Unusually cyclized triterpenes: occurrence, biosynthesis and chemical synthesis
Victoriano
Domingo
,
Jesús F.
Arteaga
,
José F.
Quílez del Moral
and
Alejandro F.
Barrero
*
Department of Organic Chemistry, Institute of Biotechnology, University of Granada,Avenida Fuentenueva, 18071, Granada, Spain. E-mail: afbarre@ugr.es; Fax: + 34-958-243318
First published on 29th October 2008
Abstract
Covering: 1998 to 2008
The biosynthetic origin of most of triterpenes lies in cascade cyclizations and rearrangements of the acyclic precursors squalene (S) and 2,3-oxidosqualene (OS), processes leading to tetra- and pentacyclic triterpene skeleta. Apart from these, a number of triterpenoid structures derived from cyclization processes, that are different from those leading to tetra- and pentacyclic triterpenes, are also found in Nature. We have defined these processes as unusual cyclizations, and grouped them in three blocks, namely, incomplete cyclizations of the corresponding S-derived precursors, cyclizations of S or OS towards polycyclic triterpenes and subsequent cleavage of the preformed ring systems, and two independent cyclizations of the S- or OS-derived precursor. Apart from the molecules obtained from intact organisms, we will also consider the compounds obtained from in vitro cyclizations promoted by enzyme systems. After establishing which compounds could unambiguously be grouped under the term ‘unusually cyclized triterpenes’, this review moves on to the advances achieved in this kind of structure during the last ten years. These advances are presented in three parts. The first one presents the structure and biological properties of the unusual triterpenes reported in the last decade. The second part considers the main biosynthetic pathways which justify the formation of these triterpenes from their corresponding acyclic precursors. Finally, we look at the achievements made in different synthetic strategies directed at some of these molecules. One hundred and twenty-three references are cited.
 Victoriano Domingo | Victoriano Domingo was born in Granada (Spain) in 1981 and graduated in Chemistry in 2006 at the University of Granada. He is a Ph.D. student at the same University under the supervision of Prof. Alejandro F. Barrero and Dr José F. Quílez. Currently, he is in the process of completing the total synthesis of various triterpenes. His interests lie in modern synthetic methodologies with application in the field of bioactive natural products. |
 Jesús F. Arteaga | Jesús F. Arteaga was born in Salamanca (Spain) in 1979 and he graduated in Chemistry in 2002 at the University of Granada. He received his Ph. D. degree in 2006 under the supervision of Prof. Alejandro F. Barrero at the same university. From 2006–2007 he formed part of a post-doctoral team at the Institut de Chimie des Substances Naturelles (Gif-sur-Yvette, France) with Prof. Simeon Arseniyadis working on the total synthesis of biologically active natural products. He is currently a post-doctoral investigator at the University of Huelva (Spain). His research interests are mainly focused on the development of new applications of free-radical chemistry in the synthesis of natural products and homogeneous catalysis. |
 José F. Quílez del Moral | José F. Quílez del Moral was born in Linares (Spain) in 1967 and studied Chemistry at the University of Granada, where he graduated in 1990. He received his Ph.D. degree in 1996 under the supervision of Prof. A. F. Barrero. In 1997, he started a 20 month post-doctoral fellowship with Prof. S. Arseniyadis at the Institut de Chimie des Substances Naturelles at Gif-sur Yvette (France) working on the total synthesis of taxol. He returned to Granada as an assistant professor and joined Prof. A. F Barrero's group in 1998. He is currently a Senior Lecturer and his research interests are directed towards the development of new methods for the synthesis of molecules having biological activity. |
 Alejandro F. Barrerro | Alejandro F. Barrerro was born in Orense (Spain) in 1949. He obtained his Ph.D. degree in 1975 at the University of Salamanca under the guidance of Professors Joaquín Pascual de Teresa, Arturo San Feliciano and Inés Sánchez Bellido. After working as a research scientist at the Compañía Española de Petróleos Research Center in San Fernando de Henares (Spain), he returned to the University of Salamanca as a Lecturer. He moved to the University of Granada as Full Professor in 1983, where he is Head of the Organic Chemistry Department. He is also President of the Natural Product Group of the Royal Spanish Society of Chemistry. His work includes the direction of more than 40 doctoral theses. His current interests are the chemistry and biotechnology of terpenoids and the application of both radical cyclization reactions and new couplings catalyzed by transition metals to the synthesis of natural bioactive products. |
1 Introduction
Triterpenes are one of the most structurally diverse groups of terpenes, with more than 100 skeleta described as natural products.1–4 The origin of this diversity is the type of mechanism that takes part in their biosynthesis – the enzymatic systems generically named terpenoid synthases act on acyclic polyprene precursors generating carbocations that, by electrophilic additions on double bonds, produce processes of cyclization and rearrangements. In the specific case of the triterpenes, the triterpenoid synthases act on the most common precursors – squalene (S) and oxidosqualene (OS) – and biosynthesis is initiated by enzymatic protonation of the terminal double bond or the oxirane respectively. As those precursors contain five or six double bonds, the possibilities of cyclization accompanied by rearrangement are theoretically very great.5–7 This has generated a good deal of interest amongst scientists. In this context, the hypothesis of isoprene as a biogenetic precursor for triterpenes by Ruzicka, Eschenmoser, Arigoni et al.8,9 was a significant milestone which was complemented by the characterization of some thirty OS cyclases (OSCs) and various studies of mutations on the genes involved in these cyclization processes.5,10–12Most of the triterpenes contain tetra- or pentacyclic skeletons, but together with these a number of triterpenoid structures derived from cyclization processes different to those leading to tetra- and pentacyclic triterpenes have also been reported from Nature. We have named these structures ‘unusually cyclized triterpenes’, and have grouped them as follows:
a) Triterpenes arising via an incomplete cyclization of the corresponding S-derived precursors. These processes start at the terminal isopropylidene unit or at its corresponding epoxide and lead to mono-, bi- and tricyclic triterpenic skeletons.
b) Triterpenes arising via cyclization of S or OS towards polycyclic triterpenes and subsequent cleavage of the preformed ring systems.
c) Triterpenes arising via two independent cyclizations of the S- or OS-derived precursor.
The aim of this review is to cover the structure, biological properties, biosynthesis and chemical synthesis of the unusually cyclized triterpenes described in the last ten years. Apart from the molecules obtained from live organisms, compounds obtained from in vitro cyclizations of S or OS promoted by enzymatic systems are also included.
2 Occurrence and biological activities of unusually cyclized triterpenes
2.1 Triterpenes resulting from incomplete cyclizations of S and OS

| Compound | R2 | Source | |||
|---|---|---|---|---|---|
| Achilleol A (1) | OH | Bupleurum spinosum,17Santolina elegans,18Camellia sasanqua, C. japonica,19 wheat germ oil and rice bran oil,20 SHC mutant [Alicyclobacillus acidocaldarius ΔG600],21 OSLC mutant [S. cerevisiae H234 (M/Y/F), Y510 (A/K/W/H),22,23 V454A, V454G],24 CAS1 mutant [Arabidopsis thaliana I481 (A/G),25 Y410C Y532H26], Gene expression: CAMS1 [At1g78955],27 BARS1 [At4g15370 ]28 | |||
| Achilleol A esterified derivative (2) | CH3(CH2)10COO | B. spinosum,17S. elegans,18C. sasanqua,19C. japonica,19 wheat germ oil and rice bran oil20 | |||
| Achilleol A esterified derivative (3) | CH3(CH2)12COO | ||||
| Achilleol A esterified derivative (4) | CH3(CH2)14COO | ||||
| 3-Deoxyachilleol (5) | H | SHC Mutant [Alicyclobacillus acidocaldarius D377 (C/N), Y612A]29 | |||
| Camelliol C (6) | — | C. sasanqua,19C. japonica,19 OSLC mutant: [S. cerevisiae Y510 (K/W)],22 CAS1 [Arabidopsis thaliana I481 (A/G)],25 Gene expression: CAMS1 [At1g78955],27 BARS1[At4g15370]28 | |||
| Monocyclosqualene (7) | — | Ligularia fischeri 30 | |||
| 6α-Hydroxyachilla-9,13,17,21-tetraene (8) | — | SHC mutants [Alicyclobacillus acidocaldarius L607(F/W)]31 | |||
| Neoachillapentaene (9) | — | SHC mutants [Alicyclobacillus acidocaldarius L607(F/W), Y420W]31 | |||
Monocyclosqualene 7 was isolated from the herb Ligularia fischeri var. spiciformis (Compositae),30 and 3-deoxyachilleol A 5 from a squalene hopene cyclase (SCH) mutant of the prokaryotic bacterium Alicyclobacillus acidocaldarius.29 Only camelliol C 6 was reported to present a slight inhibitory effect on the HIV transcriptase.14 It is possible that these compounds may have a biological role as reinforcers of the cell membranes.15,16

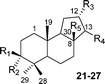
| Compound | Δ | R1 | R2 | R3 | R4 | Source | |
|---|---|---|---|---|---|---|---|
| α-Polypodatetraene (10) | Δ8(26) | H | H | — | H | SHC mutants [A. acidocaldariusY420A, Y609F]34,35 | |
| γ-Polypodatetraene (11) | Δ7 | H | H | — | H | SHC mutants [A. acidocaldarius F365A,36 Y420A,34 Y609 (A/L/C/S),37,38 Y612A,37 L607K39,40] | |
| 3β-Hydroxy γ-polypodatetraene (12) | Δ7 | OH | H | — | H | Cratoxylum cochinchinense 41 | |
| Myrrhanol A (13) | — | OH | H | α-OH | CH2OH | Balsamodendron mukul 33,42 | |
| Myrrhanone A (14) | — | O | O | α-OH | CH2OH | B. mukul 33,42 | |
| Myrrhanone A acetate (15) | — | O | O | α-OH | CH2OAc | B. mukul 43 | |
| Myrrhanol B (16) | — | OH | H | α-OH | COOH | B. mukul 43 | |
| Myrrhanone B (17) | — | O | O | α-OH | COOH | B. mukul 43 | |
| 21,22-Dihydro α-polypodatetraene (18) | Δ8(26) | H | H | — | H | Tetrahymena pyriformis 44 | |
| 21,22-Dihydro γ-polypodatetraene (19) | Δ7 | H | H | — | H | T. pyriformis 44 | |
| Neopolypodatetraene (20) | — | — | — | — | — | SHC mutant [A. acidocaldarius F365A]37 | |
| Compound | R1 | R2 | R3 | R4 | R5 | Source |
|---|---|---|---|---|---|---|
| Malabarica-14(27),17,21-trien-3-ol (21) (13R or 13S) | OH | H | H |
|
Me | SHC mutant [A. acidocaldarius ΔG600],21 Gene expression: BARS1[At4g15370]28 |
| 13β-Malabaricatriene (22) | H | H | H |

|
Me | SHC mutant [A. acidocaldarius F601A]45 |
| 13α-Malabaricatriene (23) | H | H | H |

|
Me | SHC mutants [A. acidocaldarius F601A, Y420A, I261A],31,45,46 sediment lake Cadagno47 |
| Arabidiol (24) | OH | H | H |

|
Me | Gene expression: [At4g15340],48 SHC mutant [A. acidocaldarius ΔG600]21 |
| Arabidiol 20,21-epoxide (25) | OH | H | H |

|
Me | Gene expression: [At4g15340]48 |
| 26 | O | O | H |

|
Me | Caloncoba echinata 49 |
| 27 | O | O | H |

|
Me | C. echinata 49 |
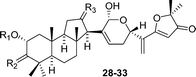
| Compound | R1 | R2 | R3 | Source |
|---|---|---|---|---|
| Sodagnitin A (28) | H | O | H2 | Cortinarius sodagnitus, C. fulvoincarnatus, C. arcuatorum50 |
| Sodagnitin B (29) | H | O | O | C. sodagnitus, C. fulvoincarnatus, C. arcuatorum50 |
| Sodagnitin C (30) |

|
O | H2 | C. sodagnitus, C. fulvoincarnatus, C. arcuatorum, C. cf. calochrous50 |
| Sodagnitin D (31) |
|
O | O | C. sodagnitus, C. fulvoincarnatus, C. arcuatorum50 |
| Sodagnitin E (32) | H | H, β-OH | H2 | C. sodagnitus, C. fulvoincarnatus, C. arcuatorum50 |
| Sodagnitin F (33) | H | H, β-OH | O | C. sodagnitus, C. fulvoincarnatus, C. arcuatorum50 |
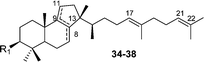
| Compound | R1 | Source |
|---|---|---|
| Thalianol (34), podioda-8,17,21-trien-3β-ol | OH | Gene expression: [At5g48010],51 SHC mutant [A. acidocaldarius ΔG600]21 |
| (21S)-21,22-Epoxy-malabarica-8,17-dien-3-ol (35) | OH | Gene expression: [At5g48010]51 |
| Podioda-9(11)-17,21-trien-3β-ol (36) | OH | SHC mutant [A. acidocaldarius ΔG600]21 |
| 7,17,21-Podiodatriene (37) | H | SHC mutant [A. acidocaldarius F605A],52,53 Gene expression: BARS1 [At4g15370 ]28 |
| 8,17,21-Podiodatriene (38) | H | SHC mutant [A. acidocaldarius F605A]52,53 |

| Compound | Δ13 | R1 | R2 | R3 | R4 | R5 | Source |
|---|---|---|---|---|---|---|---|
| 3-Epi-29-hydroxystelliferin A (39) | Z | H | OH | OH | OH | H | Stelleta globostellata 55 |
| Stelliferin G (40) | Z | H | H | OH | OAc | H | Jaspis species 54 |
| 29-Hydroxystelliferin E (41) | Z | OAc | H | OH | OAc | H | Jaspis species,54S. globostellata55 |
| 29-Hydroxystelliferin A (42) | Z | OAc | H | OH | OH | H | Jaspis species54 |
| 29-Hydroxystelliferin D (43) | Z | OH | H | OH | OH | H | S. globostellata 55 |
| Stelliferin riboside E (44) | Z | H | OAc | H | H or O-ribose | O-ribose or H | R. globostellata 56 |
| 3-O-Deacetyl-13Z-stelliferin riboside (45) | Z | H | OH | H | H or O-ribose | O-ribose or H | R. globostellata 56 |
| Stelliferin riboside (46) | E | H | OAc | H | H or O-ribose | O-ribose or H | R. globostellata,56G. globostellifera57 |
| 3-Hydroxy-13E-stelliferin riboside (47) | E | H | OH | H | H or O-ribose | O-ribose or H | R. globostellata,56G. globostellifera57 |
| 13αH-Isomalabarica-14(27),17E,21-trien-3β-ol (48) | — | — | — | — | — | — | OSLC [S. cerevisiae H234 (L/M/N/D),58,59 F445 (C/M/N/T/D),60 Y510(F/H)23,61,62] |

| Compound | Δ13 | R1 | R2 | R3 | R4 | Source |
|---|---|---|---|---|---|---|
| Globostellatic acid F (49) | Z | H | OH | COOH |

|
R. globostellata 56 |
| Globostelletin (50) | Z | H | OH | CH3 |

|
R. globostellata 56 |
| Globostellatic acid G (51) | Z | H | OH | COOH |
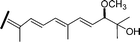
|
R. globostellata 56 |
| Globostellatic acid I (52) | Z | H | OH | COOH |

|
R. globostellata 56 |
| Globostellatic acid K (53) | Z | H | OAc | COOH |

|
R. globostellata 56 |
| Globostellatic acid M (54) | Z | H | OH | COOH |

|
R. globostellata 56 |
| Globostellatic acid H (55) | E | H | OH | COOH |

|
R. globostellata 56 |
| Globostellatic acid J (56) | E | H | OAc | COOH |

|
R. globostellata 56 |
| Globostellatic acid L (57) | E | H | OH | COOH |
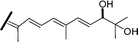
|
R. globostellata 56 |
| Globostellatic acid E (58) | Z | H | OAc | COOCH3 |
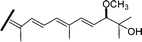
|
Jaspis species63 |
| 13Z,17Z-Globostellatic acid X methyl ester (59) | Z | H | OAc | COOCH3 |

|
R. globostellata 64 |
| 13Z,17E-Globostellatic acid X methyl ester (60) | Z | H | OAc | COOCH3 |

|
R. globostellata 64 |
| 13E,17Z-Globostellatic acid X methyl ester (61) | E | H | OAc | COOCH3 |

|
R. globostellata 64 |
| 13E,17E-Globostellatic acid X methyl ester (62) | E | H | OAc | COOCH3 |

|
R. globostellata 64 |
| Globostellatic acid F methyl ester (63) | E | H | OAc | COOCH3 |

|
R. globostellata 64 |
| Globostellatic acid B methyl ester (64) | E | H | OAc | COOCH3 |

|
R. globostellata 64 |
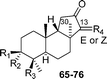
| Compound | Δ13 | R1 | R2 | R3 | R4 | Source |
|---|---|---|---|---|---|---|
| 3-O-Acetyl-jaspiferal B methyl ester (65) | Z | H | OAc | COOCH3 |
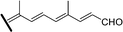
|
Jaspis species63 |
| Jaspolide A (66) | Z | OH | H | CH3 |

|
Jaspis species65 |
| Jaspolide B (67) | E | OH | H | CH3 |

|
Jaspis species 65 |
| Stelletin J (68) | Z | OH | H | CH2OH |

|
R. globostellata 66 |
| Stelletin K (69) | Z | OH | H | COOH |

|
R. globostellata 66 |
| Stelletin I (70) | Z | OAc | H | CH3 |
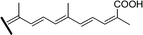
|
R. globostellata 67 |
| Stelletin L (71) | E | OH | H | CH3 |

|
Stelletta tenuis 68 |
| Stelletin M (72) | Z | OH | H | CH3 |

|
S. tenuis 68 |
| Isogeoditin A (23Z) (73) | Z,E | O | O | CH3 |

|
R. aff. distincta69 |
| Isogeoditin B (23Z) (74) | Z | OAc | H | CH3 |

|
R. aff. Distincta69 |
| Geoditin A (23E) (75) | Z | O | O | CH3 |

|
R. aff. distincta,69G. japonica70 |
| Geoditin B (23E) (76) | Z | OAc | H | CH3 |

|
R. aff. distincta,69G. japonica70 |
Isomalabaricanes have been found exclusively in marine sponges, mostly from the Pacific, and in many cases form their yellow pigmentation. The substances possessing this skeleton are characterised structurally by a carbonyl group on C-12, an E or Z double bond on C-13 (which can undergo light-induced isomerization), and highly unsaturated systems on the side chain, which in many cases are four and five double bonds conjugated with the carbonyl on C-12. Moreover, oxygenated functions are frequently present on the ring at C-3 and on the C-29 methyl group.
They have been classified into three groups according to their structure and origin. Stelliferins (Table 6) have a methylene on C-23, and an oxygenated function on C-22. The second group comprises globostellatic acids (Table 7), isolated almost exclusively from Rhabdastrella globostellata. Most of these compounds contain a carboxylic acid or a methyl ester on C-29 and a highly unsaturated side chain with 4 or 5 conjugated double bonds. The third group – jaspolides and stelletins (Table 8) – either contain side chains with terminal pyrone rings (66–67) or a carboxylic acid in place of one of the terminal methyls of the side chain (70–72), or are nor-triterpenes that have lost the last carbon of the side chain, forming methylketones (73–76).
Numerous examples of isomalabaricanes have been found with cytotoxic activity. Mixtures of 29-hydroxystelliferin A/29-hydroxystelliferin B and stelliferin G/13 E-stelliferin G showed antiproliferative activity against melanoma cells (MALME-3M),104 with an IC50 value of 0.11 and 0.23 µg/mL respectively. Stelliferin riboside 44 and 3-O-deacetyl-13, 14 Z-stelliferin riboside 45 showed potent activity against the mouse lymphoma cell line L5178Y.56
Stelliferin riboside 44 and the 3-epi-acetate derivative of 29-hydroxystelliferin E41 were shown to induce 29% and 23% DNA-polymerase β binding, respectively, at 28 µg/mL. These compounds displayed varying levels of activity toward the A2780 ovarian cancer cell line, revealing structure-based effects on both the level of cytotoxicity and DNA-polymerase β binding.66
Globostellatic acids E–M showed potent activity against the mouse lymphoma cell lne L5178Y, with ED50 values of 0.3–10.4 nM. They were weakly active or inactive against a human cervix carcinoma HeLa and rat pheochromocytoma PC-12 cell lines.56 Six globostellatic acids methyl esters 59–64, especially those with 13E geometry 61–64, exhibited a high selective index value in antiangiogenic activity, inhibiting proliferation of human umbilical vein endothelial cells selectively in comparison with other cell lines.64
Stelletins L and M (71–72) also showed interesting cytotoxic activity against stomach cancer.68 Some isomalabaricanes possess antimicrobial activities, thus stelliferin ribosides 44–46 show activity against E. coli, presenting an inhibition zone of 12 mm at a loading concentration of 10 µg. Globostelletin (50) and globostellatic acids G and I (51–52) show moderate activities against E. coli, and globostelletin also inhibits Bacillus subtilis, with inhibition zones of 12 and 13 mm at concentrations of 5 and 10 µg.56
2.2 Triterpenes derived from processes of cyclization of S or OS towards polycyclic triterpenes and subsequent retrocyclization reactions from pentacyclic carbocation intermediates
Oxidosqualene cyclases (OSCs) have been recognized to catalyze carbon-carbon bond formations and rearrangements including hydride and methyl groups to finally produce different polycyclic structures. However, recent reports by Matsuda and Ezibuka's groups of the identification of a marneral synthase yielding the A-ring-seco-triterpene marneral on one hand, and an OSCs yielding seco-amyrins revealed that OSCs have the ability to cleave preformed ring systems in addition to forming multiring systems. Bearing these findings in mind, we have established a new group of triterpenes which includes those structures derived from an OSC-mediated annulation of S or OS to give, in an earlier part of the reaction, a carbenium ion intermediate possessing bi-, tetra- or pentacyclic systems, which in a later process undergo fragmentations of the previously generated rings to produce seco-bi-, tetra- and pentacyclic triterpenes. However, those seco-triterpenoids arising from postcyclization steps,71,72 are not included here.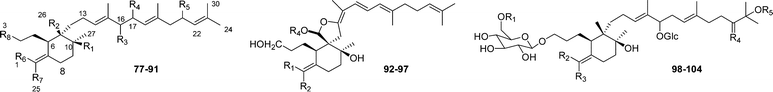
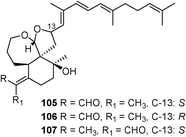
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | Source |
|---|---|---|---|---|---|---|---|---|---|
| Marneral (77) | H | CH3 | H | H | H | CH3 | CH3 | CHO | Gene expression: [At5g42600]72 |
| Marnerol (78) | H | CH3 | H | H | H | CH3 | CH3 | CH2OH | Gene expression: [At5g42600]72 |
| 23-Hydroxyiridal (79) | OH | CH3 | H | H | Δ21 | CHO | CH3 | CH2OH | I. variegata 73 |
| 18,19-Epoxy-10- deoxyiridal (80) | H | CH3 | H | H | H | CHO | CH3 | CH2OH | I. germanica 74 |
| 18,19-Epoxyiridal (81) | OH | CH3 | H | H | H | CHO | CH3 | CH2OH | I. versicolor 75 |
| 16,26-Dihydroxyiridal (82) | OH | CH2OH | OH | H | H | CHO | CH3 | CH2OH | I. versicolor 75 |
| 22,23-Epoxi-21-hydroxyiridal (83) | OH | CH3 | H | H | OH | CHO | CH3 | CH2OH | I. cristata 76 |
| 22,23-Epoxyiridal (84) | OH | CH3 | H | H | H | CHO | CH3 | CH2OH | I. cristata 76 |
| 22,23-Epoxy-10-deoxy-21-hydroxyiridal (85) | H | CH3 | H | H | OH | CHO | CH3 | CH2OH | I. cristata 76 |
| Iritectol A (86) | OH | CH3 | OH | H | H | CHO | CH3 | CH2OH | I. tectorum 77 |
| Iritectol B (87) | OH | CH3 | O | H | H | CHO | CH3 | CH2OH | I. tectorum 77 |
| Iridobelamal A (88) | OH | CH3 | α-OH | H | H | CH3 | CHO | CH2OH | B. chinensis,79I. tectorum77 |
| 3-O-Decanoyl-16-O-acetylisoiridogermanal (89) | OH | CH3 | β-OAc | H | H | CHO | CH3 | Myristic acid | B. chinensis 78 |
| 3-O-Tetradecanoyl-16-O-acetylisoiridogermanal (90) | OH | CH3 | β-OAc | H | H | CHO | CH3 | Capric acid | B. chinensis 78 |
| Iristectorone K (91) | — | — | — | — | — | — | — | — | I. germanica 79 |
| Iridotectoral A (92) | CH3 | CHO | — | H | — | — | — | — | I. tectorum,80 |
| (13R)-Iridotectoral C (93) | CHO | CH3 | — | CH3 | — | — | — | — | I. tectorum 81 |
| (13R)-Iridotectoral D (94) | CH3 | CHO | — | CH3 | — | — | — | — | I. tectorum 81 |
| (13S)-Iridobelamal B (95) | CHO | CH | — | CH3 | — | — | — | — | B. chinensis 80 |
| Belachinal (96) | CHO | CH3 | — | H | — | — | — | — | B. chinensis 78 |
| Tigridal (97) | CHO | CH3 | — | H | — | — | — | — | Tigridia pavonea 82 |
| 98 | Glc | CHO | CH3 | O | H | — | — | — | I. spuria 83 |
| 22-Oxo-23-hydroxy-iridal-3,16-di-β-D-glucopyranoside (99) | H | CHO | CH3 | O | H | — | — | — | I. spuria 83 |
| 22,23-Dihydroxy-iridal-3,16-di-β-D-glucopyranoside (100) | H | CHO | CH3 | H,OH | H | — | — | — | I. spuria 83 |
| 22-Oxo-isoiridal-3,16,23-tri-β-D-glucopyranoside (101) | H | CH3 | CHO | O | Glc | — | — | — | I. spuria 83 |
| 102 | Glc | CH3 | CHO | O | H | — | — | — | I. spuria 83 |
| 22-Oxo-23-hydroxy-isoiridal-3,16-di-β-D-glucopyranoside (103) | H | CH3 | CHO | O | H | — | — | — | I. spuria 83 |
| 22,23-Dihydroxy-isoiridal-3,16-di-β-D-glucopyranoside (104) | H | CH3 | CHO | H,OH | H | — | — | — | I. spuria 83 |
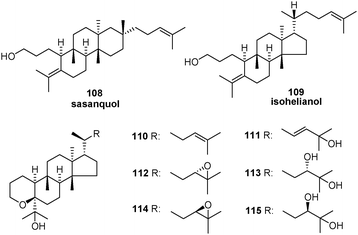

| Compound | Δ | R1 | R2 | R3 | R4 | R5 | R6 | Source |
|---|---|---|---|---|---|---|---|---|
| 8,14-Seco-oleana-8(26),13-dien-3β-ol (116) | Δ14(27) | OH | H | CH3 | CH3 | CH3 | H | Stevia viscida 86 |
| 8,14-Seco-oleana-8(26),13-dien-3β-acetyl (117) | Δ14(27) | OAc | H | CH3 | CH3 | CH3 | H | S. eupatoria 86 |
| β-Seco-amyrin (118) | Δ7,Δ14(27) | OH | H | CH3 | CH3 | CH3 | H | Gene expression: [At1g78500]87 |
| α-Seco-amyrin (119) | Δ7,Δ14(27) | OH | H | CH3 | H | CH3 | CH3 | Gene expression: [At1g78500]87 |
| 8,14-Secotaraxastane (120) | Δ8(26),Δ12 | OH | H | H | CH3 | CH3 | CH3 | Koelpinia linearis 88 |
| 8,14-Secotaraxastane (121) | Δ8(26),Δ13 isomer | OH | H | H | CH3 | CH3 | CH3 | K. linearis 88 |
| 8,14-Seco-20-hydroxy-taraxastane (122) | Δ12 | OH | H | OH | CH3 | H | CH3 | K. linearis 88 |
| Achilleol B (123) | Δ6(25) | — | — | — | — | — | — | Achillea odorata 89 |
| Camelliol A (124) | Δ1(6) | — | — | — | — | — | — | Camellia sasanqua 19 |
| Camelliol B (125) | — | — | — | — | — | — | — | C. sasanqua 19 |
2.3 Triterpenes deriving from two independent cyclization processes of S or OS derivatives
Bis-(6,11-cyclofarnesa-2,7(14)-diene) 126, isolated from an anoxic sulfur-rich sediment, is a unique example of the cycloachillane skeleton.90

The oxidative degradation of hoogianal yielded β-irone, a compound of interest in perfumery due to its violet aroma.91

These triterpenoids exhibited mild toxicity against brine shrimp (Artemia salina).92



A number of sodwanones have been found to possess cytotoxic activity against different cell lines. The cytotoxic activities of sodwanone S (145) were evaluated against 13 human tumor lines.98 Sodwanone V (148) inhibited hypoxia-induced HIF-1 activation in T47D breast tumor cells and PC-3 prostate tumor cells (IC50 15 µM).99

| Compound | R1 | R2 | R3 | Source | |
|---|---|---|---|---|---|
| Yardenone (153) | O | O | H | Ptilocaulis spiculifer,97Axinella species,99Axinella cf. bidderi102 | |
| Yardenone A (154) | O | O | OH | A. cf. bidderi 102 | |
| Yardenone B (155) | H | OH | H | A. cf. bidderi 102 | |
| Dihydroyardenone (156) | H | OH | H | P. spiculifer 97,101 | |
| 12R-Hydroxyyardenone (157) | O | O | OH | Axinella species99 | |


| Compound | R1 | R2 | R3 | R4 | R5 | ||
|---|---|---|---|---|---|---|---|
| 21-Deacetyl-21-oxoraspacionin (161) | OH | H | — | — | OAc | ||
| 15-Deacetyl-21-oxoraspacionin (162) | OH | H | — | — | OH | ||
| 4,21-Dioxoraspacionin (163) | O | O | — | — | OAc | ||
| 10-Acetoxy-4-acetyl-21-oxo-28-hydroraspacionin (164) | OAc | H | O | O | — | ||
| 10-Acetoxy-4-acetyl-28-hydroraspacionin (165) | OAc | H | H | OAc | — | ||
| 10-Acetoxy-28-hydroraspacionin (166) | OH | H | H | OAc | — | ||
| 10-Acetoxy-21-deacetyl-4-acetyl-21-oxo-28-hydroraspacionin (167) | OAc | H | H | OH | — | ||
| 10-Hydroxy-4,21-dioxo-28-hydroraspacionin (168) | — | — | — | — | — | ||
3 Biosynthesis of unusually cyclized triterpenes
Since the postulation of the isoprene biogenetic rule by Ruzicka et al.,8,9 considerable progress has been made in understanding the biogenesis of the more than 100 known triterpene skeletons.4–7,10,105,106 The cyclization mechanism is initiated by protonation of S or OS or other related initiators. The carbocation thus formed triggers a cascade of cyclizations by electrophilic addition to olefins, generating carbocations. The corresponding cyclic cations are stabilized by loss of a proton or by addition of water or other nucleophiles, just after the cyclization or after going through different 1,2-antiperiplanar rearrangements. The interest in this type of mechanism has led to the involvement of molecular biology, genetics, biochemistry, etc. in this topical scientific field, thus more than 30 OSCs and SCs have been characterized. Cloning of different OSCs and SCs has led to the identification of poly- and monofunctional cyclases that yield a single product or various, and experiments of directed mutagenesis have shown that changes in a single amino acid can cause profound alterations in their specificity.27,87,105,107It is currently accepted that in the mechanism of cyclizations105 mediated by OSC and SC, the cyclases provide a template that folds the flexible acyclic substrate and the carbocation intermediates in a series of conformations which define the regio- and stereospecificity of the process. The carbocation intermediates generated are stabilized with electron-rich environments, namely cation-π interactions with aromatic residues of Phe, Tyr, and Trp. This stabilization favours the cyclization kinetics, making the process more selective.52,108,109 Finally, the terminal carbocations are neutralised either by the selective loss of a proton from a basic center of the enzyme or by addition of water.
In this review, we have highlighted summarized schemes of the main biosynthetic routes, either established or proposed, towards unusually cyclized triterpenes. In this context, the most recent advances in the enzymatic synthesis of cyclic triterpenes have been reviewed by Abe.105
3.1 Biosyntheses mediated by OSC and SC. Triterpenes originating from a single cyclization
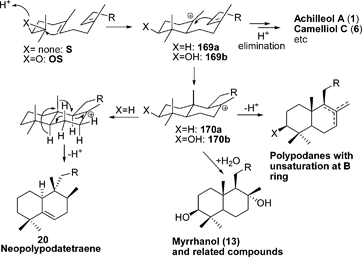 | ||
| Scheme 1 Proposed biosynthesis of achillanes and polypodanes. | ||
It has also been found that several mutants of SHC,21 lanosterol synthase,24 and cycloartenol synthase25,26 produce achilleol A 1 or camelliol C 6. Mutants of Alicyclobacillus acidocaldarius generate achillanes 5, 7 and 8.31
While it has not been demonstrated experimentally that an OSC makes polypodanes, SHC mutants from A. acidocaldarius29,34,38,40,110 produce polypodatetraenes 10 and 11, as well as from the rearranged derivative 20.
 | ||
| Scheme 2 Proposed biosynthesis of malabaricanes. | ||
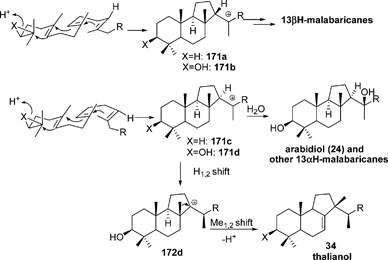
It has been found that two A. thaliana genes, namely At4g15340 and At5g4810, encode OSCs that produce arabidiol 24 and its 20,21-epoxide 25, and thalianol 34 and its 21,22 epoxide 35, respectively.48,51 The mechanism of their formation is rationalized via the tricyclic carbenium ion 168d, which either rearranges to 172d and, by a subsequent H-9 elimination, affords 34, or undergoes stereospecific addition of water to give 24. In an A. acidocaldarius SHC elimination of the residue Gly600, which is located close to the place of formation of ring D, generated a mutant that was incubated with OS to produce malabaricanes 21 and 24 and rearranged malabaricanes 34 and 36, together with achilleol A 1.21 Similarly, a number of malabaricanes and rearranged malabaricanes have been obtained by site-directed mutagenesis of A. acidocaldarius SCH, which includes point mutants of Phe601, Ile261, Tyr420, etc.11,105
Isomalabaricanes, the other large group of 6,6,5 tricyclic triterpenes, have the opposite stereochemistry at C-8 and C-9 to that of the malabaricanes. The biogenetic origin of these compounds is thought to involve cyclizations of chair-boat conformations of S or OS leading to the tricyclic carbocations 173a,b. Deprotonation of H-13, oxidation on C-12 to carbonyl group, and different dehydrogenations and oxidations, mainly on the side chain, generate the great variety of natural isomalabaricanes (Scheme 3). Tyr510 mutants were proved to afford isomalabaricatrienol 48, which is considered a putative precursor of isomalabaricane triterpenoids in sponges.23
 | ||
| Scheme 3 Proposed biosynthesis of isomalabaricanes. | ||
3.2 Biosynthesis of triterpenes via processes of cyclization of S or OS towards polycyclic triterpenes and subsequent retrocyclization reactions
 | ||
| Scheme 4 Proposed biosynthesis of iridals. | ||
Reduction of the aldehyde group, oxidation by monooxygenases at C-1 and/or C-25, C-10, C-16, C-17, C-26, and C-13, or dehydrogenation on the side chain, would give rise to many of the iridals described. In support of Marner's hypothesis, it has recently been established that the A. thaliana gene At5g42600 encodes a new OSC (called marneral synthase)72 that catalyses the formation of the iridal marneral 77 and its reduction product marnerol 78. Analysis of the sequence alignments for marneral synthase showed that this cyclase contains Gly at the position corresponding to Cys456 in other OSCs. The crystal structure of HLC showed that Cys456 is linked by a hydrogen bond to the Asp455 catalytic center which protonates the oxirane. This change may facilitate the mobility of Asp455 and would then help the deprotonation of H-5 and the fragmentation of the A ring.
 | ||
| Scheme 5 Proposed seco-tetracyclic triterpene biosynthesis. | ||
 | ||
| Scheme 6 Proposed biosynthesis of seco-pentacyclic triterpenes. | ||
3.3 Biosynthesis of triterpenes involving two independent cyclization processes
Cycloiridals are considered to be derived from iridals. It has been suggested that the biosynthesis of hoogianal proceeds via the corresponding iridals, which suffer a S-adenosylmethionine-mediated monocyclization initiated on the terminal double bond of the farnesyl side chain (Scheme 7). This proposal may well account for the presence of the additional carbon in these homotriterpenes.111 | ||
| Scheme 7 Proposed biosynthesis of cycloiridals. | ||
 | ||
| Scheme 8 Proposed biosynthesis of yardenones. | ||
 | ||
| Scheme 9 Proposed biosynthesis of onoceranes. | ||
 | ||
| Scheme 10 Proposed biosynthesis of siphonellinol B and dahabinone. | ||
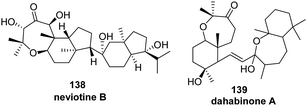
A more complex pathway seems to be involved in the biogenesis of neviotine B (138) (Scheme 11).94 Thus, starting from the same diepoxide (180), a first cyclization could occur to give the intermediate tricyclic structure 181. Then, the epoxide resulting derived from the methylene at C-15 could undergo a proton-induced opening, leading after a cascade cyclization process to the neviotane skeleton.
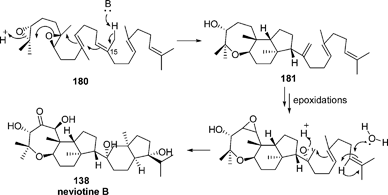 | ||
| Scheme 11 Proposed biosynthesis of neviotine B. | ||
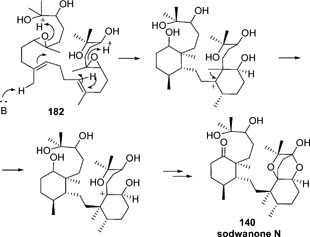 | ||
| Scheme 12 Proposed biosynthesis of sodwanone N. | ||
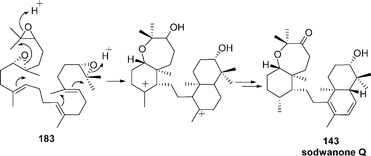 | ||
| Scheme 13 Proposed biosynthesis of sodwanone Q. | ||
 | ||
| Scheme 14 Proposed shipolenol G biosynthesis. | ||
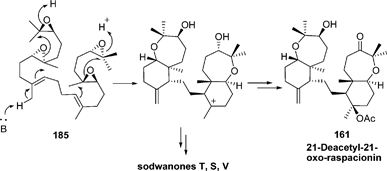 | ||
| Scheme 15 Proposed biosynthesis of raspacionin. | ||
It has been proposed that the biosynthesis of abudinols involves the same tetraepoxyderivative of squalene (185). Once the first tandem cyclization takes place, abstraction of H-14 could trigger the generation of the tricyclic moiety (Scheme 16).
 | ||
| Scheme 16 Proposed biosynthesis of abudinol. | ||
Kashman et al. proposed that abudinol B (159) could be involved in the biosynthesis of muzitone (160).94
4 Chemical syntheses of unusually cyclized triterpenes
Although a number of impressive syntheses of unusually cyclized triterpenes has been achieved in the last ten years,113 we have included here only the syntheses of compounds whose occurrence has been reported since 1998 (shown in section 2).4.1 Achillanes
The synthesis of achilleol A 1 was planned on the basis of a C15-C15 convergent strategy (Scheme 17).114 The monocyclic sesquiterpenic elegansidiol (187) was obtained through a Ti-mediated radical cyclization of oxirane 186, prepared from commercially available geranylacetone. Alcohol 175 was converted into allylic bromide which was then coupled with farnesylsulfone. Desulfonation with NaHg in MeOH and deprotection of the silyl ether with TBAF led to achilleol A 1.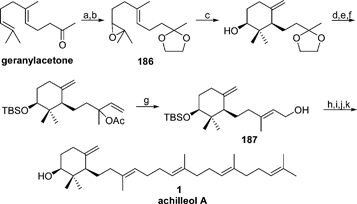 | ||
| Scheme 17 Reagents and conditions: (a) Ethylene glycol, TsOH, 93%; (b) NBS, K2CO3, 71%; (c) Cp2TiCl, 65%; (d) CeCl3, NaI, 95%; (e) TBSCl, imidazole, DMAP, 72%; (f) i. allylmagnesium bromide; ii. Ac2O, 68%; (g) i. PdCl2(MeCN)2; ii. K2CO3, MeOH, 88%; (h) PBr3, DMAP, 75%; (i) nBuLi, PhSO2-farnesyl, 74%; (j) NaHg, MeOH, 55%; (k) TBAF, 95%. | ||
Very recently, our research group carried out the first total synthesis of (−)-achilleol B,115 an achievement that led us to reassign the relative stereochemistry of the interannular junction as cis. The key steps employed in our synthesis were an enantioselective Robinson annelation for the construction of the bicyclic moiety,116 and a Ti(III)-mediated cyclization of a chiral monoepoxide to construct the monocyclic moiety.
Scheme 18 summarizes the formation of the bicyclic synthon. Thus, based on the work of Heathcock,117 ketone 188 was prepared from dimedone. Treatment of 188 with (S)-phenylethylamine led to enamine 189 which reacts with freshly prepared Nazarov reagent to obtain a mixture of the desired keto ester and its acyclic precursor 190, which was converted into the keto ester by treatment with KF in MeOH. Reduction with H2-Pd/C and treatment with the corresponding enol triflate with MeLi in the presence of CuBr·SMe2 gave the cis-decalin 192. Subsequent reduction with LiAlH4 and treatment with PBr3 led to the allylic bromide 193.
 | ||
| Scheme 18 Reagents and conditions: (a) i. NaOH, CH3I, H2O; ii. LAH, Et2O; (b) PDC, DCM; (c) H2, Pd/C, EtOAc; (d) (S)-phenylethylamine, PhH, 74%; (e) methyl 3-oxo-4-pentenoate, PhH, 41%; (f) KF, MeOH, 99%; (g) H2, Pd/C, EtOAc, 91%; (h) i. Tf2O, iPr2EtN; ii. CuBr·SMe2, MeLi, 91%; (i) i. DIBALH, 94%; ii. PBr3, 90%. | ||
The monocyclic synthon was synthesized starting from geranylacetone (Scheme 19). Sharpless asymmetric dihydroxylation of this acyclic precursor led to the corresponding asymmetric diol. Protection of the carbonyl group, and treatment of the corresponding diol with mesyl chloride and subsequent treatment with base gave rise to oxirane 194. Ti(III)-mediated cyclization of this epoxide led after deprotection of the ketyl group led to ketoalcohol 195. Application of the Horner–Wadsworth–Emmons protocol to the corresponding silyl derivative provided the requisite two-carbon homologation. The corresponding unsaturated ester was converted to bicyclic sulfone 196 following straightforward transformations.
 | ||
| Scheme 19 Reagents and conditions: (a) i. AD-mix-β, CH3SO2NH2, tBuOH/H2O, 0 °C, 67%, ii. ethylene glycol, TsOH, 82%; iii. MsCl, Py, DMAP, K2CO3, MeOH, 74%, (b) i. Cp2TiCl, 77%; ii. CeCl3, NaI, 95%; (c) i. TBSCl, imidazole, DMAP, 91%; ii. diethyl phosphonoacetate, NaH, DIBALH, 74%; iii. PBr3, DMAP, 75%; iii. NaSO2Ph, DMF, 72%. | ||
Finally, the target (−)-achilleol B was efficiently assembled as shown in Scheme 20.
4.2 Polypodanes
Akita et al. reported the synthesis of (+)-α-polypodatetraene 10 and (+)-γ-polypodatetraene 11.118 Starting from (+)-albicanol and (−)-drimenol respectively, the hydrocarbon chain was lengthened following straightforward transformations, including a Wittig reaction (Scheme 21). The subsequent formation of sulfone 197 enabled coupling with geranyl bromide to give, after desulfonation, the desired final products.119 | ||
| Scheme 20 Reagents and conditions: (a) i. nBuLi, THF, 71%; (b) i. Li, EtNH2, THF; ii. TBAF, 86%. | ||
 | ||
| Scheme 21 | ||
4.3 Malabaricanes
In our laboratories malabaricanes 22 and 23 were prepared through a titanocene-catalyzed cyclization of 2,3-oxidosqualene, mimicking the enzyme lanosterol synthase.120 Treatment of OS with a catalytic quantity of Cp2TiCl2 led to a mixture of 13α- and 13β- malabaricanes in 39% yield (Scheme 22).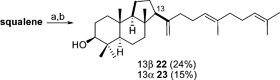 | ||
| Scheme 22 Reagents and conditions: (a) i. NBS, MeOH/H2O; ii. K2CO3, MeOH; (b) Cl2TiCp2, Mn, TMSCl, collidine, THF, 39%. | ||
4.4 Marneral
In 2008, Arseniyadis et al. reported the convergent synthesis of (+)-marneral (Scheme 23).121 This achievement was based on the B-alkyl Suzuki–Miyaura coupling of vinyl iodide 201 and the corresponding monocyclic counterpart 200. Central to the elaboration of the monocyclic moiety was the mercury-catalyzed Claisen rearrangement of the allylic alcohol 199, efficiently prepared from commercially available (R)-pulegone. X-Ray analysis of intermediate 198a established the relative configuration of the final product. | ||
| Scheme 23 Reagents and conditions: (a) LDA, ClCO2Et; (b) NaH-MeI (40%, 2 steps); (c) LiAlH4; (d) AcOH (75%); (e) TBSCl (88%); (f) vinyl ether, Hg(OAc)2, sealed tube, 190 °C, 2 d (80%); (g) Wittig (92%); (h) 9-BBN, NaOH-H2O2 (80%); (i) Ac2O-DMAP (90%); (j) HF-MeCN; (k) Swern; (l) Wittig (79%, 3 steps); (m) K2CO3, MeOH, (n) TBSCl, DMAP (77%, 2 steps), (o) 9-BBN, THF, (p) Pd(dppf)Cl2, AsPh3, Cs2CO3, H2O, DMF, 25 °C, 12 h (67%), (q) TBAF (87%); (r) Swern (96%). | ||
4.5 ent-Abudinol B
McDonald et al.122 carried out the first synthesis of ent-abudinol B inspired by the biosynthesis proposed for abudinols A (158) and B (159), using cascade cyclizations of di-epoxides coupled to the corresponding enol-silane 202 or ene-propargylsilane 204 to construct the different carbocycles of the structure (Scheme 24). The key step was the Pd-catalyzed cross-coupling of ABC (205) and DE (203) units, which proceeded in 70% yield. Hydrogenation of the diene occurred in low yield (30%). Final desilylation provided the structure corresponding to ent-abudinol B. | ||
| Scheme 24 Reagents and conditions: (a) NaBH4, MeOH, 0 °C, 90%; (b) Shi catalyst, Oxone, K2CO3, Na2EDTA, DMM/MeCN/H2O, 0 °C, 95%; (c) SO3-pyridine, DMSO, Et3N, DCM, 0 °C, 95%; (d) KHMDS, TBSCl, THF, −78 °C, 87%; (e) TBSOTf, DTBMP, DCM, −78 °C, 60%; (f) KHMDS, THF, −78 °C, then PhNTf2, −78 °C, 95%; (g) Ph3P, NBS, THF, 0 °C, then cat. Bu4NI, NaSO2Tol, 99%; (h) nBuLi, THF, −78 to −40 °C, then 1-bromo-4-trimethylsilyl-2-butyne, −78 to 20 °C, 92%; (i) Shi catalyst, Oxone, K2CO3, Na2EDTA, DMM/MeCN/H2O, 0 °C, 76%; (j) Cl2Pd(dppp), LiEt3BH, THF, 0 °C, 71%; (k) TMSOTf, DTBMP, DCM, −78 °C, 75%; (l) Bu4NF, THF, 99%; (m) O3, DCM, −78 °C, then Me2S, −78 to 20 °C, 88%; (f) 95%; (n) 205, Cl2Pd(PPh3)2, Ph3P, PhOK, bis(neopentylglycolato)diboron, toluene, 50 °C, then 203, Cl2Pd(dppf), K3PO4, DMF, 80 °C, 70%; (o) 10% Pd/C, toluene, H2, 0 °C, 30%; (p) Bu4NF, THF, 60 °C, 84%. | ||
In 2008 the same research group reported a total synthesis of the same triterpene ent-abudinol (Scheme 25).123 The synthesis relies on Lewis acid promoted tandem oxa- and carbacyclizations of the squalene-like substrate 209. This 29-carbon substrate was prepared by coupling of diepoxybromide 206 with the Brook rearrangement product of the lithium alkoxide of 207. Compounds 206 and 208 were prepared from farnesyl acetate and geranyl bromide, respectively (Scheme 25). Me3SiOTf-promoted tandem cyclization of 209 afforded tricyclic ketone 210 as the major product. Ketone 210 was converted to diepoxide 211 by methylenation and double enantioselective epoxidation of the trisubstituted double bonds. Again, trimethylsilyl triflate promoted a tandem oxa- and carbocyclization of 211 to provide ent-abudinol in modest yield.
 | ||
Scheme 25
Reagents and conditions: a) D-Epoxone (0.5 equiv), Oxone, pH 10.5 buffer, DMM/MeCN/H2O, −5 °C, 54%; b) K2CO3, MeOH, 99%; c) MsCl, Et3N, −40 °C, then LiBr, THF, 0 °C, 60%; d) LDA, THF, −30 °C, then 207; e) NaOAc, HOAc, 84% (2 steps); f) H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHMgCl, Et2O, 0 °C, then HCl, 88%; g) nBuLi, hexane/THF, −78 °C, then 206, 50%. DMM = dimethoxymethane; h) Me3SiOTf, DTBMP, CH2Cl2, −78 °C, then Bu4NF, 50%; i) Ph3PMeBr, KOtBu, benzene, 80 °C, 30% (+55% C-14 epimer); j) D-Epoxone (0.5 equiv), Oxone, pH 10.5 buffer, DMM/MeCN/H2O,−5 °C, 50%; k) Me3SiOTf, DTBMP, CH2Cl2, −78 °C, then Bu4NF. CHMgCl, Et2O, 0 °C, then HCl, 88%; g) nBuLi, hexane/THF, −78 °C, then 206, 50%. DMM = dimethoxymethane; h) Me3SiOTf, DTBMP, CH2Cl2, −78 °C, then Bu4NF, 50%; i) Ph3PMeBr, KOtBu, benzene, 80 °C, 30% (+55% C-14 epimer); j) D-Epoxone (0.5 equiv), Oxone, pH 10.5 buffer, DMM/MeCN/H2O,−5 °C, 50%; k) Me3SiOTf, DTBMP, CH2Cl2, −78 °C, then Bu4NF. | ||
5 Conclusions
There has been an increase in the description of new natural triterpenes derived from partial cyclization of squalene or 2,3-oxidosqualene in the last few years. In addition, the confirmation of the existence of OSCs specific for the formation of these compounds seems to indicate that these molecules are more widely distributed in Nature than might be expected a priori. At the same time, the biosynthetic proposals that suggest new retrocyclization mechanisms are involved in the biosynthesis of these molecules seem to widen the range of structural diversity in these irregular triterpenes, a possibility that should be explored in the future.Marine organisms, mainly sponges, prove to be a remarkable source of triterpenes, which are structurally distant from those having a terrestrial origin. Many of these compounds, which often possess interesting biological properties, derive from cyclization of polyepoxides of squalene or 2,3-oxidosqualene. In our opinion, the scientific research into these unusually cyclized triterpenes constitutes a valid strategy for the advance in the understanding of the metabolism of natural products. This research should also contribute to the discovery of new bioactive molecules with potential applications.
6 References
- J. D. Connolly and R. A. Hill, Dictionary of Terpenoids, Chapman & Hall, London, 1991 Search PubMed.
- J. Buckingham, in Dictionary of Natural Products, Chapman & Hall, London, 1996 Search PubMed.
- J. D. Connolly and R. A. Hill, Triterpenoids, Nat. Prod. Rep., 2007, 24, 465 RSC and previous reviews.
- R. Xu, G. C. Fazio and S. P. Matsuda, Phytochemistry, 2004, 65, 261 CrossRef CAS.
- I. Abe, M. Rohmer and G. D. Prestwich, Chem. Rev., 1993, 93, 2189 CrossRef CAS.
- I. Abe and G. D. Prestwich, ‘Squalene epoxidase and oxidosqualene: lanosterol cyclase. Key enzymes in cholesterol biosynthesis’, in Comprehensive Natural Products, ed. D. H. R. Barton and K. Nakanishi, Elsevier, Oxford, UK, 1999, p. 267 Search PubMed.
- K. Poralla, ‘Cycloartenol and other triterpene cyclases’, in Comprehensive Natural Products, ed. D. H. R. Barton and K. Nakanishi, Elsevier, Oxford, UK, 1999, p. 299 Search PubMed.
- A. Eschenmoser, L. Ruzicka, O. Jeger and D. Arigoni, Helv. Chim. Acta, 1955, 38, 1890 CrossRef CAS.
- A. Eschenmoser and D. Arigoni, Helv. Chim. Acta, 2005, 88, 3011 CrossRef CAS.
- K. U. Wendt, G. E. Schulz, E. J. Corey and D. R. Liu, Angew. Chem., Int. Ed., 2000, 39, 2812 CrossRef CAS.
- T. Hoshino and T. Sato, Chem. Commun., 2002, 291 RSC.
- M. J. R. Segura, B. E. Jackson and S. P. T. Matsuda, Nat. Prod. Rep., 2003, 20, 304 RSC.
- A. F. Barrero, E. J. Alvarez-Manzaneda and R. Alvarez-Manzaneda, Tetrahedron Lett., 1989, 30, 3351 CrossRef CAS.
- T. Akihisa, J. Ogihara, J. Kato, K. Yasukawa, M. Ukiya, S. Yamanouchi and K. Oishi, Lipids, 2001, 36, 507 CAS.
- S. Streiff, N. Ribeiro, Z. Wu, E. Gumienna-Kontecka, M. Elhabiri, A. Albrecht-Gary, G. Ourisson and Y. Nakatani, Chem. Biol., 2007, 14, 313 CrossRef CAS.
- N. Ribeiro, S. Streiff, D. Heissler, M. Elhabiri, A. Albrecht-Gary, M. Atsumi, M. Gotoh, L. Desaubry, Y. Nakatani and G. Ourisson, Tetrahedron, 2007, 63, 3395 CrossRef CAS.
- A. F. Barrero, A. Haídour, M. Muñoz-Dorado, M. Akssira, A. Sesqui and I. Mansour, Phytochemistry, 1237, 1998, 48 Search PubMed.
- A. F. Barrero, E. J. Alvarez-Manzaneda, M. M. Herrador, R. Alvarez-Manzaneda, J. Quilez, R. Chahboun, P. Linares and A. Rivas, Tetrahedron Lett., 1999, 40, 8273 CrossRef CAS.
- T. Akihisa, K. Arai, Y. Kimura, K. Koike, W. C. M. C. Kokke, T. Shibata and T. Nikaido, J. Nat. Prod., 1999, 62, 265 CrossRef CAS.
- T. Akihisa, K. Koike, Y. Kimura, N. Sashida, T. Matsumoto, M. Ukiya and T. Nikaido, Lipids, 1999, 34, 1151 CrossRef CAS.
- T. Hoshino, K. Shimizu and T. Sato, Angew. Chem., Int. Ed., 2004, 43, 6700 CrossRef CAS.
- T. K. Wu and C. H. Chang, ChemBioChem, 2004, 5, 1712 CrossRef CAS.
- S. Lodeiro, W. K. Wilson, H. Shan and S. P. Matsuda, Org. Lett., 2006, 8, 439 CrossRef CAS.
- B. M. Joubert, L. Hua and S. P. T. Matsuda, Org. Lett., 2000, 2, 339 CrossRef CAS.
- S. P. T. Matsuda, L. B. Darr, E. A. Hart, J. B. R. Herrera, K. E. McCann, M. M. Meyer, J. Pang and H. G. Schepmann, Org. Lett., 2000, 2, 2261 CrossRef CAS.
- M. M. Meyer, R. Xu and S. P. T. Matsuda, Org. Lett., 2002, 4, 1395 CrossRef CAS.
- M. D. Kolesnikova, W. K. Wilson, D. A. Lynch, A. C. Obermeyer and S. P. T. Matsuda, Org. Lett., 2007, 9, 5223 CrossRef CAS.
- S. Lodeiro, Q. Xiong, W. K. Wilson, M. D. Kolesnikova, C. S. Onak and S. P. T. Matsuda, J. Am. Chem. Soc., 2007, 129, 11213 CrossRef CAS.
- T. Sato and T. Hoshino, Biosci., Biotechnol., Biochem., 2001, 65, 2233 CrossRef CAS.
- K. T. Lee, S. J. Koo, S. H. Jung, J. Choi, H. J. Jung and H. J. Park, Arch. Pharmacol Res., 2002, 25, 820 Search PubMed.
- T. Sato, S. Sasahara, T. Yamakami and T. Hoshino, Biosci., Biotechnol., Biochem., 2002, 66, 1660 CrossRef CAS.
- K. Shiojima, Y. Arai, K. Masuda, T. Kamada and H. Ageta, Tetrahedron Lett., 1983, 24, 5733 CrossRef CAS.
- I. Kimura, M. Yoshikawa, S. Kobayashi, Y. Sugihara, M. Suzuki, H. Oominami, T. Murakami, H. Matsuda and V. Doiphode, Bioorg. Med. Chem. Lett., 2001, 11, 985 CrossRef CAS.
- C. Pale-Grosdemange, T. Merkofer, M. Rohmer and K. Poralla, Tetrahedron Lett., 1999, 40, 6009 CrossRef CAS.
- T. Sato and T. Hoshino, Biosci., Biotechnol., Biochem., 2001, 65, 2233 CrossRef CAS.
- T. Hoshino and T. Sato, Chem. Commun., 1999, 2005 RSC.
- T. Sato and T. Hoshino, Biosci., Biotechnol., Biochem., 2001, 65, 2233 CrossRef CAS.
- C. Full and K. Poralla, FEMS Microbiol. Lett., 2000, 183, 221 CrossRef CAS.
- T. Sato, S. Sasahara, T. Yamakami and T. Hoshino, Biosci., Biotechnol., Biochem., 2002, 66, 1660 CrossRef CAS.
- S. Schmitz, C. Full, T. Glaser, K. Albert and K. Poralla, Tetrahedron Lett., 2001, 42, 883 CrossRef CAS.
- L. Nguyen and L. Harrison, Phytochemistry, 1999, 50, 471 CrossRef CAS.
- H. Matsuda, T. Morikawa, S. Ando, H. Oominami, T. Murakami, I. Kimura and M. Yoshikawa, Chem. Pharm. Bull., 2004, 52, 1200 CrossRef CAS.
- H. Matsuda, T. Morikawa, S. Ando, H. Oominami, T. Murakami, I. Kimura and M. Yoshikawa, Bioorg. Med. Chem., 2004, 12, 3037 CrossRef CAS.
- J. L. Giner, S. Rocchetti, S. Neunlist, M. Rohmer and D. Arigoni, Chem. Commun., 2005, 3089 RSC.
- T. Hoshino, M. Kouda, T. Abe and S. Ohashi, Biosci., Biotechnol., Biochem., 1999, 63, 2038 CAS.
- T. Merkofer, C. Pale-Grosdemange, K. U. Wendt, M. Rohmer and K. Poralla, Tetrahedron Lett., 1999, 40, 2121 CrossRef CAS.
- A. Behrens, P. Schaeffer, S. Bernasconi and P. Albrecht, Org. Geochem., 1999, 30, 379 CrossRef CAS.
- T. Xiang, M. Shibuya, Y. Katsube, T. Tsutsumi, M. Otsuka, H. Zhang, K. Masuda and Y. Ebizuka, Org. Lett., 2006, 8, 2835 CrossRef CAS.
- H. L. Ziegler, D. Stark, J. Christensen, C. E. Olsen, A. A. Sittie and J. W. Jaroszewski, J. Nat. Prod., 2002, 65, 1764 CrossRef CAS.
- B. Sontag, R. Froede, M. Bross and W. Steglich, Eur. J. Org. Chem., 1999, 255 CrossRef CAS.
- G. C. Fazio, R. Xu and S. P. T. Matsuda, J. Am. Chem. Soc., 2004, 126, 5678 CrossRef CAS.
- N. Morikubo, Y. Fukuda, K. Ohtake, N. Shinya, D. Kiga, K. Sakamoto, M. Asanuma, H. Hirota, S. Yokoyama and T. Hoshino, J. Am. Chem. Soc., 2006, 128, 13184 CrossRef CAS.
- T. Hoshino, M. Kouda, T. Abe and T. Sato, Chem. Commun., 2000, 1485 RSC.
- K. M. Meragelman, T. C. McKee and M. R. Boyd, J. Nat. Prod., 2001, 64, 389 CrossRef CAS.
- N. Oku, S. Matsunaga, S. Wada, S. Watabe and N. Fusetani., J. Nat. Prod., 2000, 63, 205 CrossRef CAS.
- M. Fouad, R. A. Edrada, R. Ebel, V. Wray, W. E. G. Muller, W. H. Lin and P. Proksch, J. Nat. Prod., 2006, 69, 211 CrossRef CAS.
- J. N. Tabudravu and M. Jaspars, J. Nat. Prod., 2001, 64, 813 CrossRef CAS.
- T. K. Wu, Y. T. Liu, C. H. Chang, M. T. Yu and H. J. Wang, J. Am. Chem. Soc., 2006, 128, 6414 CrossRef CAS.
- T. K. Wu, Y. T. Liu and C. H. Chang, ChemBioChem, 2005, 6, 1177 CrossRef CAS.
- T. K. Wu, Y. T. Liu, F. H. Chiu and C. H. Chang, Org. Lett., 2006, 8, 4691 CrossRef CAS.
- T. K. Wu and C. H. Chang, ChemBioChem, 2004, 5, 1712 CrossRef CAS.
- S. Lodeiro, W. K. Wilson, H. Shan and S. P. Matsuda, Org. Lett., 2006, 8, 439 CrossRef CAS.
- A. Zampella, M. V. D'Auria, C. Debitus and J. Menou, J. Nat. Prod., 2000, 63, 943 CrossRef CAS.
- S. Aoki, M. Sanagawa, Y. Watanabe, A. Setiawan, M. Arai and M. Kobayashi, Bioorg. Med. Chem., 2007, 15, 4818 CrossRef CAS.
- S. Tang, Y. Pei, H. Fu, Z. Deng, J. Li, P. Proksch and W. Lin, Chem. Pharm. Bull., 2006, 54, 4 CrossRef CAS.
- J. A. Clement, M. Li, S. M. Hecht and D. G. I. Kingston, J. Nat. Prod., 2006, 69, 373 CrossRef CAS.
- D. Tasdemir, G. C. Mangalindan, G. P. Concepcion, S. M. Verbitski, S. Rabindran, M. Miranda, M. Greenstein, J. N. A. Hooper, M. K. Harper and C. M. Ireland, J. Nat. Prod., 2002, 65, 210 CrossRef CAS.
- H. Lin, Z. Wang, J. Wu, N. Shi, H. Zhang, W. Chen, S. L. Morris-Natschke and A. Lin, J. Nat. Prod., 2007, 70, 1114 CrossRef CAS.
- F. Lu, Z. Deng, J. Li, H. Fu, R. W. M. van Soest, P. Proksch and W. Lin, J. Nat. Prod., 2004, 67, 2033 CrossRef.
- W. Zhang and C. Che, J. Nat. Prod., 2001, 64, 1489 CrossRef CAS.
- N. Wang, Z. Li, D. Song, W. Li, H. Fu, K. Koike, Y. Pei, Y. Jing and H. Hua, J. Nat. Prod., 2008, 71, 990 CrossRef CAS.
- Q. Xiong, W. K. Wilson and S. P. Matsuda, Angew. Chem., Int. Ed., 2006, 45, 1285 CrossRef CAS.
- M. Lamshoeft and F. J. Marner, Nat. Prod. Res., 2005, 19, 57 CrossRef.
- J. Bonfils, F. J. Marner and Y. Sauvaire, Phytochemistry, 1998, 48, 751 CrossRef CAS.
- M. Lamshoft, H. Schmickler and F. Marner, Eur. J. Org. Chem., 2003, 4, 727 CrossRef.
- L. Taillet, J. Bonfils, F. Marner and Y. Sauvaire, Phytochemistry, 1999, 52, 1597 CrossRef CAS.
- R. Fang, P. J. Houghton, C. Luo and P. J. Hylands, Phytochemistry, 2007, 68, 1242 CrossRef CAS.
- H. Ito, S. Onoue, Y. Miyake and T. Yoshida, J. Nat. Prod., 1999, 62, 89 CrossRef CAS.
- I. Orhan, B. Sener, T. Hashimoto, Y. Asakawa, M. Özgüven and F. Ayanoglu, Fitoterapia, 2002, 73, 316 CrossRef CAS.
- K. Takahashi, Y. Hoshino, S. Suzuki, Y. Hano and T. Nomura, Phytochemistry, 2000, 53, 925 CrossRef CAS.
- K. Takahashi, S. Suzuki, Y. Hano and T. Nomura, Biol. Pharm. Bull., 2002, 25, 432 CrossRef CAS.
- K. Effers, B. Scholz, C. Nickel, B. Hanisch and F. Marner, Eur. J. Org. Chem., 1999, 11, 2793 CrossRef.
- F. J. Marner, B. Singab Abdel Nasser, M. Al-Azizi Mohamed, A. El-Emary Nasr and M. Schafer, Phytochemistry, 2002, 60, 301 CrossRef CAS.
- T. Akihisa, K. Yasukawa, Y. Kimura, S. Yamanouchi and T. Tamura, Phytochemistry, 1998, 48, 301 CrossRef.
- M. Ukiya, T. Akihisa, H. Tokuda, K. Koike, Y. Kimura, T. Asano, S. Motohashi, T. Nikaido and H. Nishino, J. Nat. Prod., 2003, 66, 1476 CrossRef CAS.
- L. U. Roman, D. Guerra-Ramirez, G. Moran, I. Martinez, J. D. Hernandez, C. M. Cerda-Garcia-Rojas, J. M. Torres-Valencia and P. Joseph-Nathan, Org. Lett., 2004, 6, 173 CrossRef.
- M. Shibuya, T. Xiang, Y. Katsube, M. Otsuka, H. Zhang and Y. Ebizuka, J. Am. Chem. Soc., 2007, 129, 1450 CrossRef CAS.
- W. A. Shah, M. Y. Dar and M. A. Qurishi, Chem. Nat. Compd., 2004, 40, 30 CrossRef CAS.
- A. F. Barrero, R. E. A. Manzaneda, R. R. A. Manzaneda, R. S. Arseniyadis and E. Guittet, Tetrahedron, 1990, 46, 8161 CrossRef CAS.
- A. Behrens, P. Schaeffer, S. Bernasconi and P. Albrecht, Geochim. Cosmochim. Acta, 2000, 64, 3327 CrossRef CAS.
- F. J. Marner and B. Hanisch, Helv. Chim. Acta, 2001, 84, 933 CrossRef CAS.
- T. Tanaka, M. Ishibashi, H. Fujimoto, E. Okuyama, T. Koyano, T. Kowithayakorn, M. Hayashi and K. Komiyama, J. Nat. Prod., 2002, 65, 1709 CrossRef CAS.
- U. Shmueli, S. Carmely, A. Groweiss and Y. Kashman, Tetrahedron Lett., 1981, 22, 709 CrossRef CAS.
- Y. Kashman and A. Rudi, Phytochemistry Rev., 2004, 3, 309 Search PubMed.
- Y. Kashman, T. Yosief and S. Carmeli, J. Nat. Prod., 2001, 64, 175 CrossRef CAS.
- S. Jain, S. Lapookhieo, Z. Shi, L. Fu, S. Akiyama, Z. Chen, D. T. Youseff, R. W. M. van Soest and K. A. El Sayed, J. Nat. Prod., 2007, 70, 928 CrossRef CAS.
- A. Rudi, T. Yosief, M. Schleyer and Y. Kashman, Tetrahedron, 1999, 55, 5555 CrossRef CAS.
- C. Funel-Le Bon, F. Berrue, O. P. Thomas, F. Reyes and P. Amade, J. Nat. Prod., 2005, 68, 1284 CrossRef.
- J. Dai, J. A. Fishback, Y.-D. Zhou and D. G. Nagle, J. Nat. Prod., 2006, 69, 1715 CrossRef CAS.
- A. Rudi, I. Goldberg, Z. Stein, Y. Kashman, Y. Benayahu, M. Schleyer and M. D. García-Gravalos, J. Nat. Prod., 1995, 58, 1702 CrossRef CAS.
- A. Rudi, Z. Stein, I. Goldberg, T. Yosief, Y. Kashman and M. Schleyer, Tetrahedron Lett., 1998, 39, 1445 CrossRef CAS.
- I. Carletti, C. Long, C. Funel and P. Amade, J. Nat. Prod., 2003, 66, 25 CrossRef CAS.
- M. L. Ciavatta, G. Scognamiglio, E. Trivellone, T. Bisogno and G. Cimino, Tetrahedron, 2002, 58, 4943 CrossRef CAS.
- R. Puliti, E. Trivellone, A. Crispino, G. Cimino, C. M. Mattia and L. Mazzarella, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, C47, 2609 CrossRef.
- I. Abe, Nat. Prod. Rep., 2007, 24, 1311 RSC.
- P. Manitto, Biosynthesis of Natural Products, Ellis Harwood, Chichester, 1981, pp. 213–313 Search PubMed.
- D. R. Phillips, J. M. Rasbery, B. Bartel and S. P. T. Matsuda, Curr. Opin. Plant Biol., 2006, 9, 305 CrossRef CAS.
- D. A. Dougherty, Science, 1996, 271, 163 CrossRef CAS.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS.
- C. Full, FEBS Lett., 2001, 509, 361 CrossRef CAS.
- F. J. Marner, Curr. Org. Chem., 1997, 1, 153 CAS.
- L. Jaenicke and F. J. Marner, Pure Appl. Chem., 1990, 62, 1365 CAS.
- Y. Mi, J. V. Schreiber and E. J: Corey, J. Am. Chem. Soc., 2002, 124, 11290 CrossRef CAS.
- A. F. Barrero, J. M. Cuerva, E. J. Álvarez-Manzaneda, J. E. Oltra and R. Chahboun, Tetrahedron Lett., 2002, 43, 2793 CrossRef CAS.
- J. F. Arteaga, V. Domingo, J. F. Quílez del Moral and A. F. Barrero, Org. Lett., 2008, 10, 1723 CrossRef CAS.
- A. F. Barrero, S. Arseniyadis, J. F. Quílez del Moral, M. M. Herrador and A. Rosellón, Synlett, 2005, 5, 789 CrossRef.
- J. E. Ellis, J. S. Dutcher and C. H. Heathcock, J. Org. Chem., 1976, 41, 2670 CrossRef CAS.
- M. Kinoshita, M. Ohtsuka, D. Nakamura and H. Akita, Chem. Pharm. Bull., 2002, 50, 930 CrossRef CAS.
- M. Mohri, H. Kinoshita, K. Inomata and H. Kotake, Chem. Lett., 1985, 4, 451 CrossRef.
- J. Justicia, A. Rosales, E. Buñuel, J. L. Oller-López, M. Valdivia, A. Haïdour, J. E. Oltra, A. F. Barrero, D. J. Cárdenas and J. M. Cuerva, Chem. Eur. J., 2004, 10, 1778 CrossRef CAS.
- A. Corbu, M. Perez, M. Aquino, P. Retailleau and S. Arseniyadis, Org. Lett., 2008, 10, 2853 CrossRef CAS.
- R. Tong, J. C. Valentine, F. E. McDonald, R. Cao, X. Fang and K. I. Hardcastle, J. Am. Chem. Soc., 2006, 129, 1050.
- R. Tong and F. E. McDonald, Angew. Chem., Int. Ed., 2008, 47, 4377 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |