DOI:
10.1039/B9NR00162J
(Review Article)
Nanoscale, 2009,
1, 16-39
Mechanised nanoparticles for drug delivery
Received
27th June 2009
, Accepted 24th August 2009
First published on 4th September 2009
Abstract
Time and time again humanity is faced with a unifying global crisis that crosses the many great divides in different societies and serves to bring once segregated communities back together as a collective whole. This global community instinctively turns to science to develop the means of addressing its most pressing problems. More often than not, these forces dictate the direction that scientific research takes. This influence is no more apparent than in the field of supramolecular chemistry where, for decades now, its responsibility to tackle such issues has been put on the back burner as a consequence of a lack of platforms with which to deliver this contemporary brand of chemistry to meaningful applications. However, the tide is slowly turning as new materials emerge from the field of nanotechnology that are poised to host the many attractive attributes that are inherent in the chemistry of these supermolecules and also in the mechanostereochemistry of mechanically interlocked molecules (MIMs), which can be reused as a sequel to supramolecular chemistry. Mesoporous silica nanoparticles (SNPs) have proven to be supremely effective solid supports as their surfaces are easily functionalised with either supermolecules or MIMs. In turn, the blending of supramolecular chemistry and mechanostereochemistry with mesoporous SNPs has led to a new class of materials – namely, mechanised SNPs that are effectively biological nanoscale ‘bombs’ that have the potential to infiltrate cells and then, upon the pulling of a chemical trigger, explode! The development of these materials has been driven by the need to devise new therapies for the treatment of cancer. Recent progress in research promises not only to control the acuteness of this widespread and insidious disease, but also to make the harsh treatment less debilitating to patients. This global scourge is the unifying force that has brought together supramolecular chemistry, mechanostereochemistry and nanotechnology, uniting these three communities for the common good. At the nanoscale level, the mechanism for the release of cargos from the confines of the nanopores in the SNPs is accomplished by way of mechanical modifications made on the surface of these functionalised supports. These mechanical motions rely on both supramolecular, i.e., host–guest complexes, and mechanostereochemical phenomena (e.g., bistable rotaxanes), which are often stimulated by changes in pH, light and redox potentials, in addition to enzymatic catalysis. The future of this field lies in the development of ‘smart bombs’ wherein the loaded mechanised SNPs are endocytosed selectively by cancer cells, whereupon an intracellular trigger causes release of a cytotoxin, effectively leading to apoptosis. This review serves to highlight (1) the evolution of surface-functionalisation of SNPs with supermolecules and also with MIMs, (2) the mechanisms through which controlled-release of cargo from mechanised SNPs occurs, and (3) results from the in vitro application of these mechanised SNPs.
1. Introduction
The application of nanotechnology to the treatment of degenerative diseases – their diagnosis, their monitoring, and their control – has garnered attention in recent years. More specifically, research into the targeting and delivery of diagnostic, therapeutic, and pharmaceutical agents is right at the forefront of nanomedicine as scientists and engineers strive, along with physicians, to meet a long list of requirements from the laboratory to the pharmacy. Presently, drug delivery systems are being designed to alter the pharmacokinetics and pharmacodynamics of the drugs they deliver, as well as to function as reservoirs for the drugs themselves. The majority of drug delivery systems currently approved by the Food and Drug Administration (FDA), fall either into the category of liposomal, lipid-based formulations or into the collection of therapeutic compounds that are linked in some way to polyethyleneglycol (PEG). The mechanised silica nanoparticles (MSNPs), which will be brought into the spotlight in this review, offer a fresh approach to drug delivery, which combines the robustness of the drug delivery vehicle with the preciseness of molecular machinery.
Some of the nanotechnologies that can be accessed with MSNPs, which range in diameter from approximately 50 to 200 nm, are illustrated in Fig. 1. The molecular machinery on the surfaces of the silica nanoparticles (SNPs) falls broadly into two categories – that is, molecular on the right-hand side, and supramolecular on the left-hand side. While the latter are much easier to come by synthetically than the former, their operation is irreversible. On the other hand, the molecular nanovalves lend themselves to robustness in addition to being much more amenable to precise control than the supramolecular ones. The difference is that, while the former behave like [2]rotaxanes – where one of the stoppers on the dumbbell component is the SNP itself – the latter are more akin to semirotaxanes – that is, they are pseudorotaxanes with one stopper which is once again the SNP. Two different types of molecular nanovalves have been identified and investigated: in one type, the covalent rupture of the terminal stoppers allows the rings to forsake their recognition sites and depart from the stalks. This process can be likened to a beverage can with a snap-top, i.e., break it once, for example, by the enzymatic cleavage of a particular covalent bond and then the ring will depart from the stalk, followed immediately thereafter by the spilling out of the cargo. The other molecular type is based on switchable bistable [2]rotaxane molecules where, under redox or pH control, for example, the ring can be switched from one recognition site to the other and vice versa. By contrast, in the supramolecular varieties of nanovalves, there are no stoppers and so the rings owe their association with the stalks to a single strong binding site which can be modified, such as to kill the binding interactions, thus permitting the rings to depart from the stalks. In addition to enzyme activation of the molecular machinery, situations involving light-activation, or changes in pH, to bring about the activation of acid- and base-responsive nanovalves have all been harnessed. It should be stressed that these are only some of the possible nanotechnologies that can be assessed: there are more!
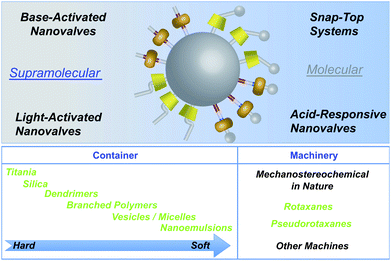 |
| | Fig. 1 Top: A graphical representation of the mechanised silica nanoparticle-based technologies highlighted in this review. Mesoporous SNPs, in particular MCM-41, provide an outstanding platform on which molecular machinery, be it supramolecular or molecular in nature, can be attached to the surfaces of the SNPs. Bottom: The range of containers and machines that compete today in the arena of drug delivery systems. | |
In this review, we will highlight the molecular machinery, which can be incorporated onto the surfaces of silica mesostructures to make MSNPs, and the different release mechanisms employed to unload the cargo on demand. We begin by discussing how silica thin films and nanoparticles are synthesised, functionalised with organic and metallic substrates, as well as how to assess the SNPs' cellular uptake and cytotoxicity as a result of investigations conducted on mammalian cell lines . Following these discussions, the incorporation of supramolecular and molecular machinery – such as [2]pseudorotaxanes and [2]rotaxanes nanovalves – into MSNPs, will be reviewed. This exploration is followed by the introduction of various snap-top MSNP systems which are activated by chemical or light stimulus. We conclude by reviewing the progress being made in making MSNPs more biocompatible as a result of introducing semirotaxanes as the nanovalves which incorporate water-soluble cyclodextrin and cucurbit[n]uril rings.
2. Mesoporous silica structures
In this section, we will focus on the synthesis and modification of mesoporous silica thin films and nanoparticles as supports for the formation of functional materials. We will describe various strategies that have been used to place active molecules into specific regions of the mesostructures, leading to the development of operational molecular machines. A discussion of the different types of cargo molecules that have been encapsulated inside the nanopores of SNPs will also be covered. The discussion will conclude with cell intake and cytotoxicity studies carried out on SNPs and MSNPs.
2.1. Mesoporous silica
Research on the surfactant-templated synthesis of mesostructures has been an active field in materials chemistry ever since the development1 of mesoporous silicate structures (e.g., MCM-41) by the Mobil Corporation. The production of these materials combines the sol –gel process2 of metal oxides, a technique that is widely used to make inorganic glass, with cationic surfactants, as templating agents to form the ordered structures. Although the original synthetic methods yield large particles of hexagonally packed mesoporous silicate, the process can be applied to create various forms, including thin films , nanoparticles, or monoliths.3 Furthermore, this approach has been extended to non-silica oxides, such as TiO2 and ZrO2, using various types of templating agents, including non-ionic surfactants and block copolymers .4
2.2. Synthesis of silica thin films
Mesostructured silica thin films and particles are highly versatile frameworks for functional materials where a desired function – such as energy transfer, electron transfer, or operating molecular machines – is induced by molecules deliberately placed in specific regions of the structure.5 Highly ordered mesostructured thin films can be prepared from a solution at room temperature by a process called “evaporation-induced self-assembly.” This process employs a one-pot sol , comprised of a silica precursor and a templating surfactant in aqueous ethanol.6 A glass or silicon substrate is dipped into the solution and a thin film of liquid is pulled along with the substrate as it is retracted. Preferential evaporation of the ethanol during the film deposition drives the formation of surfactant micelles, which further assemble into a liquid crystalline mesophase. Condensation of the silica monomers around the surfactant micelles eventually leads to the formation of mesostructured silica films. Various techniques for functionalisation with molecular machines, and preliminary studies on these systems, have been performed using thin films as the silica supports.7
Synthetic procedures that rely upon base have been employed3a–b to catalyse the silica condensation and small cationic templating surfactants in order to yield mesoporous SNPs that are suitable for biomedical applications. These SNPs have been used successfully as imaging agents and carriers for drugs and proteins.8 In a typical synthesis (Fig. 2), the silicate source, tetraethylorthosilicate, is mixed with the surfactantcetyltrimethylammonium bromide in a heated basic (pH 11) aqueous solution. The nanoparticles are formed using the micellar template during the base-catalysed sol –gel process. On refluxing the nanoparticles in acidic alcohol, the electrostatic interaction between the cationic surfactant head groups and the anionic silicates is broken, allowing the surfactants to be removed from the mesopores. Electron microscopy and X-ray diffraction analyses show that the shape of the nanoparticles, with their hexagonal arrays of pores, remains intact after the surfactant removal process. The nanoparticles (Fig. 3) are roughly spherical, smaller than 200 nm in diameter, and contain pores with an average diameter of around 2 nm.
2.4. Functionalisation of silica nanostructures
The possibility of introducing organic appendages onto the walls of the pores and onto the surfaces of the nanoparticles makes mesoporous silica highly suitable as a host material for the incorporation of guest molecules.9 A uniform distribution of active molecules and functional groups can be obtained in an efficient manner using co-condensation methods.10,11 In co-condensation reactions, the functional silane molecules are mixed with the silicate precursors and added directly to the solution in the expectation that they will arrange themselves within the developing mesostructure as the materials form. Alternatively, post-synthetic approaches to functionalisation are also useful for derivatising the surfaces of the nanoparticles. It is often desirable to functionalise accessible silica surfaces, such as the surfaces of nanoparticles or areas near the pore orifices.12 Post-synthetic grafting involves the attachment of functional groups to the surface of the mesopores through silylation, either before or after the surfactant removal. Organic functional groups that are introduced upon modification with a silane linker are typically used for further derivatisations.
2.5. Incorporation of metal nanocrystals into mesoporous silica
Metal nanocrystals can be incorporated into the mesostructured silica in order to provide additional functionalities.13,14 Mesoporous silica spheres can be formed around the nanocrystals by mixing the silica source with an aqueous solution of surfactant-coated nanocrystals.15 The electrostatic interactions between the hydrolysed silicate molecules, surfactant-coated nanocrystals, and free surfactant molecules help to promote the base-catalysed silica condensation to form the mesostructure. This general procedure has been used to embed iron oxide, gold, and silver nanocrystals at the centers of mesoporous SNPs (Figs. 3c and d). By incorporating metal nanocrystals into the mesostructured silica, the nanoparticles can be used for imaging, as well as for the delivery of biologically active molecules.16
2.6. Cargo molecules loaded into mesoporous silica nanostructures and mechanised silica nanoparticles
One of the most attractive and exploitable properties of mesoporous SNPs and MSNPs is their ability to encapsulate different types of cargo molecules. In addition to incorporating metal nanocrystals, as described in Section 2.5., and loading of fluorescent molecules – e.g., Rhodamine B, fluorescein, calcein, and coumarins – into SNPs and MSNPs, various drugs and biomolecules have been infiltrated into MCM-41 nanostructures.
Of particular importance is the ability of SNPs to sequester drug molecules and protect the payload from enzymatic degradation, while reducing the undesired side-effects associated with many drugs. The drug-absorbing and drug-releasing behaviour of mesoporous silica nanostructures has been studied9b,17,18 extensively with both hydrophobic drugs – such as ibuprofen,17a–gpentagastrin,17herythromycin,17i and camptothecin8c – and water-soluble ones – e.g., aspirin,18aalendronate,18b and captopril.18c These studies showed that even though conventional silica nanostructures exhibit sustained-release properties, the drugs are not released in a controlled manner. Additionally, their irregular bulk morphology makes these silica nanostructures inadequate as drug delivery systems. To overcome these disadvantages, MSNPs are made from SNPs with regular morphology. Recent reports of such systems include the encapsulation of paclitaxel,17j,19doxorubicin,20vancomycin21 and cholestane22 inside stimuli-responsive MSNPs.
Aside from storing drugs, increasing the pore diameter allows proteins to be stored inside SNPs.8e,23 The robust silica framework protects the proteins from denaturation and hydrolysis, thus appropriately constructed SNPs are able to deliver membrane-impermeable proteins into the cells. Other biomolecules have also been incorporated into SNPs, such as nucleotides.24 Surface functionalisation with cationic groups allows the binding of negatively charged nucleotides to the nanoparticles. These methods prove advantageous over traditional transfection systems since the nanoparticles can store different cargo molecules within the pores. The resulting nanoparticles are used to deliver biologically active molecules and nucleotides simultaneously. Additionally, mesoporous silica has been demonstrated to store MRI contrast agents,8g fluorescent cell tags8a and DNAplasmids.8d
2.7. Cellular uptake and cytotoxicity studies of silica nanoparticles and mechanised silica nanoparticles
The cellular uptake and cytotoxicity of SNPs have been investigated in order to establish the foundation for MSNPs as effective drug delivery systems. The cellular uptake mechanism of the nanoparticles can be studied by, first of all, attaching fluorescent dye molecules to the surface of the SNPs using the co-condensation method8a,25 without affecting the size or shape of the SNPs, then by monitoring their uptake with fluorescence microscopy. The mesoporous SNPs are taken up8c by cancer cells (Fig. 4) within 30 min and they do not cause any observed cytotoxicity. The intracellular location of the SNPs was determined by staining the cells with Acridine Orange. This dye specifically stains acidic organelles – such as lysosomes – red, while the rest of the cell stains green. The green fluorescence of the SNPs overlaps with the red fluorescence of Acridine Orange exhibiting yellow fluorescence, a situation which indicates that the SNPs are mainly taken up by the acidic organelles.
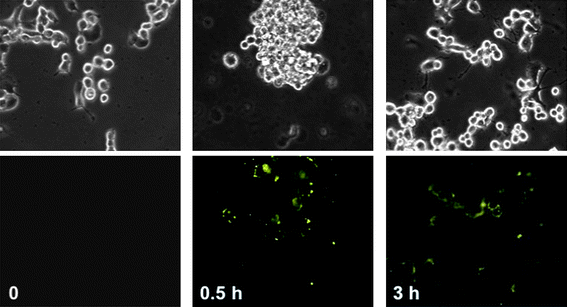 |
| | Fig. 4 Cellular uptake of fluorescent mesoporous SNPs by PANC-1, human pancreatic carcinoma, epithelial-like cell line . The upper panels show phase contrast microscope images of the cancer cells. The lower panels show fluorescence microscope images of the cells incubated with SNPs for the indicated number of hours. As a control, PANC-1 cells were incubated in the absence of SNPs and the fluorescence microscope image, lower left image, does not show any fluorescence. | |
A study by Linden et al.26 used 400 nm sized-SNPs functionalised with polyethyleneimine (PEI) and further modified by introducing fluorescein isocyanate (FITC) and folic acid (FA) as fluorescent and targeting moieties, respectively, to target cancer cells. The folate receptor is overexpressed27 on the surface of several cancer cell lines – such as colorectal, endometrial, ovarian, lung, breast, and renal cell carcinoma – and has become an attractive targeting receptor for selective drug delivery. The cellular uptake of PEI/FITC-functionalised SNPs (SNPs-PEI/FITC) and PEI/FITC/FA-functionalised SNPs (SNPs-PEI/FITC/FA), by HeLa cancer cells, after 24 h of incubation, was found by flow cytometry to be 24 and 40%, respectively. In comparison, the fluorescence of internalised FITC-functionalised SNPs (SNPs-FITC) was equal to the background fluorescence. These results are in contrast to what was reported earlier by Lin's group,25 which showed cellular uptake of 100 nm-sized FITC-labeled SNPs. The difference in the cellular uptake of these two SNPs is explained by their different sizes – smaller SNPs can lead to an enhancement of nonspecific cellular uptake.
Competition experiments,26 using different concentrations of free FA from 0 to 3 mM, were conducted to verify that the uptake of FA-functionalised-SNPs is mediated via the folate receptor. Free FA was introduced into the cell medium, together with SNPs-PEI/FITC/FA during the cell incubation period. The percentage of internalised SNPs-PEI/FITC/FA decreased as the concentration of free FA was increased. At the maximum free FA concentration of 3 mM, the percentage of internalised SNPs-PEI/FITC/FA declined to about one-third of the value measured in absence of free FA. From these results, it can be concluded that, although the primary cellular uptake mechanism of the SNPs-PEI/FITC/FA is mediated by the folate receptor, there is a different nonspecific uptake mechanism that leads to the internalisation of the SNPs-PEI/FITC/FA, even at 3 mM of free FA.
In order to confirm that the SNPs-PEI/FITC/FA are capable of targeting the folate receptor,26 the internalisation of these hybrid SNPs by embryonic kidney epithelial 293 cells – which express a much lower level of the folate receptor – was measured by fluorescence using confocal microscopy and flow cytometry. HeLa cells uptook five-to-six times more SNPs-PEI/FITC/FA than 293 cells and the number of HeLa cells containing the hybrid SNPs were consistently two-fold higher than the 293 cells. Additionally, the SNPs-PEI/FITC/FA showed no apparent cytotoxicity during incubation with the HeLa cells for 24 h.
Mesoporous SNPs can be used to carry and deliver water-insoluble anticancer drugs into human cancer cells.8c,8e,9b,16c,17j,20 SNPs have been loaded with either camptothecin or paclitaxel by immersing them in concentrated dimethylsulfoxide or ethanolic solutions of the drug molecules.8c The drug-loaded SNPs were collected by centrifugation to remove the supernatant and dried under vacuum before being resuspended in aqueous solution, upon which only 4% of the stored drug molecules were released into the supernatant. However, when the drug-loaded nanoparticles were once again redispersed in organic solvents, all of the drug molecules became dispersed into solution. Based on absorption measurements, it has been calculated8c that SNPs can store between 2 to 5 weight percent of drug molecules. The cytotoxic effect of drug-loaded SNPs was tested on several cancer cell lines and was found to cause apoptosis. A control study in which unloaded SNPs were used did not inhibit cell proliferation or lead to cell death.8c
Some examples of light-driven activation as the stimulus to release drugs inside azobenzene-modified28 and gold-capped19 SNPs have been demonstrated. In the former case,28 the drug molecules are expelled out of the pores and into the cells as a consequence of the cis–trans isomerisation of the azobenzene components conjugated along the pore walls. In the latter case,19 the gold nanocrystals, functionalised with a photocleavable group, block the pore openings and trap the drug molecules within the nanoparticles. Upon irradiation at 365 nm, the gold nanocrystals are cleaved and the cargo molecules are released into the external environment. In both experiments, cell death only occurs after the light irradiation in which the drug molecules are released into the cells. Further demonstration of the controlled release of cargo molecules carried out in vitro utilises the chemically labile disulfide bonds between the pore openings and inorganicnanocrystals, i.e., cadmium sulfide or iron oxide.21,29 Upon addition of a reducing agent, the disulfide bonds are cleaved and the inorganicnanocrystals blocking the pore openings dissociate from the particle surface, thus causing the release of cargo molecules. These results indicate that mesoporous silica is promising as a biocompatible drug-carrier that can be tailored to release cargo molecules on demand.
3. Molecular machines as nanovalves for mechanised silica nanoparticles
In the previous sections, we described (i) the ease with which mesoporous silica materials can be designed, synthesised and functionalised with organic precursors, (ii) the different cargo molecules that have been loaded into the SNPs and MSNPs, and (iii) how the SNPs' and MSNPs' lack of cytotoxicity to mammalian cells demonstrates the potential of these nanoparticles to act as drug delivery vehicles. The section which follows next will be dedicated to the design and synthesis of mesoporous silica thin films and nanoparticles functionalised with molecular machinery – such as azobenzenes and MIMs, e.g., bistable [2]rotaxanes and their supramolecular analogues, the [2]pseudorotaxanes. These MSNPs operate in organic solvents and can be activated either photochemically or chemically.
Photoisomerisation of azobenzene derivatives has enabled the development of many useful applications. While the operational range of molecular motion is too restricted to create macroscopic phenomena, in the small spaces of mesopores, molecular movement on the nanometer level is sufficient to dominate the physical and chemical behaviour of guest molecules. Numerous investigations have demonstrated that, when azobenzenes are incorporated into an amorphous silica gel matrix, photo-induced cis–trans isomerisation can lead to an increased elution rate for column chromatography.30 One of the most successful methods of controlling molecular transport into, through, and out of mesoporous silica has been to use (Fig. 5) the cis–trans isomerisation of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds in azobenzene derivatives tethered to the interiors of the mesopores. The wagging motion resulting from this isomerisation resembles that of a molecular impeller where the release of the cargo molecules from the pores can be regulated photochemically. Detailed photophysical studies of azobenzene derivatives attached to mesostructured silica frameworks suggests31 the optimal excitation wavelength for azobenzene is ∼450 nm. The reason why 450 nm is optimal is that both trans-to-cis and cis-to-trans isomerisations can occur at this wavelength with their respective quantum yields being 0.36 and 0.64. Therefore, continuous excitation of azobenzene-derivatised SNPs – call them nanoimpeller-MSNPs – at 450 nm induces isomerisation and generates nanoimpeller-type motion.
N bonds in azobenzene derivatives tethered to the interiors of the mesopores. The wagging motion resulting from this isomerisation resembles that of a molecular impeller where the release of the cargo molecules from the pores can be regulated photochemically. Detailed photophysical studies of azobenzene derivatives attached to mesostructured silica frameworks suggests31 the optimal excitation wavelength for azobenzene is ∼450 nm. The reason why 450 nm is optimal is that both trans-to-cis and cis-to-trans isomerisations can occur at this wavelength with their respective quantum yields being 0.36 and 0.64. Therefore, continuous excitation of azobenzene-derivatised SNPs – call them nanoimpeller-MSNPs – at 450 nm induces isomerisation and generates nanoimpeller-type motion.
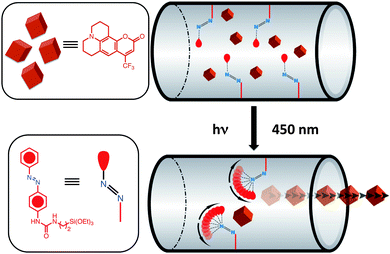 |
| | Fig. 5 Nanoimpeller-MSNPs with 4-phenylazoaniline incorporated into the mesopores and loaded with cargo molecules. Continuous excitation of the nanoimpellers at 450 nm, induces trans-to-cis and cis-to-trans isomerisations of the azobenzenes, generating a nanoimpeller-type motion, thus causing the cargo molecules to be propelled out of the mesopores. | |
These nanoimpeller-MSNPs can be synthesised32 by, first of all coupling azobenzene derivatives, such as 4-phenylazoaniline, to ethoxysilane molecules, and later by co-condensing them into the mesostructure during an evaporation-induced self-assembly process.3d Prior to photoexcitation, cargo molecules are loaded inside the nanoparticles. Since the interiors of the pores are teeming with tethered azobenzenes, brisk and unhindered diffusion pathways are not available. Excitation at 450 nm causes a continual wagging motion of the untethered termini of the bound azobenzene molecules. Such motion encourages (Fig. 5) cargo molecules to be propelled and eventually expelled out of the mesopores. In order to verify that azobenzene isomerisation drives the release, nanoimpeller-MSNPs were excited and the resulting luminescence intensity of the cargo molecules – at its emission maximum – was plotted as a function of time. The resulting release profile (Fig. 6) indicates that these SNPs are capable of holding cargo molecules prior to any photoexcitation, and expelling them when stimulated. In an analogous manner, Fujiwara et al.33 have co-condensed N-(3-triethoxysilyl)propyl-4-phenylazobenzamide into silica nanoparticles and report an exceptionally fast release rate of cholesterol molecules (from mesopores) when the grafted azobenzene derivatives undergo cis–trans isomerisation in the presence of light. The low cytotoxicity of these nanoimpeller-SNPs has also established their potential value in phototherapeutic drug delivery. Lu et al.28 have used these nanoimpeller-SNPs to deliver and release the anti-cancer drug campothecin into cancer cells, successfully inducing apoptosis.
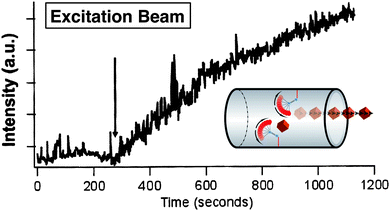 |
| | Fig. 6 Luminescence intensities of cargo molecules (Coumarin 540A), propelled out of the nanoimpeller MSNPs mesopores and into solution, as a function of time measured at 1s intervals. The arrow indicates the moment at which the excitation beam was directed onto these nanoimpeller-MSNPs. Minimal leakage has been shown by this system prior to any photoexcitation. | |
3.2. Mechanically interlocked molecules as nanovalves for mechanised silica nanoparticles
3.2.1. Mechanically interlocked molecules.
Mechanically Interlocked Molecules (MIMs), such as catenanes and rotaxanes34 have attracted considerable attention because they are commensurate with the construction of sophisticated functional molecular devices. These MIMs rely on noncovalent bonding interactions, such as donor–acceptor,35 hydrogen bonding,36 metal–ligand,37 and hydrophobic38 interactions, not only for their function but also for their existence in the first place. [2]Rotaxanes are composed of a dumbbell component encircled around its rod section by a ring: bulky stoppers at the ends of the rods prevent the ring from slipping off the dumbbell component. In bistable [2]rotaxanes, the dumbbell component has two recognition sites, both of which are complementary to recognition sites located on the ring component: preferentially at equilibrium, the ring spends much more of its time at one recognition site on the dumbbell than on the other one. Ideally, the ring responds to a stimulus, be it chemical, electrochemical, or optical, causing it to switch from its preferred recognition site to its less preferred one. By taking advantage of this kind of switching mechanism, bistable [2]rotaxanes have been incorporated into NanoElectro-Mechanical Systems (NEMS)39 and Molecular Electronic Devices (MEDs),37c,40 as well as into MSNPs.5,41
At the outset of our mission to create MSNPs, by decorating mesoporous silica nanoparticles (MCM-41) with MIMs, we aimed to establish proof of principle. In the beginning, our research was directed towards a systematic understanding of the principles governing the operation of these nanovalves, as we call them, on the surfaces of the SNPs. Our first generation of MSNPs were made by functionalising SNPs with [2]pseudorotaxanes12a,42 and bistable [2]rotaxanes12b,12c that rely on donor–acceptor interactions for the operation of their nanovalves. These MSNPs were shown to operate under redox control that is initiated chemically,12a,42 electrochemically,12b,12c and photochemically.12a At the same time, we also began to explore MSNPs with nanovalves, based on the complexation of secondary dialkylammonium ions and dibenzo[24]crown-8 (DB24C8), where switching can be controlled43 by changes in pH.
3.2.2. Donor–acceptor [2]pseudorotaxane-tethered MSNPs.
Initially, we took advantage of the redox- and photo-switching properties of [2]pseudorotaxanes, composed of 1,5-bis[2-(2-hydroxyethoxy)ethoxy]naphthalene (BHEEN) stalks, encircled by cyclobis(paraquat-p-phenylene) (CBPQT4+) rings, which act as gatekeepers to encapsulate or release cargo molecules – e.g., tris(2,2′-phenylyridyl)iridium(III), Ir(ppy)3 – within the nanopores of mesoporous silica MCM-41 nanoparticles.42
[2]Pseudorotaxanes in a sol –gel matrix and tethered to silica thin films .
The first devices we tested7a were [2]pseudorotaxane-based supramolecular machines powered by photosensitisers. They were either trapped physically inside a rigid nanoporous sol –gel framework or were attached covalently onto silica surfaces. The former device is constructed by irradiating a condensed silica sol –gel matrix containing (i) the BHEEN⊂CBPQT4+pseudorotaxane, (ii) 9-anthracenecarboxylic acid (ACA) as a photosensitiser, and (iii) a sacrificial donor, ethylenediamine-tetraacetate (EDTA) – which prevents the back electron transfer process – with a 365 nm Hg lamp. The reduction of the CBPQT4+ rings to CBPQT2(•+) bisradical dications can be monitored by the disappearance of the original pink color (λmax = 520 nm) – corresponding to the charge transfer (CT) interactions between the π-accepting CBPQT4+ rings and π-electron rich BHEEN stalks – and the rise in the intensity of the pale-blue color (λmax = 650 nm) of the dication. In the latter device, the BHEEN homolog, BHEEEN, with an extra bismethyleneoxy unit, was attached covalently to a silica surface. The BHEEEN-functionalised surface was dipped into an aqueous solution of CBPQT4+, forming the corresponding [2]pseudorotaxane on the surface. A second immersion of the [2]pseudorotaxane-coated silica surface into an ACA and EDTA solution, followed by irradiation at 365 nm, led to an increase in the naphthalene-based fluorescence intensity, similar to what was observed in the sol –gel matrix device. After these proof-of-principle studies involving the successful trapping of [2]pseudorotaxanes in a silica monolith and mounting them on a silica film, we proceeded to attach the molecular machinery onto the surfaces of mesoporous SNPs.
[2]Pseudorotaxane-tethered MSNPs.
The first supramolecular machine that we attached to the surfaces of SNPs was the donor–acceptor [2]pseudorotaxane that we had previously mounted onto a silica thin film – the BHEEEN⊂CBPQT4+pseudorotaxane. When these [2]pseudorotaxanes are tethered to the orifices of the nanopores (Fig. 7), the gates remain closed when CBPQT4+ rings encircle the BHEEEN units on the stalks on account of [π⋯π] and [C–H⋯O] noncovalent bonding interactions.42 Reducing the CBPQT4+ rings with a reducing agent (NaCNBH3) opens (Fig. 7b) the gates on account of the fact that the CBPQT2(•+) bisradical dication no longer interacts with BHEEEN and so the rings dethread, allowing the cargo, Ir(ppy)3, to be released (Fig. 7c). In order to confirm that the operation of the MSNPs depends on the dethreading of the CBPQT2(•+) rings from the BHEEEN gateposts, the MCM-41 nanoparticles were functionalised only with BHEEEN – that is, without threading with CBPQT4+ rings – and loaded with Ir(ppy)3. When the nanoparticles were dispersed in acetonitrile solution, the iridium complex diffused immediately into solution. The [2]pseudorotaxanes could also be activated photochemically by reducing the CBPQT4+ rings12a in the presence of ACA and the sacrificial reagent, triethanolamine (TEOA). When MSNPs, derivatised with BHEEEN⊂CBPQT4+[2]pseudorotaxanes and loaded with Ir(ppy)3, are immersed in an acetonitrile solution containing ACA and TEOA and irradiated at 365 nm, an increase in intensity corresponding to the release of Ir(ppy)3 is observed.
![Structural formulae and graphical representations of supramolecular [2]pseudorotaxane MSNPs constructed by tethering BHEEEN to the orifices of the nanopores, loading with Ir(ppy)3 and threading the BHEEEN gateposts with CBPQT4+ rings. Supramolecular MSNPs in the (a) closed and (b) open positions. The luminescent cargo is released only upon reduction of the CBPQT4+ rings by NaCNBH3. (c) Controlled release profile of Ir(ppy)3 in MeCN solution, as a function of time. A flat baseline is obtained prior to the addition of NaCNBH3 (indicated by the arrow) demonstrating that there is no leakage of the cargo molecules when the MSNPs are closed.](/image/article/2009/NR/b9nr00162j/b9nr00162j-f7.gif) |
| | Fig. 7 Structural formulae and graphical representations of supramolecular [2]pseudorotaxane MSNPs constructed by tethering BHEEEN to the orifices of the nanopores, loading with Ir(ppy)3 and threading the BHEEEN gateposts with CBPQT4+ rings. Supramolecular MSNPs in the (a) closed and (b) open positions. The luminescent cargo is released only upon reduction of the CBPQT4+ rings by NaCNBH3. (c) Controlled release profile of Ir(ppy)3 in MeCN solution, as a function of time. A flat baseline is obtained prior to the addition of NaCNBH3 (indicated by the arrow) demonstrating that there is no leakage of the cargo molecules when the MSNPs are closed. | |
These results demonstrate that the cargo inside the nanopores of the MSNPs can be released on demand when exposed to an external stimulus, be it either chemical or photochemical. Once these MSNPs have been opened, they cannot be easily closed, because, after the CBPQT4+ rings are reduced, they dethread from the BHEEEN gateposts and become dispersed in solution. A disadvantage of the supramolecular nanovalves is that their operation is irreversible. Reversibility can be introduced by replacing these supramolecular nanovales with switchable bistable donor–acceptor [2]rotaxanes which can also be operated with chemical, electrical or optical stimuli.
3.2.3. Reversible, redox-active [2]rotaxane-tethered MSNPs.
Reversibly operating MSNPs that can be opened or closed under redox control have also been constructed (Fig. 8) using bistable [2]rotaxanes12b,12c as the nanovalves. Switchable donor–acceptor [2]rotaxanes with a CBPQT4+ ring, that can be induced to shuttle between the two recognition units – namely, tetrathiafulvalene (TTF) and 1,5-dioxynaphthalene (DNP) – have been employed12c as the nanovalves on MSNPs. The bistable [2]rotaxanes are attached to the silica surface of the MSNPs such that the free DNP units are closer to the orifices while the bulky CBPQT4+ rings are associated with the more distant TTF units (Fig. 8a). This ground state co-conformation constitutes the open position of the MSNPs and allows luminescent cargos, e.g., Ir(ppy)3 or Rhodamine B (RhB), to diffuse (Fig. 8b, Step 1) into the nanopores. After these MSNPs have been loaded, the gates of the molecular nanovalves are closed by oxidizing (Fig. 8b, Step 2) the TTF units to TTF2+ dication with Fe(ClO4)3. As a consequence of Coulombic repulsion, the CBPQT4+ rings move from the dication to the DNP unit (Fig. 8b, Step 3), thus preventing the cargo from flowing out of the nanopores. The operation of the MSNPs was monitored over time by measuring the emission intensity of the cargo in solution by fluorescence spectroscopy. When the nanovalves on the MSNPs are in the closed position, a flat baseline in the fluorescence spectrum is obtained, a characteristic which attests to the absence of any leakage. It is only after the reduction of the TTF2+ dications with ascorbic acid that the luminescence intensity increases, as the CBPQT4+ rings moves back to the neutral TTF units.
![A graphical representation of switchable bistable [2]rotaxane-based MSNPs. (a) The bistable [2]rotaxanes are tethered to the orifices of the SNPs through an isocyanation between the isocyanate-functionalised SNPs and the bistable rotaxane carrying a hydroxymethyl group for reaction with the isocyanate-terminated stalks. (b) Proposed mechanism of operation of the MSNPs. In Step 1, the ground state co-conformation of the bistable [2]rotaxanes, where the CBPQT4+ rings encircles the TTF units, constitutes the open position of the MSNPs and allows the cargo molecules to be loaded under a concentration gradient into the nanopores. In Step 2, the nanovalves are closed by oxidizing the TTF units with Fe(ClO4)3 to the TTF2+ dications and as a consequence of Coulombic repulsion, the tetracationic CBPQT4+ rings move to the DNP units. In Steps 3 and 4, the nanovalves are opened by reducing the dications back to the neutral TTF units whereupon the CBPQT4+ rings move back to the TTF units, thus releasing the cargo.](/image/article/2009/NR/b9nr00162j/b9nr00162j-f8.gif) |
| | Fig. 8 A graphical representation of switchable bistable [2]rotaxane-based MSNPs. (a) The bistable [2]rotaxanes are tethered to the orifices of the SNPs through an isocyanation between the isocyanate -functionalised SNPs and the bistable rotaxane carrying a hydroxymethyl group for reaction with the isocyanate -terminated stalks. (b) Proposed mechanism of operation of the MSNPs. In Step 1, the ground state co-conformation of the bistable [2]rotaxanes, where the CBPQT4+ rings encircles the TTF units, constitutes the open position of the MSNPs and allows the cargo molecules to be loaded under a concentration gradient into the nanopores. In Step 2, the nanovalves are closed by oxidizing the TTF units with Fe(ClO4)3 to the TTF2+ dications and as a consequence of Coulombic repulsion, the tetracationic CBPQT4+ rings move to the DNP units. In Steps 3 and 4, the nanovalves are opened by reducing the dications back to the neutral TTF units whereupon the CBPQT4+ rings move back to the TTF units, thus releasing the cargo. | |
To verify that the movement of the CBPQT4+ ring is responsible for the release of the cargo molecules from the nanopores of the MSNPs,12c the dumbbell component of the [2]rotaxane, without the CBPQT4+ ring, was attached to the SNPs. Since there was no gate to block the orifices of these SNPs, the cargo molecules were not trapped inside the nanopores and leaked out into solution immediately. In order to confirm that the electrostatic interactions between the charged guest, Ir(ppy)3, and the inside of the silica nanopores are not the reason for retaining the cargo inside the MSNPs, a neutral compound, RhB, was chosen as the cargo. A similar release profile to that obtained with Ir(ppy)3 was observed for RhB.
3.2.4. Optimisation of [2]rotaxane-based MSNPs.
We have also investigated, in a systematic manner, the outcome12b of constructing MSNPs with different lengths of the silane linkers – which are used to attach the bistable [2]rotaxane molecules covalently to the silica substrate – on the rate of release of different-sized cargo molecules. These MSNPs can be categorised (Fig. 9) as having IN and OUT locations, depending on the position of the tethered bistable [2]rotaxane relative to the opening of the orifices. The MSNPs are more efficient with the bistable [2]rotaxanes anchored deep into the pores (IN position) than when the rotaxanes are tethered closer to the pores' openings (OUT position). When the bistable [2]rotaxane gatekeepers are in the IN position, both large-sized – Ir(ppy)3 and RhB with molecular dimensions of 1.0 × 1.0 and 1.0 × 1.5 nm2, respectively – and small-sized molecules, i.e., coumarins, are retained inside the nanopores. In the OUT position, however, the gatekeepers only retain the large-sized molecules, whereas the smaller coumarins leak, even when the MSNPs are closed. This difference provides great versatility, controlled very simply by the choice of where the gateposts are tethered relative to the pores' orifices and depending on the size of the cargo molecules. In order to expand the structure–property relationship of MSNPs further, the length of the spacer between the two recognition sites of the bistable [2]rotaxane was altered. It was discovered, however, that different spacer lengths do not have any significant influence on the MSNPs' operation. These results demonstrate that by fine-tuning the MSNPs' molecular properties in a modular fashion, the operation of the MSNPs can be tailored for future applications in drug delivery systems.
![A systematic study of the effect of varying the lengths of the silane linkers and the spacers between the two recognition units in the bistable [2]rotaxanes on the leakiness of bistable [2]rotaxane-based MSNPs. Top: Bistable [2]rotaxane nanovalves with different spacer lengths between the two recognition units. Middle: The luminescent probe molecule, coumarin. Lower left-hand side: Three different silane linkers a, b and c used in this study. Right-hand side: A graphical representation of MSNPs with the possible positions (IN and OUT) of the linker relative to the nanopore orifices.](/image/article/2009/NR/b9nr00162j/b9nr00162j-f9.gif) |
| | Fig. 9 A systematic study of the effect of varying the lengths of the silane linkers and the spacers between the two recognition units in the bistable [2]rotaxanes on the leakiness of bistable [2]rotaxane-based MSNPs. Top: Bistable [2]rotaxane nanovalves with different spacer lengths between the two recognition units. Middle: The luminescent probe molecule, coumarin. Lower left-hand side: Three different silane linkers a, b and c used in this study. Right-hand side: A graphical representation of MSNPs with the possible positions (IN and OUT) of the linker relative to the nanopore orifices. | |
3.2.5. Supramolecular nanovalves controlled by proton abstraction and competitive binding.
Another recognition motif43 – that is the mutual recognition between secondary dialkylammonium ions and DB24C8 – was investigated as a release mechanism (Fig. 10). DB24C8 is a sufficiently large macrocyclic polyether to be able to encircle a naphthalene-containing dialkylammonium ion center (–CH2NH2+CH2–), thus forming a [2]pseudorotaxane. Since the non-covalent bonding interactions responsible for the formation of these 1 : 1 complexes involve primarily [N–H⋯O]+ hydrogen bonds, they can be induced to dissociate in solution by the addition of base.
![Supramolecular dibenzo[24]crown-8 (DB24C8)/dialkylammonium MSNPs. These MSNPs are constructed by attaching dialkylammonium stalks to the surfaces of the SNPs, loaded with the luminescent cargo and closed by threading of the ammonium centers with the DB24C8 rings. The hydrogen-bonding recognition motif responsible for these 1 : 1 complexes, between the secondary dialkylammonium centers and the DB24C8 rings, can be made to dissociate by (a) pH adjustment with base or (b) competitive binding of the DB24C8 rings with fluorodialkylammonium or metal ions.](/image/article/2009/NR/b9nr00162j/b9nr00162j-f10.gif) |
| | Fig. 10 Supramolecular dibenzo[24]crown-8 (DB24C8)/dialkylammonium MSNPs. These MSNPs are constructed by attaching dialkylammonium stalks to the surfaces of the SNPs, loaded with the luminescent cargo and closed by threading of the ammonium centers with the DB24C8 rings. The hydrogen-bonding recognition motif responsible for these 1 : 1 complexes, between the secondary dialkylammonium centers and the DB24C8 rings, can be made to dissociate by (a) pH adjustment with base or (b) competitive binding of the DB24C8 rings with fluorodialkylammonium or metal ions. | |
4. Snap-top mechanised silica nanoparticles
In the previous section we discussed the operation of three types of MSNPs: (i) photoresponsive azobenzene-based nanoimpellers, (ii) supramolecular, redox- and pH-active nanovalves based on [2]pseudorotaxanes, composed of BHEEEN⊂CBPQT4+ and dialkylammonium ion centers and DB24C8 rings, and (iii) molecular, switchable bistable [2]rotaxanes. In this section, a range of different snap-top nanovalves which can be activated by disulfide reduction, insulin consumption, UV-light irradiation, and enzymatic cleavage of a covalent bond, as well as by capping the SNPs with polymers and lipids, will be presented and evaluated.
4.1.
Disulfide reduction activation of snap-top mechanised silica nanoparticles
4.1.1.
Gold nanoparticle-capped MSNPs.
Another kind of MSNP, which is being explored currently by other researchers in the area, uses both metallic and magnetic nanoparticles to block the nanopores of mesoporous silica. Lin et al.19 and Amorós et al.45 have reported delivery systems which utilize gold nanoparticles to cap the nanopores on mesoporous SNPs. In the former case (Fig. 11), the gold nanoparticles were functionalised with the photocleavable linker, thioundecyltetraethyleneglycoester-o-nitrobenzylethyldimethyl ammonium bromide (TUNA), and were incorporated onto the surface of MCM-41 by virtue of an electrostatic interaction with the surface of the negatively charged silica. Upon irradiation with UV light, TUNA is converted to the negatively-charged thioundecyltetraethyleneglycolcarboxylate (TUEC), which then induces the gold nanoparticles to dissociate from the surface of the mesoporous silica on account of charge repulsion, thereby releasing the (fluorescein) cargo from the nanopores of these SNPs, as shown in Fig. 11. In the latter case, the researchers designed45 a new nanoscopic gate-like hybrid system consisting of an MCM-41 support, functionalised at their pore outlets with a saccharide capable of interacting with boronic acid-functionalised gold nanoparticles which act as nanoscopic caps.
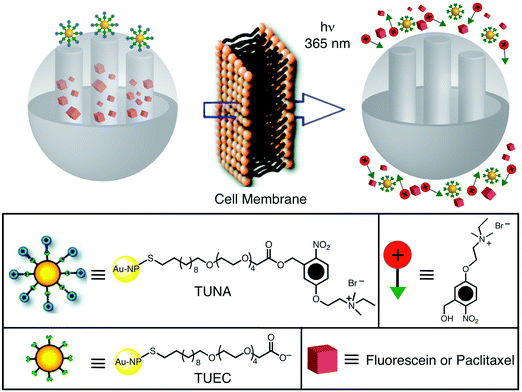 |
| | Fig. 11 Graphical representation of gold nanoparticle-capped MSNPs. The gold NPs are functionalised with a photocleavable linker, thioundecyltetra-ethyleneglycoester-o-nitrobenzylethyldimethyl ammonium bromide (TUNA), and incorporated onto the surface of the SNPs through electrostatic interactions. Upon irradiation with UV light, the TUNA molecules are converted to thioundecyltetraethyleneglycolcarboxylate (TUEC), causing the gold nanoparticles to dissociate from the surface of the SNPs as a consequence of Coulombic repulsion, and thus releasing the cargo (adapted with permission from ref. 19. © 2009 American Chemical Society). | |
4.1.2. Iron oxide-capped MSNPs.
Superparamagnetic iron oxide (Fe3O4) NPs have been utilized in a variety of biological settings. Lin et al.29 have reported the synthesis of a controlled-release delivery system based on MCM-41 nanorods, capped with Fe3O4NPs which are stimuli-responsive and chemically inert to entrapped cargo molecules. The mesoporous nanorods were functionalised with 3-(propyldisulfanyl)propionic acid, loaded with fluorescein dye and capped through an amide linkage between propionic acid on the surface and 3-aminopropyltriethoxysilyl-functionalised Fe3O4NPs. The disulfide bonds between the nanorods and the Fe3O4NPs in these MSNPs are labile and can be cleaved with disulfide reducing agents such as dithiothreitol (DTT) and dihydrolipoic acid (DHLA). After 48 h following the addition of DTT and DHLA, the total loading release of the fluorescent dyes was 40% and 31%, respectively.
4.1.3. Cadmium sulfide-capped MSNPs.
Lin et al.21 have also demonstrated the use of CdSNPs to cap the nanopores of mesoporous SNPs for the controlled release of cargo molecules. In this system (Fig. 12), mercaptoacetic acid-functionalized CdSNPs are attached within the pores of SNPs containing 2-(propyldisulfanyl)ethylamine using an amidation procedure, thus containing the cargo molecules within the mesoporous silica. The presence of disulfide bonds in the threads connecting the CdSNPs to the MCM-41 particles allows the CdS caps to be released in the presence of a disulfide-reducing agent, such as DTT or mercaptoethanol. Thus, when these MSNPs are exposed to either of these reductants, the CdSNPs are freed from the MCM-41 matrix, and the cargo molecules – in this instance, Vancomycin or adenosine triphosphate, ATP – can then escape from the pores of the mesoporous silica.
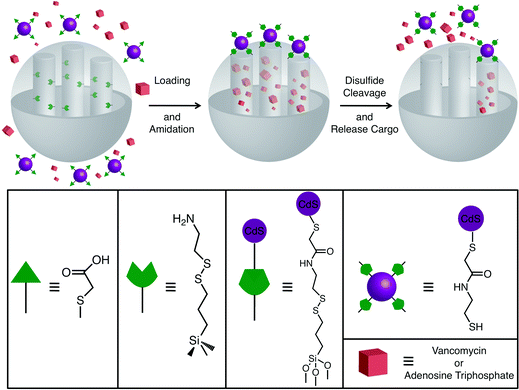 |
| | Fig. 12 A graphical representation of CdS-capped MSNPs. After loading the cargo into the SNPs, the amine-functionalised nanopores are capped by attaching mercaptoacetic acid-functionalised CdS NPs through an amidation reaction. In the presence of a disulfide reducing agent, such as dithiothreitol (DTT), the disulfide bonds break, freeing the CdSNP caps, thus opening the nanopores and releasing the cargo molecules (adapted with permission from ref. 21. © 2003 American Chemical Society). | |
Recently, the Lin group46 has also developed an MSNP system that is capable of delivering insulin and cyclic adenosine monophosphate (cAMP), sequentially. Insulin is used to cap the pores of MCM-41 and is removed upon the introduction of glucose or other saccharides, a process (Fig. 13) which then leads to the release of cAMP from these MSNPs. This dual-drug delivery system is particularly attractive for biomedical applications. After the insulin caps have been consumed in the body, the cAMP can induce insulin production by activating Ca2+ channels in pancreas beta cells.47,48
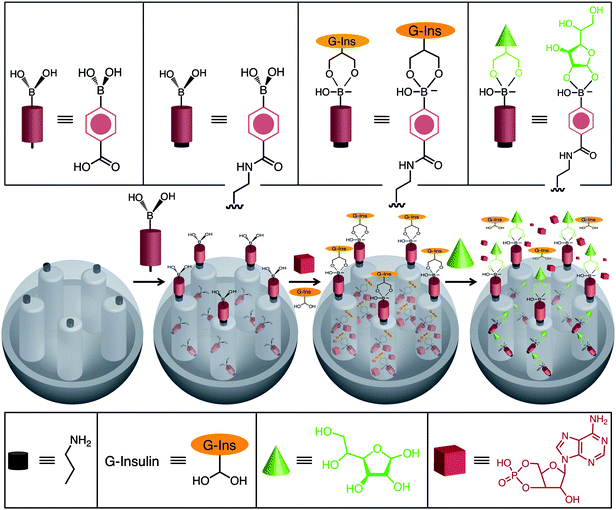 |
| | Fig. 13 An MSNP co-delivery system, for sequential release of insulin and cyclic AMP (cAMP), was constructed by the Lin group. The SNPs were loaded with cAMP and capped with G-insulin. In the presence of glucose the insulin caps are consumed, thus opening the nanopores and releasing cAMP. | |
4.3. Enzyme-activated mechanised silica nanoparticles
4.3.1.
Esterase-activated MSNPs.
Enzyme-directed cleavage of caps has been demonstrated by Bein et al.49 with biotin–avidin as a protease responsive cap. We have also developed snap-top systems (Fig. 14) based on esterasehydrolysis.50 To test the viability of our enzyme-responsive snap-top nanovalves, stoppers which could be hydrolysed by Porcine Liver Esterase (PLE) were introduced into the nanovalves where the rings are α-cyclodextrin (α-CD) tori. Mesostructured MSNPs loaded with luminescent cargo molecules (RhB) were capped with the ester-linked adamantyl stoppers. PLE catalyses the hydrolysis of adamantyl ester stoppers, resulting in dethreading of the α-CD rings, leading to the release of the cargo molecules from the nanopores. As a control, MSNPs were prepared using an adamantyl amide stopper analog, which does not undergo hydrolysis by PLE. After capping the MSNPs with adamantyl stoppers, the dye-loaded SNPs were filtered and washed to remove non-specifically adsorbed small contaminants. The enzyme-triggered release of cargo molecules was monitored using luminescence spectroscopy. The emission of RhB in the solution above the particles was measured (Fig. 15) as a function of time using a 514 nm probe beam (15 mW), both before and after addition of PLE. Prior to the addition of PLE, the emission intensity of RhB remained essentially constant, indicating that the dye remains trapped in the nanopores of these MSNPs. The emission intensity begins to increase almost immediately following the addition of PLE. The emission intensity approaches its maximum value asymptotically with a t1/2 of ∼5 min. By contrast, no such increase in emission was observed for the amide-stoppered snap-top nanovalves. These results show that the snap-top design concept is a feasible one. They open the door to a simple and responsive release nanovalve system.
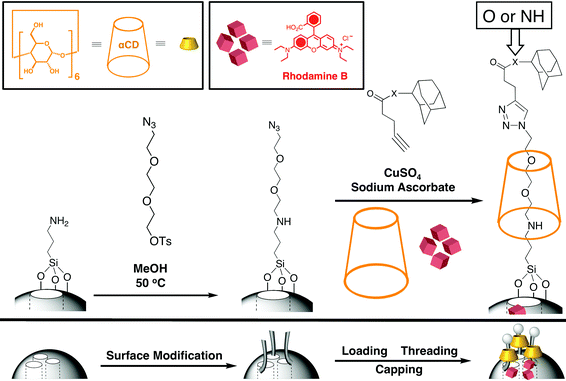 |
| | Fig. 14 Enzyme-responsive snap-top MSNPs. The nanovalves are assembled by functionalising the SNP's surface with tri(ethyleneglycol)monoazide monotosylate, followed by loading and threading of α-CD and finally by capping with alkyne-terminated adamantyl stoppers. Pig liver esterase catalyses the hydrolysis of the ester-linked adamantyl stoppers, snapping off the stopper and thus opening the MSNPs and releasing the cargo. | |
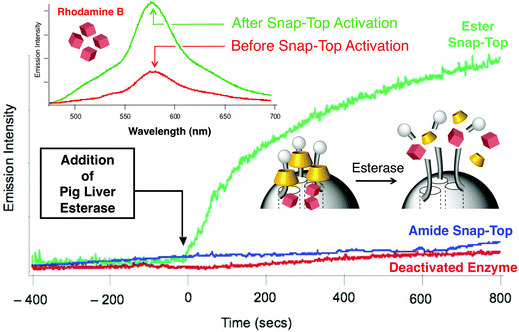 |
| | Fig. 15 Release profile from enzyme-activated MSNPs as a function of time. The emission intensity of RhB begins to increase immediately after pig liver esterase is added for the ester adamantyl-stoppered MSNPs (green trace) but not for the amide snap-top MSNPs (blue trace). Inset shows the emission intensity before and after snap-top activation as a function of wavelength. | |
4.3.2.
β-D-Galactosidase-responsive MSNPs.
Another example of enzyme-responsive controlled-release using SNPs, reported by Amarós et al.,51 uses lactose-capped MSNPs that are selectively uncapped using β-D-galactosidase, an enzyme involved in the hydrolysis of the disaccharidelactose into its two monosaccharides, galactose and glucose. In humans, β-D-Galactosidase is located predominately along the enterocytes lining the vili of the small intestine. These MSNPs were loaded with [Ru(bipy)3]Cl2, which was used as a dye for monitoring the enzyme-triggered process, and then capped with a lactose derivative tethered to the surface of the nanoparticle. The threads inhibit delivery of the cargo molecules owing to the formation of a network of disaccharides linked by hydrogen bonding. Addition of β-D-galactosidase uncapped the mesopores by hydrolysis of the 1 → 4 glycosidic bonds.
To study the enzyme-responsive controlled-release system,51 the fluorescence intensity corresponding to the released ruthenium complexes from these MSNPs was monitored by fluorescence spectroscopy at pH 7.5. This pH value was chosen as it is the pH in the intestine at which enzymatic saccharide degradation reactions typically occur. Under these conditions, negligible cargo release was observed. However, upon addition of β-D-galactosidase a sharp emission band was observed at 610 nm, on account of a decrease in the size of the appended group, thus causing the release of the entrapped dye. Furthermore, the release profile of the [Ru(bipy)3]2+dye from the MSNPs in the presence of the enzyme shows a slow but steady leakage of the cargo over at least five hours, thus avoiding the instant delivery of the ruthenium complex.
4.4. UV-light activated mechanised silica nanoparticles
4.4.1.
o-Nitrobenzyl ester snap-top MSNPs.
Cyclodextrin caps were later utilized by Kim et al.52 to develop a CD-covered nanocontainer system with o-nitrobenzyl ester moieties (Fig. 16) as the photocleavable linkers. Exposure of the MSNPs to UV light (λ = 350 nm) results in the photocleavage of o-nitrobenzyl and the release of the cargo.
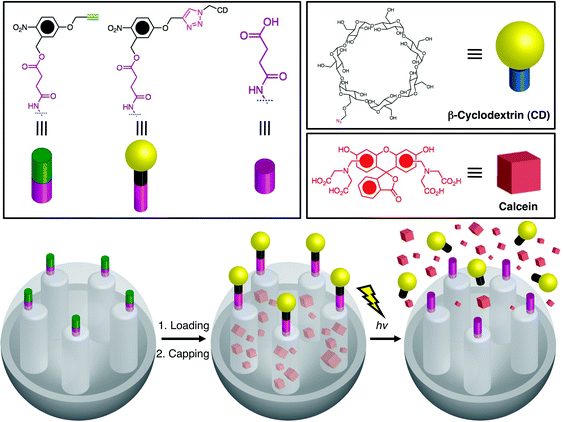 |
| | Fig. 16
Cyclodextrin-covered MSNPs with o-nitrobenzyl ester moieties as photocleavable linkers. After loading the cargo, the SNPs are capped with cyclodextrins (CDs) via a click reaction between the alkyne-functionalised linker and the azide-functionalised CDs. Irradiation of the MSNPs with UV light results in the photocleavage of the o-nitrobenzyl-bearing CD caps and release of the cargo molecules (adapted from ref. 52 with permission. © Wiley-VCH Verlag GmbH & Co. KGaA). | |
4.4.2.
Azobenzene-functionalised MSNPs.
An alternative approach53 takes advantage of the high binding affinity between β-CD and trans-azobenzene and the low, if any, binding between β-CD and cis-azobenzene to control (Fig. 17) the release of cargo molecules from MSNPs. SNPs modified with 4-(3-triethoxysilylpropylureido)azobenzene or (E)-4-((4-(benzylcarbamoyl)phenyl)-diazenyl)benzoic acid stalks provide the recognition motif for the β-CD or pyrene-functionalised-β-CD caps. Irradiation of the azobenzene stalks with 351 nm light induces isomerisation from the trans to cis geometry, causing the β-CD caps to dethread from the stalks and, as a result, the cargo is released from the nanopores.
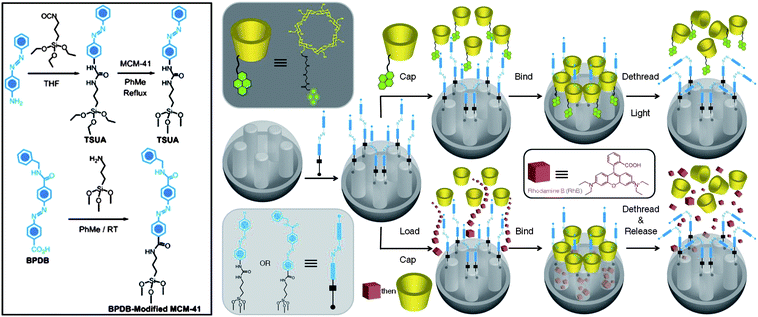 |
| | Fig. 17 β-CD-Capped and pyrene-functionalised-β-CD-capped (py-β-CD) MSNPs. SNPs are functionalised with either 4-(3-triethoxysilylpropylureido)azobene (TSUA) or E-4-((4-(benzylcarbamoyl)phenyl)diazenyl)benzoic acid (BPDB) stalks and respectively capped with py-β-CD or β-CD rings to seal the nanopores. Upon irradiation, the trans-azobenzene isomerises to cis-azobenzene leading to the dissociation of py-β-CD or β-CD rings from the stalks, opening the nanopores and releasing the cargo. | |
4.4.3. [2 + 2] Photocycloaddition of coumarins as gatekeepers.
An additional class of MSNPs currently under investigation is one which uses gatekeeper molecules to hold cargos within the pores of MCM-41 as a result of a reversible chemical reaction taking place on the surfaces of the mesoporous SNPs. In 2003, researchers developed an MSNP system, which utilises coumarin as a gatekeeper.22 The gates of the MSNP can be opened or closed upon irradiation with a specific wavelength of light. In this particular system, the MCM-41 was reacted with 7-[(3-triethoxysilyl)propoxy]coumarin in order to attach the coumarin derivative onto the surface of the SNPs. Irradiation of the coumarin-functionalised MSNPs with UV light (λ ≥ 310 nm) promotes a [2 + 2] photocycloaddition between two courmarin molecules sealing the pores of the MSNPs and enclosing the cargo. Irradiation with higher-energy UV light (λ ≈ 250 nm) cleaves the cyclobutane ring, reversing the photodimerization, opening the pores of the MCM-41 and releasing the cargo molecules.
4.5.1. Redox-activated polymer-capped MSNPs.
Capping of the pores of MCM-41 has already been established using polymers, lipids, and most recently enzyme-activated caps. Feng et al.54 have recently developed an MSNP system with gatekeeper molecules containing disulfide linkages which are responsive to redox chemistry. This system (Fig. 18) was realised by first of all functionalising MCM-41 with poly(N-acryloxysuccinimide), then cross-linking the polymer strands with cystamine to seal the pores of the MSNPs. Thus, when the cross-linked MSNP is exposed to a disulfide reducing agent such as DTT, the disulfide bonds of cystamine are reduced to thiols and the cargo escapes from the nanopores of MCM-41-based MSNPs.
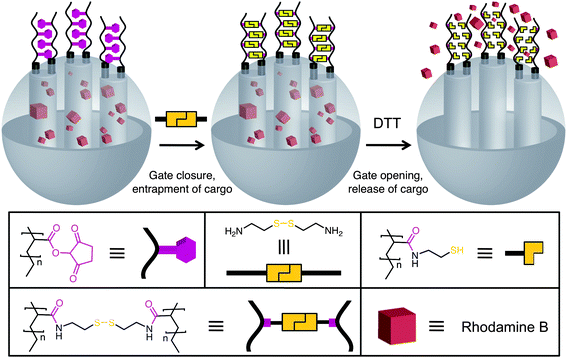 |
| | Fig. 18 Assembly of redox-responsive MSNPs with cystamine cross-linked polymers as gatekeepers. After loading the cargo into the nanopores, the poly(N-acryloxysuccinimide)-functionalised SNPs are reacted with cystamine to cross-link the polymers and close the gates. Upon addition of dithiothreitol (DTT), the disulfide bonds are reduced and the gates are opened, releasing the cargo (adapted from ref. 54 with permission. © 2008 American Chemical Society). | |
4.5.2. pH- and anion-driven supramolecular MSNPs.
Martínez-Máñez and Amorós et al.55 developed a pH- and anion-driven gate-like system by attaching polyamines onto the surface of SNPs to control the release of cargo molecules, in this case, a ruthenium complex, [Ru(bipy)3]2+. The ability of polyamines to bind anions via electrostatic forces and through hydrogen bonding is known to be pH-dependent and was used to control the release of the Ru complex in these MSNPs. The pH-control of the open–closed positions of these MSNPs arises from the hydrogen-bonding interactions between the amines at neutral pH (open-position) and the Coulombic repulsion between the ammonium centers at acidic pH, constituting the closed-position. Close to neutral pH (pH 6), the polyamines are only partially protonated and the amines hydrogen bond with each other, thus opening the nanopores. In contrast, in acidic conditions (pH 2 or 4) as a result of the Coulombic repulsion between the polyammonium centers, the polymers adopt a rigid-like conformation which pushes the polyammoniums away from each other and toward the nanopore openings.
The anion-controlled release of the cargo molecules was studied55b–c at different pH values (2, 4, 6 and 7.8) and in the presence of various anions – such as fluoride, chloride, sulfate, acetate and phosphate, as well as ATP and adenosine diphosphate (ADP). The ability of different anions to block the nanopore openings can be explained by the strength of the polyammonium complexes with the anions at different pH values. At pH 2, phosphate, sulfate and ATP can block the nanopores and prevent the release of the ruthenium cargo. These anions block the nanopores by coordinating to the polyamines through hydrogen-bonding and electrostatic interactions. At pH 4, only phosphate and ATP anions inhibit the release of the cargo molecules. At this pH, the amines are not as highly protonated as they are at pH 2 and so the coordinating complexes are not as strong as they are at pH 2. At pH 6, however, ATP is the only anion capable of blocking the nanopores, thus preventing the release of the ruthenium complex. The high selectivity of ATP to block the nanopores at both acidic and neutral pH can be related to the ability of ATP to form strong coordination complexes with polyamines over a wide range of pH values. Lastly, at pH 7.8, ATP and ADP are the only anions that can block the nanopores, thereby retaining the cargo molecules.54b
4.5.3. Liposome-fused MSNPs.
Brinker et al.20 reported recently electrostatic-mediated liposome fusion on silica cores to mimic the cell's protective lipid membranes (Fig. 19). These “protocell” MSNPs were synthesised by first of all coating positively charged SNPs with anionic 1,2-dioleoyl-sn-glycero-3-[phospho-L-serine] (DOPS) liposomes, resulting in silica-supported bilayer NPs, which undergo successive steps of electrostatically mediated lipid-exchange between the silica-supported bilayer NPs and oppositely charged free liposomes, cationic 1,2-dioleoyl-3-trimethylammoniumpropane (DOTAP), resulting in DOPS-DOTAP-fused liposomes MSNPs. These “protocell” MSNPs showed reduced bilayer defects and better control of surface charge, properties which allowed greater cargo retention, delivery, and release inside cells.
![An electrostatic mediated liposome-fused MSNP system. The negatively charged drugs are absorbed into the pores of the SNPs. To increase cargo retention, the positively charged SNPs are fused with an anionic liposome, 1,2-dioleoyl-sn-glycero-3[phospho-l-serine] (DOPS), followed by further lipid exchange with a cationic liposome, 1,2-dioleoyl-3-trimethylammoniumpropane (DOTAP). These liposome-fused MSNPs showed better cargo retention, delivery and release inside cells than with a single cationic liposome coating of the SNPs (adapted from ref. 20 with permission. © 2009 American Chemical Society).](/image/article/2009/NR/b9nr00162j/b9nr00162j-f19.gif) |
| | Fig. 19 An electrostatic mediated liposome-fused MSNP system. The negatively charged drugs are absorbed into the pores of the SNPs. To increase cargo retention, the positively charged SNPs are fused with an anionic liposome, 1,2-dioleoyl-sn-glycero-3[phospho-L-serine] (DOPS), followed by further lipid exchange with a cationic liposome, 1,2-dioleoyl-3-trimethylammoniumpropane (DOTAP). These liposome-fused MSNPs showed better cargo retention, delivery and release inside cells than with a single cationic liposome coating of the SNPs (adapted from ref. 20 with permission. © 2009 American Chemical Society). | |
4.5.4. PAMAM dendrimer-capped MSNPs.
The development of vectors that can deliver large biomacromolecules selectively has also attracted much attention. Polyamidoamine (PAMAM) dendrimers can be incorporated onto the surface of mesoporous SNPs to complex plasmidDNA efficiently, yielding a gene transfection agent.8d This objective was achieved by first of all synthesising a second-generation PAMAM dendrimer, and attaching it covalently onto the surface of MCM-41 SNPs containing Texas Red in the pores (Fig. 20) in order to track the distribution of the MSNPs throughout the cell. The PAMAM-functionalised MCM-41 was then complexed with plasmidDNA, pEGFP-C1, which codes for an enhanced green-fluorescent protein, confirming the successful delivery of the plasmid to the cell nucleus.
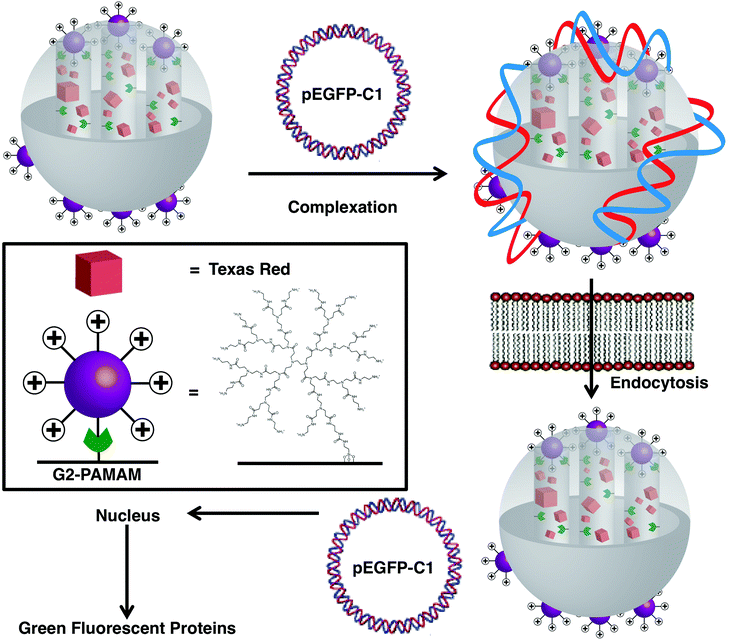 |
| | Fig. 20 Delivery of DNAplasmid pEGP-C1 in cells via a second generation PAMAM-functionalised MSNP. PAMAM-functionalised SNPs, loaded with Texas Red, were complexed with a DNAplasmid, pEGP-C1, that codes for green-fluorescent proteins. Once the MSNPs were endocytosed into the cells, the pEGP-C1 was delivered into the nucleus and GFPs were produced (adapted from ref. 8d with permission. © 2004 American Chemical Society). | |
5. Towards biocompatible mechanised silica nanoparticles
The majority of the MSNPs discussed in the previous sections operate in organic media. Much effort has been dedicated to increase the biocompatibility of the MSNPs and use them as drug delivery systems. In this section, we will discuss the MSNPs which incorporate amine and ferrocene stalks which form inclusion complexes with either cyclodextrin or cucurbit[n]uril (CB[n]) rings to act as caps on the nanovalve systems in order to make these MSNPs more biocompatible.
Cyclodextrins
56 and cucurbit[n]urils57 have been of particular interest as a consequence of their ability to form inclusion complexes, which can be reversibly dissociated by external stimuli, with a variety of guest molecules in water. These families of macrocycles offer several advantages as drug delivery agents, given their additional ability (i) to protect the drug from physical, chemical, and enzymatic degradation and (ii) to enhance cell-membrane permeability.
5.2. Polyethyleneimine-CD and -CB[n] pseudorotaxane-based mechanised silica nanoparticles
One of the simplest means of controlling molecular recognition between a stalk component and either a CD or CB[n] cap is by a change in pH, thereby making it an excellent release mechanism for drug delivery systems. Kim et al.58 reported one (Fig. 21) of the first pH-responsive pseudorotaxane-based MSNP systems. It consists of three biocompatible components, namely, SNPs, polyethyleneimine (PEI) and CD. Guest molecules – calcein in this instance – are entrapped in the nanopores of mesoporous SNPs which are blocked by the surface-grafted pH-responsive PEI/CD polypseudorotaxanes. Two complexes, one utilising α-CD and the other γ-CD, were investigated. In the former case (Fig. 21b), a single PEI polymer threads itself through a series of macrocycles forming a binary 1 : 1 complex. In the latter case (Fig. 21c), the larger cavity size of γ-CD accommodates two PEI polymer threads and therefore forms a ternary 2 : 1 complex. In both instances, the cargo remains trapped in the nanopores of the MSNPs so long as the PEI polymer remains deprotonated (pH ≥ 11). By lowering the pH, the CDs can dethread as a result of the weak interaction between the protonated PEI chain with the hydrophobic cavity of the CDs. In order to demonstrate clearly the pH-responsive release mechanism of these systems, the pH of the solutions of calcein-loaded α-CD/PEI (and γ-CD/PEI) complex was adjusted from 11 to 5.5 and monitored by fluorescence spectroscopy over time. At pH 11 weak emission is observed owing to self-quenching of calcein molecules in the pores of the MSNPs. Upon adjusting the pH to 5.5 an immediate increase in fluorescence intensity is observed, indicating the release of calcein from the nanopores.
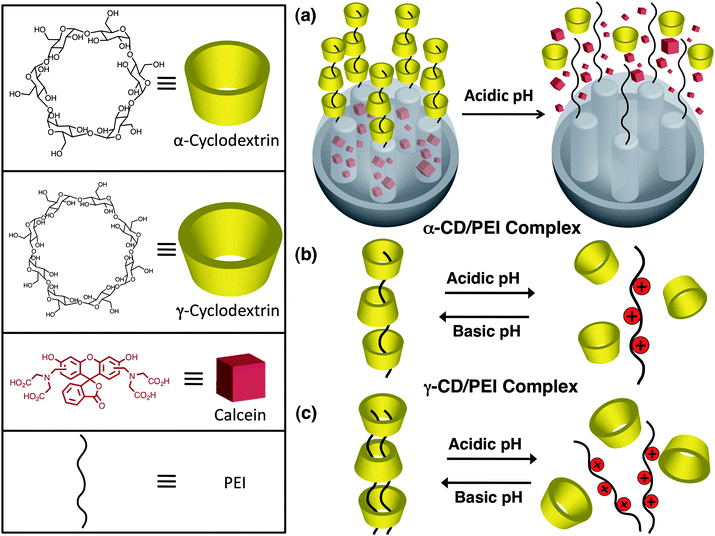 |
| | Fig. 21 (a) pH-Responsive pseudorotaxane-based MSNP systems composed of biocompatible components – SNP, polyethylenimines (PEI) and cyclodextrin (CD). With (b) α-CD, PEI forms a binary 1 : 1 complex, whereas with (c) γ-CD, the polymers form a ternary 2 : 1 complex. Upon addition of acid, the CD rings dethread from the PEI polymers, releasing the cargo molecules (adapted from ref. 58 with permission. © 2007 Wiley-VCH Verlag GmbH & Co. KGaA.). | |
5.3. CB[6]pseudorotaxane mechanised silica nanoparticles
5.3.1. Base-activated CB[6]pseudorotaxane MSNPs.
Shortly thereafter, we reported59 a pH-responsive system (Fig. 22) based on CB[6]pseudorotaxanes. Taking advantage of the ability of CB[6] to catalyse 1,3-dipolar cycloadditions, functionalised SNPs were prepared by the reaction between an azide-substituted ammonium ion and an alkyne-containing ammonium ion yielding a disubstituted 1,2,3-triazole derivative encircled by a CB[6] ring. In contrast to the CDs, CB[6] forms strong complexes with ammonium ions as a result of the [N–H⋯O] hydrogen-bonding interactions between the ion and the carbonyl groups of CB[6]. Consequently, the cargo molecules (RhB) are held tightly in the pores at pH < 10 and are released, as evidenced by fluorescence spectroscopy, upon deprotonation (pH > 10) of the ammonium stalks.
![pH-Responsive MSNPs with CB[6]. After loading the cargo molecules, CB[6] catalyses the 1,3-dipolar cycloaddition. The CB[6] rings cap the nanopores, retaining the cargo molecules inside the nanopores, on account of the strong hydrogen bonds between the secondary dialkylammonium centers in the stalks and the CB[6] rings. Upon deprotonation of the –CH2NH2+CH2– centers at high pH (pH > 10), the CB[6] rings dethread and the cargo molecules are released.](/image/article/2009/NR/b9nr00162j/b9nr00162j-f22.gif) |
| | Fig. 22 pH-Responsive MSNPs with CB[6]. After loading the cargo molecules, CB[6] catalyses the 1,3-dipolar cycloaddition. The CB[6] rings cap the nanopores, retaining the cargo molecules inside the nanopores, on account of the strong hydrogen bonds between the secondary dialkylammonium centers in the stalks and the CB[6] rings. Upon deprotonation of the –CH2NH2+CH2– centers at high pH (pH > 10), the CB[6] rings dethread and the cargo molecules are released. | |
5.3.2. Acid-activated CB[6]pseudorotaxane MSNPs.
In an effort to increase the biocompatibility of the CB[6]/bisalkylammonium [2]pseudorotaxane MSNPs previously reported,59 tunable pH-operable MSNPs were developed,60 in which biologically relevant pH changes are used to trigger the release of the cargo molecules. These MSNPs are based on trisammonium stalks in which two of the ammonium centers are tetramethyleneammoniums and the third is an anilinium unit (Fig. 23). At neutral pH, the anilinium nitrogens are deprotonated and the CB[6] rings reside on the tetramethylenediammoniums, on account of the ion–dipole interactions between the dialkylammonium centers and the occuli of the CB[6] rings, thus blocking the nanopores and encapsulating the cargo molecules. Lowering the pH to the point where the anilinium nitrogens become protonated opens the nanopores as the CB[6] rings shuttle from the two dialkylammonium centers to encapsulate one of the alkylammoniums and the anilinium ammonium center, thus opening the nanopores and releasing the cargos. The shuttling of the CB[6] rings, as the pH is lowered, is a result of the difference in the stability constant for the complexion of NH3+(CH2)6NH3+ with CB[6], which is an order of magnitude greater than that for NH3+(CH2)4NH3+.61 Since the six-carbon spacer is a better match with the inner cavity of the CB[6] ring, it allows stronger ion–dipole interactions between the ring the ammonium center. The release rate can also be adjusted by modifying the pKa of the anilinium nitrogen by changing the aniline groups with p-anisidines. The pKa of the anilinium nitrogen is approximately 1 pKa unit lower than that of the nitrogen atom in p-anisidines.62 For the MSNPs with aniline, only 13% of the encapsulated cargo molecules are released after 600 s, compared to 50% for MSNPs with p-anisidines. The operation of these MSNPs demonstrates that the pH sensitivity of these MSNPs can be rationally tuned by chemically modifying the stalks. Additionally, these MSNPs can also be operated by deprotonating the two alkylammonium centers by adjusting the pH to 10. At this pH, all of the ion–dipole interactions between the stalks and CB[6] rings are disrupted, and the CB[6] rings dethread from the stalks, opening the nanopores and releasing the cargo molecules.
![Acid-responsive CB[6]/trisammonium [2]pseudorotaxane-based MSNPs. These MSNPs operate by retaining the cargo molecules inside the nanopores at neutral pH (b), but release the contents when the pH is either lowered (a) or raised (c). (a) Upon decreasing the pH, the anilinium nitrogen is protonated and the CB[6] rings complex the upper and middle ammonium centers. (c) These MSNPs can also be operated by raising the pH to deprotonate the dialkylammonium centers, which causes the CB[6] to dethread from the trisamine stalks.](/image/article/2009/NR/b9nr00162j/b9nr00162j-f23.gif) |
| | Fig. 23 Acid-responsive CB[6]/trisammonium [2]pseudorotaxane-based MSNPs. These MSNPs operate by retaining the cargo molecules inside the nanopores at neutral pH (b), but release the contents when the pH is either lowered (a) or raised (c). (a) Upon decreasing the pH, the anilinium nitrogen is protonated and the CB[6] rings complex the upper and middle ammonium centers. (c) These MSNPs can also be operated by raising the pH to deprotonate the dialkylammonium centers, which causes the CB[6] to dethread from the trisamine stalks. | |
5.3.3. AND logic dual-controlled MSNPs.
A much more sophisticated dual-controlled MSNP system was developed63 earlier this year by our two groups, in which two different types of molecular machines – namely, azobenzene-based nanoimpellers and pH-responsive nanovalves – are attached inside and around the nanopores and it is only when both machines are activated in tandem to one another that the cargo molecules are released (Fig. 24). Two nanovalves were employed in these MSNPs, based on pH-switchable [2]pseudorotaxanes in which CB[6] rings encircle the bisammonium stalks which are tethered to the orifices of the nanopores. One of the nanovalves consists of CB[6]/bisalkylammonium [2]pseudorotaxanes and operates under basic conditions (left hand-side of Fig. 24), while the other is based on bistable CB[6]/trisammonium pseudorotaxanes and is acid-responsive (right hand-side of Fig. 24). Both dual-controlled MSNPs behave as AND logic gates. A positive output is obtained only when the dynamic wagging of the azobenzenes from the nanoimpellers – resulting from the cis–trans isomerisation of the azobenzenes – is activated by a 448 nm excitation beam and followed by a change in pH, whereupon the CB[6] rings dethread from the bisammonium stalks, thus opening the nanopores and releasing the cargo molecules.
![AND Logic, dual-controlled MSNPs composed of both nanoimpellers and pH-responsive nanovalves. These MSNPs operate only when both molecular machines operate in tandem to one another. Two different MSNPs were developed to operate at either acidic (left-hand side) or basic (right-hand side) pH. Left-hand side: The acid-responsive nanovalves are created by first of all attaching trisammonium stalks to the surfaces of the SNPs, then the cargo is loaded into the azobenzene-functionalised nanopores of the SNPs and finally capped with CB[6] rings. When the dynamic wagging of the impellers is induced by a 448 nm excitation beam, and followed by a decrease in pH, which causes the anilinium nitrogens to be protonated and the CB[6] rings to shuttle from the bisdialkylammonium centers to the distal ammonium units, the nanovalves are opened and the cargo is propelled out from the confines of the nanopores. Right-hand side: The base-responsive MSNPs use CB[6]/bisalkylammonium [2]pseudorotaxanes as the nanovalves and operate only when both the nanoimpellers are activated and the pH is raised to 10, which deprotonates both ammonium centers and the ion–dipole interactions between the CB[6] rings and the stalks are disrupted, thus opening the nanovalves and releasing the cargo molecules.](/image/article/2009/NR/b9nr00162j/b9nr00162j-f24.gif) |
| | Fig. 24 AND Logic, dual-controlled MSNPs composed of both nanoimpellers and pH-responsive nanovalves. These MSNPs operate only when both molecular machines operate in tandem to one another. Two different MSNPs were developed to operate at either acidic (left-hand side) or basic (right-hand side) pH. Left-hand side: The acid-responsive nanovalves are created by first of all attaching trisammonium stalks to the surfaces of the SNPs, then the cargo is loaded into the azobenzene-functionalised nanopores of the SNPs and finally capped with CB[6] rings. When the dynamic wagging of the impellers is induced by a 448 nm excitation beam, and followed by a decrease in pH, which causes the anilinium nitrogens to be protonated and the CB[6] rings to shuttle from the bisdialkylammonium centers to the distal ammonium units, the nanovalves are opened and the cargo is propelled out from the confines of the nanopores. Right-hand side: The base-responsive MSNPs use CB[6]/bisalkylammonium [2]pseudorotaxanes as the nanovalves and operate only when both the nanoimpellers are activated and the pH is raised to 10, which deprotonates both ammonium centers and the ion–dipole interactions between the CB[6] rings and the stalks are disrupted, thus opening the nanovalves and releasing the cargo molecules. | |
5.4. pH- and redox-active ferrocene-based mechanised silica nanoparticles
In an effort to increase biocompatibility, MSNPs were investigated that respond to a milder pH stimulus. It has been shown64,65 that CB[7] forms (Fig. 25) very stable inclusion complexes (association constants between 109 and 1011 M−1) with ferrocene (Fc) derivatives. Anionic ferrocenecarboxylates, however, are not bound at all by CB[7] as a result of the strong electrostatic repulsion between the carboxylate groups and the carbonyl groups of CB[7]. The dependence of pH on CB[7] binding towards ferrocenedicarboxylic acid was investigated by 1H NMR spectroscopy and it was observed that at pH 2, CB[7] binds the neutral ferrocenedicarboxylic acid as illustrated by an upfield shift of the resonances for the ferrocene protons. Increasing the pH to 10 yields deprotonated ferrocene dianion, a species which does not bind to the electron-rich interior of CB[7]. Furthermore, fluorescence confirmed the release of the RhB cargo upon adjusting from an aqueous solution of pH 3 to a solution of pH 10.
In addition to pH-controlled release, ferrocenedicarboxylic acids have been used to functionalise SNPs utilising yet another release mechanism – e.g., redox change. In general, Fc residues have been shown to complex β-CD, whereas the oxidised, positively charged ferrocenium ions do not form stable complexes.66 This recognition motif has been utilised by us64 to control (Fig. 26) the release of cargo from the nanopores of MSNPs. Thus, in a neutral aqueous solution, β-CD covers the nanopores of these MSNPs. Applying a voltage (+1 V) to the solution oxidises the Fc stalks to their respective ferrocenium ions, resulting in dethreading of the β-CDmacrocycles and release of the cargo.
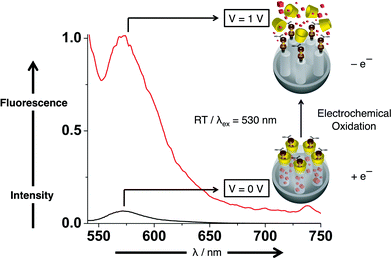 |
| | Fig. 26 Normalized fluorescence spectra demonstrating the controlled release of Rhodamine B under redox conditions, in an aqueous solution, using 530 nm excitation wavelength. By applying a voltage (1 V) – red emission spectrum – ferrocene molecules are oxidised to ferrocenium ions, causing the β-CD rings to dethread from the stalk and releasing the cargo. For comparison, a small trace of fluorescence of the cargo molecules dispersed in solution is observed at 0 V, black spectrum. | |
![A graphical representation of a viologen-based MSNP system in which the stalks have a carboxylic acid terminal. The viologen threads are tethered to the nanopores through an amide bond, followed by loading of Rhodamine B cargo molecules and subsequent capping with CB[6] rings. Initially, at neutral aqueous solution, the carboxylic acids are deprotonated and the CB[6] rings form a strong complex with the viologen units, thus closing the nanopores and retaining the cargo molecules inside the pores. Upon lowering the pH of the solution to pH 4, the carboxylates are protonated and the CB[6] rings move from the viologen units to the carboxylic acid, thus opening the nanopores and releasing the cargo.](/image/article/2009/NR/b9nr00162j/b9nr00162j-f27.gif) |
| | Fig. 27 A graphical representation of a viologen -based MSNP system in which the stalks have a carboxylic acid terminal. The viologen threads are tethered to the nanopores through an amide bond, followed by loading of Rhodamine B cargo molecules and subsequent capping with CB[6] rings. Initially, at neutral aqueous solution, the carboxylic acids are deprotonated and the CB[6] rings form a strong complex with the viologen units, thus closing the nanopores and retaining the cargo molecules inside the pores. Upon lowering the pH of the solution to pH 4, the carboxylates are protonated and the CB[6] rings move from the viologen units to the carboxylic acid, thus opening the nanopores and releasing the cargo. | |
6. Conclusion
In this review we have discussed some of the steps taken towards realising the vision of developing MSNPs for therapeutic-delivery systems. The lack of cytotoxicity of mesoporous SNPs, along with their ease of synthesis and functionalisation with organic molecules – such as nanovalves and snap-tops – have made SNPs an attractive platform on which to construct smart devices for drug delivery. The community has witnessed tremendous progress since the first proof-of-principle studies – where silica surfaces were functionalised with supermolecules – to the encapsulation of different molecules inside SNPs and nowadays to the development of MSNPs that only release the entrapped cargo in response to an external stimulus. Much effort has been devoted to create MSNPs that not only operate in organic solvents, but also in aqueous solutions, as a prelude to achieving the biocompatibility necessary for drug delivery. We realise that more biological studies, especially with MSNPs, need to be completed before these MSNP systems are fit for clinical use. We believe, however, that given the pace at which research is being conducted in the field as a whole, it will not be all that long before therapeutic treatments are delivered inside MSNPs functionalised selectively with targeting agents in order to minimise undesirable side-effects.
References
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
-
C. J. Brinker and G. W. Scherer, Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing, Academic Press, New York, 1990 Search PubMed.
-
(a) Q. Cai, Z.-S. Luo, W.-Q. Pang, Y.-W. Fan, X.-H. Chen and F.-Z. Cui, Chem. Mater., 2001, 13, 258–263 CrossRef CAS;
(b) J. Kobler, K. Moller and T. Bein, ACS Nano, 2008, 2, 791–799 CrossRef CAS;
(c) Y. Lu, H. Fan, A. Stump, T. L. Ward, T. Rieker and C. J. Brinker, Nature, 1999, 398, 223–226 CrossRef CAS;
(d) Y. Lu, R. Ganguli, C. A. Drewien, M. T. Anderson, C. J. Brinker, W. Gong, Y. Guo, H. Soyez, B. Dunn, M. H. Huang and J. I. Zink, Nature, 1997, 389, 364–368 CrossRef CAS;
(e) S. P. Naik, W. Fan, T. Yokoi and T. Okubo, Langmuir, 2006, 22, 6391–6397 CrossRef CAS.
-
(a) A. Sayari, B.-H. Han and Y. Yang, J. Am. Chem. Soc., 2004, 126, 14348–14349 CrossRef CAS;
(b) P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152–155 CrossRef CAS;
(c) J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56–77 CrossRef CAS.
- S. Angelos, M. Liong, E. Choi and J. I. Zink, Chem. Eng. J., 2008, 137, 4–13 CrossRef CAS.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
-
(a) S. Y. Chia, J. G. Cao, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2001, 40, 2447–2451 CrossRef CAS;
(b) E. Johansson, E. Choi, S. Angelos, M. Liong and J. I. Zink, J. Sol-Gel Sci. Technol., 2008, 46, 313–322 CrossRef CAS;
(c) P. N. Minoofar, B. S. Dunn and J. I. Zink, J. Am. Chem. Soc., 2005, 127, 2656–2665 CrossRef CAS.
-
(a) Y.-S. Lin, C.-P. Tsai, H.-Y. Huang, C.-T. Kuo, Y. Hung, D.-M. Huang, Y.-C. Chen and C.-Y. Mou, Chem. Mater., 2005, 17, 4570–4573 CrossRef CAS;
(b) Y.-S. Lin, S.-H. Wu, Y. Hung, Y.-H. Chou, C. Chang, M.-L. Lin, C.-P. Tsai and C.-Y. Mou, Chem. Mater., 2006, 18, 5170–5172 CrossRef CAS;
(c) J. Lu, M. Liong, J. I. Zink and F. Tamanoi, Small, 2007, 3, 1341–1346 CrossRef CAS;
(d) D. R. Radu, C.-Y. Lai, K. Jeftinija, E. W. Rowe, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2004, 126, 13216–13217 CrossRef CAS;
(e) I. I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem. Soc., 2007, 129, 8845–8849 CrossRef CAS;
(f) K. M. L. Taylor, J. S. Kim, W. J. Rieter, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2008, 130, 2154–2155 CrossRef CAS;
(g) C.-P. Tsai, Y. Hung, Y.-H. Chou, D.-M. Huang, J.-K. Hsiao, C. Chang, Y.-C. Chen and C.-Y. Mou, Small, 2008, 4, 186–191 CrossRef CAS;
(h) S.-H. Wu, Y.-S. Lin, Y. Hung, Y.-H. Chou, Y.-H. Hsu, C. Chang and C.-Y. Mou, ChemBioChem, 2008, 9, 53–57 CrossRef CAS.
-
(a) A. Stein, B. J. Melde and R. C. Schroden, Adv. Mater., 2000, 12, 1403–1419 CrossRef CAS;
(b) M. Vallet-Regí, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef CAS;
(c) D. S. Shephard, W. Zhou, T. Maschmeyer, J. M. Matters, C. L. Roper, S. Parsons, B. F. G. Johnson and M. J. Duer, Angew. Chem., Int. Ed., 1998, 37, 2719–2723 CrossRef CAS.
- S. L. Burkett, S. D. Sims and S. Mann, Chem. Commun., 1996, 1367–1368 RSC.
- D. J. Macquarrie, Chem. Commun., 1996, 1961–1962 RSC.
-
(a) T. D. Nguyen, K. C.-F. Leung, M. Liong, Y. Liu, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 2101–2110 CrossRef CAS;
(b) T. D. Nguyen, Y. Liu, S. Saha, K. C.-F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626–634 CrossRef CAS;
(c) T. D. Nguyen, H.-R. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. F. Stoddart and J. I. Zink, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029–10034 CrossRef CAS.
- I. Gorelikov and N. Matsuura, Nano Lett., 2008, 8, 369–373 CrossRef CAS.
- J. Kim, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C.-H. Shin, J.-G. Park, J. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 688–689 CrossRef CAS.
- H. Fan, K. Yang, D. M. Boye, T. Sigmon, K. J. Malloy, H. Xu, G. P. Lopez and C. J. Brinker, Science, 2004, 304, 567–571 CrossRef CAS.
-
(a) J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS;
(b) M. Liong, B. France, K. A. Bradley and J. I. Zink, Adv. Mater., 2009, 21, 1684–1689 CrossRef CAS;
(c) M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS.
-
(a) M. Vallet-Regí, A. Rámila, R. P. del Real and J. Pérez-Pariente, Chem. Mater., 2001, 13, 308–311 CrossRef CAS;
(b) B. Muñoz, A. Rámila, J. Pérez-Pariente, I. Díaz and M. Vallet-Regí, Chem. Mater., 2003, 15, 500–503 CrossRef CAS;
(c) J. Andersson, J. Rosenholm, S. Areva and M. Lindén, Chem. Mater., 2004, 16, 4160–4167 CrossRef;
(d) P. Horcajada, A. Rámila, J. Pérez-Pariente and M. Vallet-Regí, Microporous Mesoporous Mater., 2004, 68, 105–109 CrossRef CAS;
(e) C. Charnay, S. Bégu, C. Tourné-Péteilh, L. Nicole, D. A. Lerner and J.-M. Devoisselle, Eur. J. Pharm. Biopharm., 2004, 57, 533–540 CrossRef CAS;
(f) C. Tourné-Péteilh, D. Brunel, S. Bégu, B. Chiche, F. Fajula, D. A. Lerner and J.-M. Devoisselle, New J. Chem., 2003, 27, 1415–1418 RSC;
(g) A. Rámila, B. Muñoz, J. Pérez-Pariente and M. Vallet-Regí, J. Sol-Gel Sci. Technol., 2003, 26, 1199–1202 CrossRef CAS;
(h) C. Tourné-Péteilh, D. A. Lerner, C. Charnay, L. Nicole, S. Bégu and J.-M. Devoisselle, ChemPhysChem, 2003, 4, 281–286 CrossRef CAS;
(i) J. C. Doadrio, E. M. B. Sousa, I. Izquierdo-Barba, A. L. Doadrio, J. Pérez-Pariente and M. Vallet-Regí, J. Mater. Chem., 2006, 16, 462–467 RSC;
(j) J. Lu, M. Liong, S. Sherman, T. Xia, M. Kovochich, A. E. Nel, J. I. Zink and F. Tamanoi, NanoBiotechnology, 2007, 3, 89–95 CrossRef CAS;
(k) Y. Klichko, M. Liong, E. Choi, S. Angelos, A. E. Nel, J. F. Stoddart, F. Tamonoi and J. I. Zink, J. Am. Ceram. Soc., 2009, 92, S2–S10 CrossRef CAS.
-
(a) W. Zeng, X.-F. Qian, Y.-B. Zhang, J. Yin and Z.-K. Zhu, Mater. Res. Bull., 2005, 40, 766–772 CrossRef CAS;
(b) F. Balas, M. Manzano, P. Horcajada and M. Vallet-Regí, J. Am. Chem. Soc., 2006, 128, 8116–8117 CrossRef CAS;
(c) F. Qu, G. Zhu, S. Huang, S. Li, J. Sun, D. Zhang and S. Qiu, Microporous Mesoporous Mater., 2006, 92, 1–9 CrossRef CAS.
- J. L. Vivero-Escoto, I. I. Slowing, C.-W. Wu and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–2463 CrossRef CAS.
- J. Liu, X. Jiang, C. Ashley and C. J. Brinker, J. Am. Chem. Soc., 2009, 131, 7567–7569 CrossRef CAS.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS.
- N. K. Mal, M. Fujiwara and Y. Tanaka, Nature, 2003, 421, 350–353 CrossRef CAS.
- Y.-J. Han, G. D. Stucky and A. Butler, J. Am. Chem. Soc., 1999, 121, 9897–9898 CrossRef CAS.
- F. Torney, B. G. Trewyn, V. S. Lin and V. S.-Y. Wang, Nat. Nanotechnol., 2007, 2, 295–300 Search PubMed.
- I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem.
Soc., 2006, 128, 14792–14793 CrossRef CAS.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lidén, ACS Nano, 2009, 3, 197–206 CrossRef CAS.
-
(a) H. Elnakat and M. Ratnam, Adv. Drug Delivery Rev., 2004, 56, 1067–1084 CrossRef CAS;
(b) J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147–162 CrossRef CAS.
- J. Lu, E. Choi, F. Tamanoi and J. I. Zink, Small, 2008, 4, 421–426 CrossRef CAS.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S.-Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038–5044 CrossRef CAS.
- M. Fujiwara, M. Akiyama, M. Hata, K. Shiokawa and R. Nomura, ACS Nano, 2008, 2, 1671–1681 CrossRef CAS.
- P. Sierocki, H. Maas, P. Dragut, G. Richardt, F. Vögtle, L. De Cola, F. A. M. Brouwer and J. I. Zink, J. Phys. Chem. B, 2006, 110, 24390–24398 CrossRef CAS.
- S. Angelos, E. Choi, F. Vögtle, L. De Cola and J. I. Zink, J. Phys. Chem. C, 2007, 111, 6589–6592 CrossRef CAS.
- Y. Zhu and M. Fujiwara, Angew. Chem., Int. Ed., 2007, 46, 2241–2244 CrossRef CAS.
-
(a)
J.-P. Sauvage and C. Dietrich-Buchecker, Molecular Catenanes, Rotaxanes and Knots, Wiley-VCH, Weinheim, 1999 Search PubMed;
(b)
G. Schill, Catenanes, Rotaxanes and Knots, Academic Press, New York, 1971 Search PubMed.
-
(a) M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, C. Hamers, G. Mattersteig, M. Montalti, A. N. Shipway, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 333–337 CrossRef CAS;
(b) V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Org. Chem., 2000, 65, 1924–1936 CrossRef CAS;
(c) C. P. Collier, G. Mattersteig, E. W. Wong, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172–1175 CrossRef CAS;
(d)
J.-P. Collin, J.-M. Kern, L. Raehm and J.-P. Sauvage, Molecular Switches, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed;
(e) Y. Luo, C. P. Collier, J. O. Jeppesen, K. A. Nielsen, E. DeIonno, G. Ho, J. Perkins, H. R. Tseng, T. Yamamoto, J. F. Stoddart and J. R. Heath, ChemPhysChem, 2002, 3, 519–525 CrossRef CAS.
-
(a) A. Altieri, G. Bottari, F. Dehez, D. A. Leigh, J. K. Y. Wong and F. Zerbetto, Angew. Chem., Int. Ed., 2003, 42, 2296–2300 CrossRef CAS;
(b) G. Bottari, D. A. Leigh and E. M. Perez, J. Am. Chem. Soc., 2003, 125, 13360–13361 CrossRef CAS;
(c) A. M. Brouwer, S. M. Fazio, C. Frochot, F. G. Gatti, D. A. Leigh, J. K. Y. Wong and G. W. H. Wurpel, Pure Appl. Chem., 2003, 75, 1055–1060 CrossRef;
(d) F. G. Gatti, S. Lent, J. K. Y. Wong, G. Bottari, A. Altieri, M. A. F. Morales, S. J. Teat, C. Frochot, D. A. Leigh, A. M. Brouwer and F. Zerbetto, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10–14 CrossRef CAS;
(e) E. M. Pérez, D. T. F. Dryden, D. A. Leigh, G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2004, 126, 12210–12211 CrossRef CAS.
-
(a) J. P. Collin, A. C. Laemmel and J. P. Sauvage, New J. Chem., 2001, 25, 22–24 RSC;
(b) L. Raehm, J. M. Kern and J. P. Sauvage, Chem.–Eur. J., 1999, 5, 3310–3317 CrossRef CAS.
- H. Murakami, A. Kawabuchi, R. Matsumoto, T. Ido and N. Nakashima, J. Am. Chem. Soc., 2005, 127, 15891–15899 CrossRef CAS.
- Y. Liu, A. H. Flood, P. A. Bonvallett, S. A. Vignon, B. H. Northrop, H. R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard, C. M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS.
-
(a) A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 433–444 CrossRef CAS;
(b) C. P. Collier, J. O. Jeppesen, Y. Luo, J. Perkins, E. W. Wong, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2001, 123, 12632–12641 CrossRef CAS;
(c) A. Star, Y. Liu, K. Grant, L. Ridvan, J. F. Stoddart, D. W. Steuerman, M. R. Diehl, A. Boukai and J. R. Heath, Macromolecules, 2003, 36, 553–560 CrossRef CAS;
(d) D. W. Steuerman, H. R. Tseng, A. J. Peters, A. H. Flood, J. O. Jeppesen, K. A. Nielsen, J. F. Stoddart and J. R. Heath, Angew. Chem., Int. Ed., 2004, 43, 6486–6491 CrossRef CAS;
(e) J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H. R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS;
(f) E. DeIonno, H. R. Tseng, D. D. Harvey, J. F. Stoddart and J. R. Heath, J. Phys. Chem. B, 2006, 110, 7609–7612 CrossRef CAS;
(g) S. S. Jang, Y. H. Jang, Y. H. Kim, W. A. Goddard, A. H. Flood, B. W. Laursen, H. R. Tseng, J. F. Stoddart, J. O. Jeppesen, J. W. Choi, D. W. Steuerman, E. DeIonno and J. R. Heath, J. Am. Chem. Soc., 2005, 127, 1563–1575 CrossRef CAS;
(h) A. H. Flood, A. J. Peters, S. A. Vignon, D. W. Steuerman, H. R. Tseng, S. Kang, J. R. Heath and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 6558–6564 CrossRef CAS;
(i) M. R. Diehl, D. W. Steuerman, H. R. Tseng, S. A. Vignon, A. Star, P. C. Celestre, J. F. Stoddart and J. R. Heath, ChemPhysChem, 2003, 4, 1335–1339 CrossRef CAS;
(j) H. B. Yu, Y. Luo, K. Beverly, J. F. Stoddart, H. R. Tseng and J. R. Heath, Angew. Chem., Int. Ed., 2003, 42, 5706–5711 CrossRef CAS;
(k) J. W. Choi, A. H. Flood, D. W. Steuerman, S. Nygaard, A. B. Braunschweig, N. N. P. Moonen, B. W. Laursen, Y. Luo, E. DeIonno, A. J. Peters, J. O. Jeppesen, K. Xu, J. F. Stoddart and J. R. Heath, Chem.–Eur. J., 2006, 12, 261–279 CrossRef CAS.
-
(a) S. Saha, K. C.-F. Leung, T. D. Nguyen, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 685–693 CrossRef CAS;
(b) S. Angelos, E. Johansson, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 2261–2271 CrossRef CAS.
- R. Hernandez, H.-R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370–3371 CrossRef CAS.
- T. D. Nguyen, K. C.-F. Leung, M. Liong, C. D. Pentecost, J. F. Stoddart and J. I. Zink, Org. Lett., 2006, 8, 3363–3366 CrossRef CAS.
- K. C.-F. Leung, T. D. Nguyen, J. F. Stoddart and J. I. Zink, Chem. Mater., 2006, 18, 5919–5928 CrossRef CAS.
- E. Aznar, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto, P. Amorós and C. Guillem, J. Am. Chem. Soc., 2009, 131, 6833–6843 CrossRef CAS.
- Y. Zhao, B. G. Trewyn, I. I. Slowing and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 8398–8400 CrossRef CAS.
- M. A. Charles, R. Fanska, F. G. Schmid, P. H. Forsham and G. M. Grodsky, Science, 1973, 179, 569–571 CrossRef CAS.
- O. Dyachok, O. Idevall-Hagren, J. Sågetorp, G. Tian, A. Wuttke, C. Arrieumerlou, G. Akusjärvi, E. Gylfe and A. Tengholm, Cell Metab., 2008, 8, 26–37 CrossRef CAS.
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef CAS.
- K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y.-W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382–2383 CrossRef CAS.
- A. Bernardos, E. Aznar, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto, J. M. Barat and P. Amorós, Angew. Chem., Int. Ed., 2009, 48, 5884–5887 CrossRef CAS.
- C. Park, K. Lee and C. Kim, Angew. Chem., Int. Ed., 2009, 48, 1275–1278 CrossRef CAS.
- D. P. Ferris, Y.-L. Zhao, N. M. Khashab, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 1686–1688 CrossRef CAS.
- R. Liu, X. Zhao, T. Wu and P. Y. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef CAS.
-
(a) R. Casasús, M. D. Marcos, R. Martínez-Máñez, J. V. Ros-Lis, J. Soto, L. A. Villaescusa, P. Amorós, D. Beltrán, C. Guillem and J. Latorre, J. Am. Chem. Soc., 2004, 126, 8612–8613 CrossRef CAS;
(b) R. Casasús, E. Aznar, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto and P. Amorós, Angew. Chem., Int. Ed., 2006, 45, 6661–6664 CrossRef CAS;
(c) R. Casasús, E. Climent, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto, P. Amorós, J. Cano and E. Ruiz, J. Am. Chem. Soc., 2008, 130, 1903–1917 CrossRef CAS.
-
(a)
Comprehensive Supramolecular Chemistry, Volume 3, Pergamon, New York, 1996 Search PubMed;
(b) C. M. Cardona, T. D. McCarley and A. E. Kaifer, J. Org. Chem., 2000, 65, 1857–1864 CrossRef CAS;
(c) L. A. Godínez, S. Patel, C. M. Criss and A. E. Kaifer, J. Phys. Chem., 1995, 99, 17449–17455 CrossRef CAS;
(d) L. A. Godínez, L. Schwartz, C. M. Criss and A. E. Kaifer, J. Phys. Chem. B, 1997, 101, 3376–3380 CrossRef CAS;
(e) B. González, I. Cuadrado, B. Alonso, C. M. Casado, M. Morán and A. E. Kaifer, Organometallics, 2002, 21, 3544–3551 CrossRef CAS;
(f) A. Harada, Acc. Chem. Res., 2001, 34, 456–464 CrossRef CAS;
(g) R. Isnin, C. Salam and A. E. Kaifer, J. Org. Chem., 1991, 56, 35–41 CrossRef CAS;
(h) J. Liu, J. Alvarez, W. Ong, E. Román and A. E. Kaifer, Langmuir, 2001, 17, 6762–6764 CrossRef CAS;
(i) J. Liu, S. Mendoza, E. Román, M. J. Lynn, R. L. Xu and A. E. Kaifer, J. Am. Chem. Soc., 1999, 121, 4304–4305 CrossRef CAS;
(j) J. Liu, W. Ong, E. Román, M. J. Lynn and A. E. Kaifer, Langmuir, 2000, 16, 3000–3002 CrossRef CAS;
(k) T. Matsue, D. H. Evans, T. Osa and N. Kobayashi, J. Am. Chem. Soc., 1985, 107, 3411–3417 CrossRef CAS;
(l) A. Nelson, J. M. Belitsky, S. Vidal, C. S. Joiner, L. G. Baum and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 11914–11922 CrossRef CAS;
(m) S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1959–1976 CrossRef CAS;
(n) M. T. Rojas, R. Koniger, J. F. Stoddart and A. E. Kaifer, J. Am. Chem. Soc., 1995, 117, 336–343 CrossRef CAS;
(o) I. Tomatsu, A. Hashidzume and A. Harada, J. Am. Chem. Soc., 2006, 128, 2226–2227 CrossRef CAS;
(p) Y. Wang and A. E. Kaifer, J. Phys. Chem. B, 1998, 102, 9922–9927 CrossRef CAS;
(q) A. Diaz, P. A. Quintela, J. M. Schuette and A. E. Kaifer, J. Phys. Chem., 1988, 92, 3537–3542 CrossRef CAS;
(r) L. A. Godínez, J. Lin, M. Muñoz, A. W. Coleman and A. E. Kaifer, Electroanalysis, 1996, 8, 1072–1074 CAS;
(s) R. Isnin and A. E. Kaifer, J. Am. Chem. Soc., 1991, 113, 8188–8190 CrossRef;
(t) A. E. Kaifer, Acc. Chem. Res., 1999, 32, 62–71 CrossRef CAS;
(u) J. Liu, R. Castro, K. A. Abboud and A. E. Kaifer, J. Org. Chem., 2000, 65, 6973–6977 CrossRef CAS;
(v) A. Mirzoian and A. E. Kaifer, Chem.–Eur. J., 1997, 3, 1052–1058 CrossRef CAS;
(w) Y. Wang, J. Alvarez and A. E. Kaifer, Chem. Commun., 1998, 1457–1458 RSC.
-
(a) A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094–8100 CrossRef CAS;
(b) W. S. Jeon, A. Y. Ziganshina, J. W. Lee, Y. H. Ko, J.-K. Kang, C. Lee and K. Kim, Angew. Chem., Int. Ed., 2003, 42, 4097–4100 CrossRef CAS;
(c) J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS;
(d) J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS;
(e) K. Moon and A. E. Kaifer, Org. Lett., 2004, 6, 185–188 CrossRef CAS;
(f) W. Ong, M. Gomez-Kaifer and A. E. Kaifer, Org. Lett., 2002, 4, 1791–1794 CrossRef CAS;
(g) T. Ooya, D. Inoue, H. S. Choi, Y. Kobayashi, S. Loethen, D. H. Thompson, Y. H. Ko, K. Kim and N. Yui, Org. Lett., 2006, 8, 3159–3162 CrossRef CAS;
(h) A. M. M. Rawashdeh, A. Thangavel, C. Sotiriou-Leventis and N. Leventis, Org. Lett., 2008, 10, 1131–1134 CrossRef CAS.
- C. Park, K. Oh, S. C. Lee and C. Kim, Angew. Chem., Int. Ed., 2007, 46, 1455–1457 CrossRef CAS.
- S. Angelos, Y.-W. Yang, K. Patel, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222–2226 CrossRef CAS.
- S. Angelos, N. M. Khashab, Y.-W. Yang, A. Trabolsi, H. A. Khatib, J. F. Stoddart and J. I Zink, J. Am. Chem. Soc., 2009, 346 DOI:10.1021/ja9010157.
- W. L. Mock and J. Pierpont, J. Chem. Soc., Chem. Commun., 1990, 1509–1511 RSC.
-
R. C. Weast, CRC Handbook of Chemistry and Physics, CRC Press, Ohio, 54th edn, 1973 Search PubMed.
- S. Angelos, Y.-W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344–11346 CrossRef CAS.
- N. M. Khashab, A. Trabolsi, Y. A. Lau, M. W. Ambrogio, D. C. Friedman, H. A. Khatib, J. I. Zink and J. F. Stoddart, Eur. J. Org. Chem., 2009, 1669–1673 CrossRef CAS.
- S. Top, A. Vessières, G. Leclercq, J. Quivy, J. Tang, J. Vaissermann, M. Huché and G. Jaouen, Chem.–Eur. J., 2003, 9, 5223–5236 CrossRef CAS.
- A. Harada and S. Takahashi, J. Chem. Soc., Chem. Comm., 1984, 645–646 RSC.
-
(a) V. Sindelar, S. Silvi and A. E. Kaifer, Chem. Commun., 2006, 2185–2187 RSC;
(b) V. Sindelar, S. Silvi, S. E. Parker, D. Sobransingh and A. E. Kaifer, Adv. Funct. Mater., 2007, 17, 694–701 CrossRef CAS.
- N. M. Khashab, M. E. Belowich, A. Trabolsi, D. C. Friedman, C. Valente, Y. Lau, H. A. Khatib, J. I. Zink and J. F. Stoddart, Chem. Commun., 2009, 5371–5373 RSC.
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here. 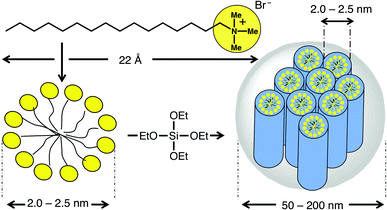

![Redox- and pH-controlled of ferrocene⊂β-CD (top route) and ferrocene⊂CB[7] (bottom route) MSNPs. The β-CD and CB[7] rings form strong inclusion complexes with ferrocene derivatives. Upon reduction of ferrocene to ferrocenium ions, the β-CD rings dethread opening the nanopores and releasing the cargo. The cargo from the ferrocene⊂CB[7] MSNPs can be released by a change in pH. By acidifying the solution in which the MSNPs are dispersed, the ferrocenecarboxylic acid is deprotonated to ferrocenecarboxylate and the CB[7] dethreads from the stalk on account of the strong electrostatic repulsion between the carboxylate groups and the carbonyl groups of CB[7].](/image/article/2009/NR/b9nr00162j/b9nr00162j-f25.gif)