and the solvent was removed under reduced pressure to afford crude ketone 13. Data of crude 13: IRνmax (liquid film)/cm−1 1726 (CO), 1612 (CC); 1H-NMR (300 MHz; CDCl3; Me4Si) δ 1.25–1.70 (10H, m), 3.60 (3H, s, 6-OMe), 3.80 (3H, s, COOMe), 3.94 (1H, dd, J = 7.3, 6.6 Hz, H-5), 4.00 (1H, dt, J = 6.6, 1.4 Hz, H-6), 4.17 (1H, dd, J = 7.3, 6.4 Hz, H-4), 4.66 (1H, ddd, J = 6.4, 3.9, 1.4 Hz, H-3), 6.68 (1H, dd, J = 3.9, 1.4 Hz, H-2); 13C-NMR (75.5 MHz; CDCl3; Me4Si) δ 24.1 (t), 25.2 (t), 30.0 (t), 35.8 (t), 37.2 (t), 52.6 (q), 59.6 (q), 74.5 (d), 77.1 (d), 113.3 (s), 133.0 (d), 134.3 (s), 164.6 (s), 200.9 (s); EIMS m/z 296 (M+, 94%); HREIMS m/z calcd for C15H20O6 (M)+ 296.1260, found 296.1262
To a suspension of NaBH4 in MeOH (0.5 mL) was added crude ketone 13 dissolved in CH2Cl2 (2 mL) at 0 °C with stirring. After 1 hr, the reaction mixture was treated with aq. NH4Cl and extracted with CH2Cl2. The organic layer was dried over MgSO4 and filtered. The solvent was removed under reduced pressure to afford a crude residue that was purified by column chromatography (eluent: EtOAc: hexane = 1: 1) to give 14 (11.9 mg, 86% in 2 steps). 14: Colorless oil; [α]D25 +25.1 (c 0.12 in CHCl3); IRνmax (liquid film)/cm−1 3497 (OH), 1720 (CO), 1656 (CC); 1H-NMR (300 MHz; CDCl3; Me4Si) δ 1.25–1.70 (10H, m), 3.17 (1H, d, J = 11.5 Hz, 5-OH), 3.58 (3H, s, 6-OMe), 3.81 (3H, s, COOMe), 3.83 (1H, m, H-5), 4.28 (1H, dd, J = 4.9, 0.6 Hz, H-6), 4.48 (1H, ddd, J = 5.7, 3.4, 0.7 Hz, H-3), 4.66 (1H, dd, J = 5.7, 3.2 Hz, H-4), 6.82 (1H, br d, J = 3.4 Hz, H-2); 13C-NMR (75.5 MHz; CDCl3; Me4Si) δ 24.1(t), 24.3 (t), 25.3 (t), 36.0 (t), 37.5 (t), 52.3 (q), 61.3 (q), 68.1 (d), 72.1 (d), 73.0 (d), 74.2 (d), 111.5 (s), 129.7 (s), 137.3 (d), 166.0 (s); EIMS m/z 298 (M+, 69%); HREIMS m/z calcd for C15H22O6 (M)+ 298.1416, found 298.1419.
(−)-Pericosine B: Methyl (3R,4R,5R,6R)-6-methoxy-3,4,5-trihydroxy-1-cyclohexene-1-carboxylate 2
To a solution of alcohol 14 (13.2 mg) in MeOH (0.5 mL) was added TFA (0.5 mL, excess) at 0 °C and the reaction mixture was stirred for 1 hr. After stirring for another 4 hr at rt, the reaction mixture was condensed under reduced pressure to afford a crude residue that was purified by silica gel chromatography (eluent: 3–5% MeOH in CH2Cl2) to give (−)-2 (7.9 mg, 82%). (−)-2: Colorless crystals (hexane-EtOAc); mp 69–71 °C; [α]D25−32.6 (c 0.35 in EtOH); IRνmax (liquid film)/cm−13433 (OH), 1713 (CO), 1651 (CC); 1H-NMR (500 MHz; acetone-d6; Me4Si) δ 3.60 (3H, s, 6-OMe), 3.78 (3H, s, COOMe), 3.85 (1H, dd, J = 4.1, 2.0 Hz, H-5), 3.98 (1H, m, H-4), 4.20 (1H, m, H-3), 4.26 (1H, ddd, J = 4.1, 1.1, 0.9 Hz, H-6), 6.74 (1H, dd, J = 2.5, 1.1 Hz, H-2); 13C-NMR (125.6 MHz; acetone-d6; Me4Si) δ 52.2 (q), 61.5 (q), 69.5 (d), 70.0 (d), 72.8 (d), 77.0 (d), 130.5 (s), 141.9 (d), 166.9 (s); EIMS m/z 219 (M+, 0.8%), 186 (M+−MeOH, 8%); HREIMS m/z calcd for C9H15O6 (M + H)+ 219.0868, found 219.0860.
Methyl (3R,4R,5S,6S)-6-methoxy-3,4,5-trihydroxy-1-cyclohexene-1-carboxylate 15
Alcohol 10 (11.4 mg) was converted to (−)-15 (4.7 mg, 57%) by the same process as above. (−)-15: Colorless crystals (hexane-EtOAc); mp 94–97 °C; [α]D25−75.6 (c 0.23 in EtOH); IRνmax (KBr)/cm−1 3418 (OH), 1716 (CO), 1651 (CC); 1H-NMR (500 MHz; acetone-d6; Me4Si) δ 3.51 (3H, s, 6-OMe), 3.676 (1H, dd, J = 7.3, 4.1 Hz, H-4), 3.76 (3H, s, COOMe), 3.98 (1H, ddd, J = 4.8, 0.9, 0.7 Hz, H-6), 4.11 (1H, dd, J = 7.3, 4.8 Hz, H-5), 4.32 (1H, br dd, J = 4.1, 3.9 Hz, H-3), 6.69 (1H, ddd, J = 3.9, 0.9, 0.5 Hz, H-2); 13C-NMR (125.6 MHz; acetone-d6; Me4Si) δ 52.0 (q), 59.7 (q), 66.7 (d), 70.5 (d), 71.2 (d), 79.3 (d), 132.3 (s), 139.2 (d), 167.2 (s); EIMS m/z 219 (M+, 0.4%), 186 (M+−MeOH, 6%); HREIMS m/z calcd for C9H15O6 (M + H)+ 219.0868, found 219.0861.
Acknowledgements
We are grateful to Dr. K. Minoura and Ms. M. Fujitake of this University for NMR and MS measurements, respectively. We also thank Dr. T. Yamada of this Unversity for providing copies of 1H- and 13C-NMR spectra of natural pericosine B and its acetonide, which appear in the ESI†. This work was supported in part by a Grant-in-Aid from “Dousoukai” of Osaka University of Pharmaceutical Sciences awarded to Y. Usami.References
- A. Numata, M. Iritani, T. Yamada, K. Minoura, E. Matsumura, T. Yamori and T. Tsuruo, Tetrahedron Lett., 1997, 38, 8215–8218 CrossRef CAS.
- T. Yamada, M. Iritani, H. Ohishi, K. Tanaka, M. Doi, K. Minoura and A. Numata, Org. Biomol. Chem., 2007, 5, 3979–3986 RSC.
- Y. Usami, C. Hatsuno, H. Yamamoto, M. Tanabe and A. Numata, Chem. Pharm. Bull., 2004, 52, 1130–1133 CrossRef CAS (Erratum: Chem. Pharm. Bull. 2005, 53, 271.).
- Y. Usami and Y. Ueda, Chem. Lett., 2005, 34, 1062–1063 CrossRef CAS.
- Y. Usami and Y. Ueda, Synthesis, 2007, 3219–3225 CrossRef CAS.
- Y. Usami, Y. Horibe, I. Takaoka, H. Ichikawa and M. Arimoto, Synlett, 2006, 1598–1600 CrossRef CAS.
- Y. Usami, I. Takaoka, H. Ichikawa, Y. Horibe, T. Tomiyama, M. Otsuka, Y. Imanishi and M. Arimoto, J. Org. Chem., 2007, 68, 6127–6134 CrossRef.
- Y. Usami, K. Mizuki, H. Ichikawa and A. Arimoto, Tetrahedron: Asymmetry, 2008, 19, 1460–1463.
- T. J. Donohoe, K. Blades, M. Helliwell, M. J Warning and N. J. Newcombe, Tetrahedron Lett., 1998, 39, 8755–8758 CrossRef CAS.
- Y. Usami, H. Ichikawa and M. Arimoto, Int. J. Mol. Sci., 2008, 9, 401–421 Search PubMed.
- Y. Usami and A. Numata, Chem. Pharm. Bull., 2004, 52, 1125–1129 CrossRef CAS.
- H. Okamura, Y. Nakamura, K. Morishige, R. Ohura, H. Shimizu, T. Iwakawa, M. Nakatani, Proceedings of the 40th Tennen Yuki Kagobutsu Toronkai Koen Yoshisyu, 1988, Fukuoka, Japan, pp,187–192 Search PubMed.
- J. Garcia, Ruano, J. Lopez-Cantarero, A. M. Martin, Castro, H. Adams and J. H. Rogriguez, Ramos, Phosphorus, Sulfur and Silicon and the Related Elements, 2005, 180, 1493–1494 Search PubMed.
- G. Ulibarri, W. Nadler, T. Skrydstrup, H. Audrain, A. Chianori, C. Riche and A. A. Grierson, J. Org. Chem., 1995, 60, 2753–2761 CrossRef CAS.
- β,γ-Unsaturated enone 13 was so unstable that purification by silica gel chromatography afforded a complex mixture including more stable 16 shown in Fig. 3 formed by double bond migration. Therefore, other oxidizing agents could not be used in the preparation of 13. Dess–Martin oxidation at 40 °C to accelerate the reaction resulted in the formation of 16 and aromatized product 17. Methyl (4R,5R)-4,5-cyclohexylidene-2-methoxy-3-oxo-1-cyclohexene-1-carboxylate 16: Colorless crystals (CH2Cl2); mp 60–62 °C; [α]D25−1.8 (c 0.085 in CHCl3); IRνmax (KBr)/cm−11732 (CO), 1693 (CO), 1617 (CC);1H-NMR (500 MHz; CDCl3; Me4Si) δ 1.35–1.70 (10H, m), 2.91 (1H, dd, J = 9.2, 4.8 Hz, H-6A), 3.09 (1H, dd, J = 9.2, 2.1 Hz, H-6B), 3.79 (3H, s, -OMe), 3.85 (3H, s, COOMe), 4.36 (1H, d, J = 5.3 Hz, H-4), 4.62 (1H, ddd, J = 5.3, 4.8, 2.1 Hz, H-5);13C-NMR (125.6 MHz; CDCl3; Me4Si) δ 23.7 (t), 23.8 (t), 24.9 (t), 26.4(t), 35.2 (t), 37.1 (t), 52.4 (q), 60.6 (q), 71.4 (d), 76.3 (d), 76.5 (d), 110.5 (s), 127.4 (s), 151.2 (s), 166.4 (s), 193.1 (s); EIMS m/z 296 (M+, 76%); HREIMS m/z calcd for C15H20O6 (M)+ 296.1260, found 296.1259. Methyl 3,4-dihydroxy-2-methoxybenzoate 17: Yellow oil; IRνmax (liquid film)/cm−1 3538 (OH), 1714 (CO), 1604 (CC); 1H-NMR (CDCl3) δ 3.89 (3H, s, -OMe), 3.93 (3H, s, COOMe), 5.87 (2H, br s, -OH), 6.74 (1H, d, J = 8.9 Hz), 7.46 (1H, d, J = 8.9 Hz); 13C-NMR (CDCl3) δ 51.9 (q), 62.3 (q), 110.9 (d), 115.2 (s), 123.9 (d), 136.7 (s), 148.1 (s), 148.6 (s), 165.4 (s); EIMS m/z 198 (M+, 76%), 166 (M+−MeOH, 88%); HREIMS m/z calcd for C9H10O5 (M)+ 198.0520, found 198.0524.
- Y. Usami, K. Mizuki, K. Suzuki, E. Sakamoto, H. Ichikawa, M. Arimoto, Proceedings of the 50th Tennen Yuki Kagobutsu Toronkai Koen Yoshisyu, 2008, Fukuoka, Japan, pp, 547–552 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H- and 13C-NMR spectra of synthetic (−)-pericosine B, compounds 10, 14, natural pericosine B, and its acetonide. NOESY spectrum of compound 10. See DOI: 10.1039/b813072h |
| This journal is © The Royal Society of Chemistry 2009 |