One-pot formation of SnO2 hollow nanospheres and α-Fe2O3@SnO2 nanorattles with large void space and their lithium storage properties†
Jun Song
Chen
a,
Chang Ming
Li
a,
Wen Wen
Zhou
b,
Qing Yu
Yan
b,
Lynden A.
Archer
c and
Xiong Wen
Lou
*ac
aSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive, Singapore, 637457, Singapore. E-mail: xwlou@ntu.edu.sg
bSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore, 639798, Singapore
cKAUST-Cornell Center for Energy and Sustainability, Cornell University, Ithaca, NY 14853, USA
First published on 15th September 2009
Abstract
In this work, uniform SnO2 hollow nanospheres with large void space have been synthesized by a modified facile method. The void space can be easily controlled by varying the reaction time. The formation of interior void space is based on an inside-out Ostwald ripening mechanism. More importantly, this facile one-pot process can be extended to fabricate rattle-type hollow structures using α-Fe2O3@SnO2 as an example. Furthermore, the electrochemical lithium storage properties have been investigated. It is found that α-Fe2O3@SnO2 nanorattles manifest a much lower initial irreversible loss and higher reversible capacity compared to SnO2 hollow spheres. This interesting finding supports a general hypothesis that a synergistic effect between functional core and shell materials can lead to improved lithium storage capabilities.
Introduction
Hollow nanostructures have recently attracted enormous research interest because of their potential in many fields of applications, including drug delivery, photonics, nanoreactors, catalysis, gas sensing and energy storage.1–13 Up to now many methods have been developed for the synthesis of hollow nanostructures, and the majority of them are based on hard templating approaches. As summarized in a recent comprehensive review article,1 these templating approaches are usually associated with several synthetic drawbacks. Specifically, templates which are made of different materials need to be pre-synthesized. This not only incurs extra cost but also complicates the synthetic process. Also, it is usually difficult to remove the template while keeping the hollow structure intact. For example, when carbon (derived from glucose) or latex spheres are used as the templates, calcination in air is often employed for template removal.7,8 During this process, incidents of shell collapse will inevitably take place as a result of outward evacuation of CO2. These problems together with technical difficulties of uniform shell deposition render large-scale preparation of high-purity hollow structures by templating approaches infeasible. Thus a simple one-pot method of synthesizing hollow spheres without using any template would be appreciated in order to avoid the above-mentioned drawbacks.1 To this end, we recently observed an interesting inside-out hollowing mechanism for the fabrication of SnO2 hollow nanostructures.2 However, the control over the interior void space is rather limited. As a further development of hollow structures, it would be desirable to introduce other functional cores into the interior void space. Indeed, fabrication of rattle-type structures still represents significant conceptual challenges. As a result, there are only a limited number of examples on fabrication of nanorattles, and most of them are also based on multi-step templating approaches.1,14,15 Herein, we report an improved method of synthesizing SnO2 hollow nanospheres with large void spaces. We also demonstrate that this facile one-pot process can be extended to fabricate rattle-type hollow structures using α-Fe2O3@SnO2 as an example. To show the advantages of such binary nanorattles, we have investigated the electrochemical properties which support our hypothesis that a synergistic effect between α-Fe2O3 and SnO2 can lead to improved lithium storage capabilities.Results and discussion
Based on the inside-out Ostwald ripening process,2,16 the formation of SnO2 hollow nanospheres and α-Fe2O3@SnO2 nano-rattles is illustrated in Fig. 1. Initially, amorphous fine SnO2nanoparticles are formed rapidly through the hydrolysis of K2SnO3·3H2O, and they self-assemble to form loosely packed nanospheres. During the hydrothermal ripening process, the amorphous particles at the central part possess higher surface energy and thus are easier to dissolve and relocate to the outer regions, where crystallization takes place.2 This crystallization process is facilitated by the surrounding solvents. As reaction proceeds, more small crystallites in the interior will dissolve and re-crystallize on the outer shell, leading to an enlarged hollow interior space accompanied by construction of a compact shell. The formation of α-Fe2O3@SnO2 nano-rattles is similar to the process described above. Specifically, the SnO2nanoparticles formed by hydrolysis will first aggregate around the α-Fe2O3nanocrystals to form a solid core/shell structure. The hollowing process will initiate around the core/shell interface. With longer reaction time, the Ostwald ripening process will continue to expand the interstitial space leading to a rattle-type structure of α-Fe2O3@SnO2. | ||
| Fig. 1 Schematic illustration of the formation of SnO2 hollow nanospheres and α-Fe2O3@SnO2 nano-rattles. Step I: aggregation of SnO2nanoparticles to form loosely pack nanospheres in route A, and α-Fe2O3@SnO2 solid core/shell structure in route B. Step II: in route A, the hollowing effect starts at the central region of the SnO2 nanospheres; in route B, the ripening process starts around the α-Fe2O3/SnO2 interface. Step III: enlargement of hollow interior space by further evacuation of SnO2nanoparticles in both routes A and B. | ||
Fig. 2 shows the as-formed SnO2 hollow spheres with different reaction durations. From the field-emission scanning electron microscopy (FESEM) images (A, 4 h; D, 8 h; G, 24 h), the nanospheres are well dispersed with good uniformity (low-magnification FESEM images are provided in the ESI,† Fig. S1). The diameter is generally in the range of 300–500 nm. The transmission electron microscopy (TEM) images (B and C, 4 h; E and F, 8 h; H and I, 24 h) clearly reveal the structural evolution of the interior with reaction time. With a reaction time of 4 h only, the formation of spherical structures is already complete with a relatively uniform size distribution, but the spheres have only very small voids which are mostly located eccentrically (see Fig. 2B, C). The hollow interior space becomes more apparent when the reaction is prolonged to 8 h (see Fig. 2E, F). However, a uniform shell is still not yet formed. Only after 24 h of reaction can SnO2 hollow spheres with large void space be obtained with a uniform shell of about 100 nm in thickness. These observations suggest that the internal void space can be easily controlled to a large extent by varying the reaction time. Large void spaces can hardly be achieved previously at a reaction temperature of 150 °C (see ESI,† Fig. S2).2 Even though the internal structure evolves with time, there is no significant variation in the size of the spheres during the process. It is also worth mentioning that further prolonging the reaction to 36 or 48 h will cause collapse of the shell, which is probably caused by excessive evacuation of SnO2 crystallites from the inner surface of the shell to the outside, thus deteriorating the robustness of the wall. This implies that the optimal reaction time should be around 24 h at 200 °C.
 | ||
| Fig. 2 FESEM images (A, D, G) and TEM images (B, C, E, F, H, I) of SnO2 hollow nanospheres synthesized at 200 °C with different reaction times of 4 h (A, B, C), 8 h (D, E, F), and 24 h (G, H, I). | ||
Apart from reaction temperature and time, other synthetic conditions (e.g., the effect of surfactants) were also investigated. For example, other co-solvents like 2-propanol and ethylene glycol were used as alternatives to ethanol, but the results turned out to be unsatisfactory, as the products were either heavily interconnected with hollow spherical subunits, or very fine particles without defined morphology. Use of a higher concentration of the precursor K2SnO3·3H2O produced large aggregates composed of non-uniform hollow spheres.
The chemical composition of the as-prepared SnO2 nanospheres was confirmed by X-ray diffraction (XRD) analysis as shown in Fig. 3, and energy-dispersive X-ray (EDX) analysis (see ESI,† Fig. S3). All the identified peaks of the XRD patterns can be unambiguously assigned to tetragonal SnO2 (cassiterite, Joint Committee on Powder Diffraction Standards (JCPDS) card no. 41-1445, S.G.: P42/mnm, ao = 4.7382 Å, co = 3.1871 Å).2 It is noteworthy that the crystallite size calculated using the Scherrer formula from the (110) peak increases from 7.67 Å to 8.07 Å, then to 8.76 Å in I, II and III, respectively. This result is in good agreement with the formation mechanism proposed, as small crystallites will grow into larger ones during the ripening process.
 | ||
| Fig. 3 XRD patterns of SnO2 hollow nanospheres synthesized at 200 °C with different reaction times: I, 4 h; II, 8 h; III, 24 h. | ||
Fig. 4 shows the N2 adsorption and desorption isotherm of the SnO2 hollow spheres synthesized at 200 °C with 24 h of reaction (see Fig. 2, H and I). This characteristic type IV isotherm with a type H4 hysteresis loop, which is similar to that of SnO2 hollow spheres synthesized at 150 °C,2,17 indicates clearly a mesoporous structure of these SnO2 shells. The porous structure is created during the continuous evacuation of the interior materials through the shell. As a result, these SnO2 hollow spheres have a relatively high Brunauer-Emmett-Teller (BET) specific surface area of 101 m2/g and a total pore volume of 0.225 cm3/g. The inset shows the corresponding pore size distributions calculated by the Barrett-Joyner-Halenda (BJH) method from both adsorption and desorption branches. As pointed out previously,17 the striking peak around 4.0–4.5 nm in the pore size distribution from the desorption branch might be an artifact corresponding to capillary evaporation at the lower end of the hysteresis loop with a relative pressure of about 0.4–0.5.18 This is quite conceivable since during the ripening process, large number of open channels will be created inside the shell for matter relocation and transport. However, the packing of small crystallites is a random process, which should give rise to a variation in pore size.19 These mesopores provide access from the outer environment to the inner empty space of the spheres, which uncovers another approach for functionalization of the interior. For example, it has been demonstrated that because of the presence of these mesopores in the shell, the size of the inside core material can be tuned.20 In this regard, the as-prepared mesoporous SnO2 shells with large void spaces might provide a good platform for applications like nanoreactors.5,21
 | ||
| Fig. 4 N2 adsorption/desorption isotherm of the as-prepared SnO2 hollow nanospheres. The inset shows the pore size distributions calculated by the Barrett-Joyner-Halenda (BJH) method from both branches of the isotherm. | ||
Fig. 5 shows the α-Fe2O3@SnO2 nano-rattles with chemical composition confirmed by XRD analysis (see ESI,† Fig. S4). They are of spherical shape with size of about 450 nm in diameter. It can be observed that with a reaction time of 2 h (Fig. 5A, B), some small interior void space has been created, indicating that the ripening process has already initiated between the α-Fe2O3 core and the outer SnO2 shell. The core cannot be identified as a single α-Fe2O3nanocrystal inside the shell (Fig. 5B). Instead, it appears as a bulky dark region merged with the outer shell, indicating that the core is probably still encapsulated by SnO2nanoparticles. After 24 h of ripening, these SnO2nanoparticles evacuate to outer parts and create a much more spacious void region, and the shape of the α-Fe2O3 core can be clearly revealed (Fig. 5C, D). The above observations suggest that adding α-Fe2O3nanocrystals into the current synthetic system apparently does not affect the inside-out ripening process. Hence this facile method might be applied for fabrication of nano-rattles with SnO2 shell and cores of other functional materials. This could lead to a more flexible choice of material combinations, thus granting greater application promise of core-shell hollow structures. Also, it is important to note that the relative amounts of α-Fe2O3 core and SnO2 precursor have a significant effect on the structure of the formed particles. For example, if the initial amount of α-Fe2O3 is doubled while other conditions are kept identical, the product contains both hollow nano-rattles and solid core/shell structures consisting of one α-Fe2O3 core wrapped up by a very thin layer of SnO2 with little void space created.
 | ||
| Fig. 5 TEM images of α-Fe2O3@SnO2 nano-rattles with different reaction times of 2 h (A, B) and 24 h (C, D). | ||
SnO2 has been intensively studied as a promising anode material for next-generation lithium-ion batteries (LIB).2,13,17,22–28 In a SnO2/Li half cell, there are two principal electrochemical processes: SnO2 + 4Li+ + 4e− → Sn + 2Li2O (I); Sn + xLi+ + xe− ↔ LixSn (0 ≤ x ≤ 4.4) (II).27,28 Despite its attractive theoretical reversible capacity (ca. 790 mA h g−1) based on reversible reaction (II) compared to that of current graphitic material (ca. 372 mA h g−1), up to now its performance is still unsatisfactory because of two major issues: large initial irreversible loss and poor capacity retention over extended cycling. The former is often ascribed to the irreversible reduction of SnO2 (reaction I) in the field, although our recent evidence indicates that other irreversible processes such as formation of solid-electrolyte interface (SEI) and decomposition of electrolyte might be equally responsible.28 The latter is generally believed to be caused by a large volume change of the electrode accompanying the charge/discharge processes (reaction II), which leads to disintegration (pulverization) of the electrode and thereby loss of electrical contact.29 Recently, increasing evidence has suggested that use of hollow structures could partially mitigate the above pulverization problem.2,12,13 Here we hypothesize a synergistic effect between SnO2 and α-Fe2O3 which will reduce the initial irreversible loss. Specifically, transition metaloxides (MO, M = Fe, Co, Ni, etc.) have a completely different and unusual reaction mechanism: MO + 2Li+ + 2e− ↔ Li2O + M.30 It is nested in the electrochemically driven, in situ formation of metal nanoparticles during the first discharge, which enables the subsequent reversible formation and decomposition of the Li2O nanomatrix. The concept is that the presence of Fe nanoparticles in the nanomatrix should also promote reversibility of reaction (I) and lead to a higher reversible capacity.
Fig. 6A displays the 1st discharge-charge voltage profiles of the SnO2 hollow nanospheres and α-Fe2O3@SnO2 nano-rattles at a constant current rate of 200 mA g−1. In general, these voltage profiles are characteristic of SnO2-based materials. The first discharge capacities are found to be 1621 mA h g−1 and 1544 mA h g−1, and their corresponding charge capacities are found to be 604 mA h g−1 and 865 mA h g−1, for SnO2 hollow nanospheres and α-Fe2O3@SnO2 nano-rattles respectively. The irreversible losses can therefore be calculated to be 62.7% and 44.0% for SnO2 hollow nanospheres and α-Fe2O3@SnO2 nano-rattles respectively. The observation of a much lower irreversible loss for α-Fe2O3@SnO2 nano-rattles compared to SnO2 hollow spheres might be indeed in support of our hypothesis. One might argue that the improved overall Coulombic efficiency could simply come from the α-Fe2O3 component present in the composite. This possibility might be ruled out if one considers that the Coulombic efficiency of α-Fe2O3 is generally observed to be comparable in this potential window,31–33 and the mass fraction of α-Fe2O3 in the α-Fe2O3@SnO2 nano-rattles is relatively low (as a rough estimation, 15–25%). Fig. 6B depicts the cycling performance of both samples up to 30 cycles. Because of the initial irreversible loss, there is a large drop in capacity of the 2nd cycle for both samples. Although the capacity fading behavior is generally similar for both samples, α-Fe2O3@SnO2 nano-rattles always exhibit a much higher capacity compared to SnO2 hollow spheres. For example, the capacities of the 30th cycle are found to be 419 mA h g−1 and 260 mA h g−1 for α-Fe2O3@SnO2 nano-rattles and SnO2 hollow spheres respectively. In view of the similarity in structure and size, one possible reason for the difference in capacity is the presence of the α-Fe2O3nanocrystals inside the SnO2 hollow cavity. Thus it might be presumably concluded that the core/shell materials act together to produce the synergistic improvement of electrochemical properties. Lastly, it should be pointed out that the cycling performance is still unsatisfactory despite having the hollow structure. Nevertheless, our results indicate that the cycle life of these hollow SnO2 spheres with large void space can be significantly improved by combining with the carbon coating strategy.13,17
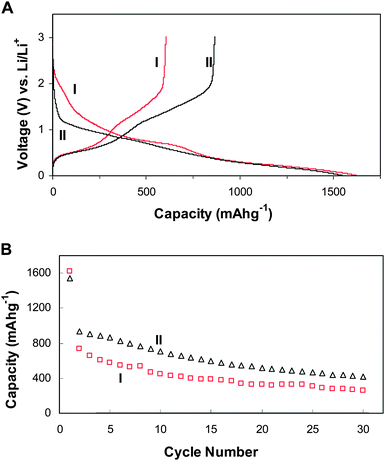 | ||
| Fig. 6 First cycle charge-discharge curves (A) and cycling performance (B) of the SnO2 hollow nanospheres (I) and α-Fe2O3@SnO2 nano-rattles (II) at a constant current rate of 200 mA g−1 with a fixed cut-off voltage window of 3 V to 10 mV. | ||
In conclusion, we have shown that uniform SnO2 hollow nanospheres with large void space can be prepared through a modified one-pot template-free method. The void space can be easily controlled by varying the reaction time. The formation of interior void space is based on an inside-out Ostwald ripening mechanism. More importantly, we have also demonstrated that the same one-pot process can be applied to fabricate more challenging functional structures like α-Fe2O3@SnO2 nano-rattles. Furthermore, we have investigated the electrochemical lithium storage properties. It has been found that α-Fe2O3@SnO2 nano-rattles manifest a much lower initial irreversible loss and higher reversible capacity compared to SnO2 hollow spheres. This interesting finding supports a general hypothesis that functional core-shell structures may produce a synergistic effect on useful properties.
Experimental
Synthesis of SnO2 hollow nanospheres
In a typical experiment, a certain amount of potassium stannate trihydrate (K2SnO3·3H2O, Sigma-Aldrich, 99.9%) was dissolved in 50 mL of an ethanol/water (2-propanol and ethylene glycol were also used as alternatives to ethanol) mixture with a volume ratio of 3:5, to achieve a concentration of 16 mM. Urea (0.3 g, 0.1 M) was usually used as an additive. After shaking by hand or vortexing for about 3 minutes, a translucent solution was obtained and then transferred into a 60 mL Teflon-lined stainless steel autoclave, followed by heating at 200 °C for a period of 4–48 h in an electric oven. After heating, the autoclave was cooled naturally to room temperature. The white precipitate was collected and washed with ultrapure water several times by centrifugation, then dried at 60 °C overnight.Synthesis of α-Fe2O3nanocrystals
2.7 g of iron(III) chloride hexahydrate (FeCl3·6H2O, Sigma-Aldrich, ≥98%) was dissolved in 500 mL of ultrapure water to form a clear yellowish solution. It was kept in a tightly sealed glass bottle and heated in an electric oven at 105 °C for 50 h. The bottle was cooled naturally after reaction. The red precipitate was collected and washed by centrifugation several times before drying at 60 °C overnight.Synthesis of α-Fe2O3@SnO2 nano-rattles
In a typical synthesis, 0.025 g of pre-synthesized α-Fe2O3nanocrystals were added into 50 mL of an ethanol/water mixture (described above) with urea as the additive. Then the solution was ultrasonicated for a few minutes to fully disperse the α-Fe2O3nanocrystals, after which a red-colored homogeneous solution was obtained. A certain amount of K2SnO3·3H2O as the precursor of SnO2 was added into the solution to achieve a concentration of 16 mM. After the K2SnO3·3H2O was fully dissolved, the mixture was transferred into a Teflon-lined stainless steel autoclave and heated in an electric oven at 200 °C for a period of 2–24 h. The autoclave was cooled naturally to room temperature. The red precipitate was collected and washed several times with ultrapure water, then dried at 60 °C overnight.Material characterization
The structure and morphology of products were examined with transmission electron microscopy (TEM; JEOL, JEM-2010, 200 kV), and field emission scanning electron microscopy (FESEM; JEOL, JSM-6700F, 5 kV). The powder X-ray diffraction (XRD) analysis was carried out with a Bruker D8 - Advance X-Ray Diffractometer (Cu Kα radiation, λ = 1.5406 Å). The N2 adsorption and desorption isotherm was obtained using a Quantachrome Instruments, Autosorb AS-6B.Electrochemical measurement
The electrochemical tests were carried out using two-electrode cells with pure lithium metal as the counter and reference electrodes at room temperature. The working electrode consists of active material (SnO2 hollow nanospheres, or α-Fe2O3@SnO2 nano-rattles), a conductive agent (carbon black, Super-P-Li), and a polymer binder (polyvinylidene difluoride, PVDF, Aldrich) in a weight ratio of 80:10:10. The electrolyte used was 1.0 M LiPF6 in a 50:50 (w/w) mixture of ethylene carbonate and diethyl carbonate. Cell assembly was carried out in an Ar-filled glovebox with concentrations of moisture and oxygen below 1.0 ppm. The charge/discharge tests were performed using a NEWARE battery tester at a constant current rate of 200 mA/g with a voltage window of 0.01–3.0 V.Acknowledgements
We are grateful to the Nanyang Technological University and to the KAUST-Cornell (KAUST-CU) Center for Energy and Sustainability for financial support.References
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987 CrossRef CAS.
- X. W. Lou, Y. Wang, C. Yuan, J. Y. Lee and L. A. Archer, Adv. Mater., 2006, 18, 2325 CrossRef CAS.
- F. Caruso, R. A. Caruso and H. Mohwald, Science, 1998, 282, 1111 CrossRef CAS.
- Y. G. Sun and Y. N. Xia, Science, 2002, 298, 2176 CrossRef CAS.
- Y. D. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711 CrossRef CAS.
- H. P. Liang, H. M. Zhang, J. S. Hu, Y. G. Guo, L. J. Wan and C. L. Bai, Angew. Chem. Int. Ed., 2004, 43, 1540 CrossRef CAS.
- X. M. Sun and Y. D. Li, Angew. Chem., Int. Ed., 2004, 43, 3827 CrossRef CAS.
- Z. Z. Yang, Z. W. Niu, Y. F. Lu, Z. B. Hu and C. C. Han, Angew. Chem., Int. Ed., 2003, 42, 1943 CrossRef CAS.
- S. W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642 CrossRef CAS.
- Y. Piao, J. Kim, H. Bin Na, D. Kim, J. S. Baek, M. K. Ko, J. H. Lee, M. Shokouhimehr and T. Hyeon, Nat. Mater., 2008, 7, 242 CrossRef CAS.
- B. X. Li, Y. Xie, M. Jing, G. X. Rong, Y. C. Tang and G. Z. Zhang, Langmuir, 2006, 22, 9380 CrossRef CAS.
- H. Ma, F. Y. Cheng, J. Chen, J. Z. Zhao, C. S. Li, Z. L. Tao and J. Liang, Adv. Mater., 2007, 19, 4067 CrossRef CAS.
- X. W. Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536 CrossRef CAS.
- K. Kamata, Y. Lu and Y. N. Xia, J. Am. Chem. Soc., 2003, 125, 2384 CrossRef CAS.
- X. W. Lou, C. Yuan and L. A. Archer, Small, 2007, 3, 261 CrossRef CAS.
- H. C. Zeng, Curr. Nanosci., 2007, 3, 177 Search PubMed.
- X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, Chem. Mater., 2008, 20, 6562 CrossRef CAS.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169 CrossRef CAS.
- X. W. Lou, C. Yuan and L. A. Archer, Adv. Mater., 2007, 19, 3328 CrossRef CAS.
- J. Li and H. C. Zeng, Angew. Chem., Int. Ed., 2005, 44, 4342 CrossRef CAS.
- X. W. Lou, C. L. Yuan, E. Rhoades, Q. Zhang and L. A. Archer, Adv. Funct. Mater., 2006, 16, 1679 CrossRef CAS.
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Science, 1997, 276, 1395 CrossRef CAS.
- Y. Wang, J. Y. Lee and H. C. Zeng, Chem. Mater., 2005, 17, 3899 CrossRef CAS.
- S. J. Han, B. C. Jang, T. Kim, S. M. Oh and T. Hyeon, Adv. Funct. Mater., 2005, 15, 1845 CrossRef CAS.
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater., 2007, 19, 2336 CrossRef CAS.
- R. Demir-Cakan, Y. S. Hu, M. Antonietti, J. Maier and M. M. Titirici, Chem. Mater., 2008, 20, 1227 CrossRef.
- M. S. Park, Y. M. Kang, G. X. Wang, S. X. Doti and H. K. Liu, Adv. Funct. Mater., 2008, 18, 455 CrossRef CAS.
- X. W. Lou, J. S. Chen, P. Chen and L. A. Archer, Chem. Mater., 2009, 21, 2868 CrossRef CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS.
- C. Z. Wu, P. Yin, X. Zhu, C. Z. OuYang and Y. Xie, J. Phys. Chem. B, 2006, 110, 17806 CrossRef CAS.
- Y. N. Nuli, P. Zhang, Z. P. Guo and H. K. Liu, J. Electrochem. Soc., 2008, 155, A196 CrossRef CAS.
- X. L. Wu, Y. G. Guo, L. J. Wan and C. W. Hu, J. Phys. Chem. C, 2008, 112, 16824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further images of nanospheres; EDX spectrum; XRD patterns. See DOI: 10.1039/b9nr00102f |
| This journal is © The Royal Society of Chemistry 2009 |