Synthesis and characterisation of bulky guanidines and phosphaguanidines: precursors for low oxidation state metallacycles†
Guoxia
Jin
b,
Cameron
Jones
*a,
Peter C.
Junk
a,
Kai-Alexander
Lippert
b,
Richard P.
Rose
ab and
Andreas
Stasch
a
aSchool of Chemistry, PO Box 23, Monash University, 3800, VIC, Australia
bSchool of Chemistry, Main Building, Cardiff University, Cardiff, UK CF10 3AT
First published on 9th October 2008
Abstract
Reactions of alkali metal amides or phosphides with the bulky carbodiimide, ArN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNAr (Ar = C6H3Pri2-2,6), followed by aqueous work-ups, have yielded several guanidines, ArNC(NR2)N(H)Ar (R = cyclohexyl (GisoH) or Pri (PrisoH); NR2 = cis-NC5H8Me2-2,6 (PipisoH)), a bifunctional guanidine, {ArNCN(H)Ar}2{μ-N(C2H4)2N} (Pip(GisoH)2), and two phosphaguanidines, ArNC(PR2)N(H)Ar (R = cyclohexyl (CyP-GisoH) or Ph (PhP-GisoH)). A very bulky guanidine, ArNC{N(Ar)SiMe3}N(H)Ar (ArSi-Giso), and an aryl coupled bifunctional guanidine, {ArN(H)C(NPri2)NC6H2Pri2-2,6-}2 (PrisoH)2, have been prepared by other routes. All compounds have been crystallographically characterised and shown to exist in a number of isomeric forms in the solid state. These appear to be largely retained in solution. The deprotonation of GisoH with BunLi in either hexane or THF led to crystallographically characterised dimeric and monomeric complexes respectively, viz. [Li{Li(κ2-N,N′-Giso)2}] and [Li(THF)(η1-N,η3-Ar-Giso)]. Deprotonation of PrisoH and Pip(GisoH)2 with K[N(SiMe3)2] gave the unsolvated polymer, [{K(η1-N,η6-Ar-Priso)}∞], and the solvated complex, [{K(THF)2}{Pip(Giso)2}{K(THF)3}], respectively.
CNAr (Ar = C6H3Pri2-2,6), followed by aqueous work-ups, have yielded several guanidines, ArNC(NR2)N(H)Ar (R = cyclohexyl (GisoH) or Pri (PrisoH); NR2 = cis-NC5H8Me2-2,6 (PipisoH)), a bifunctional guanidine, {ArNCN(H)Ar}2{μ-N(C2H4)2N} (Pip(GisoH)2), and two phosphaguanidines, ArNC(PR2)N(H)Ar (R = cyclohexyl (CyP-GisoH) or Ph (PhP-GisoH)). A very bulky guanidine, ArNC{N(Ar)SiMe3}N(H)Ar (ArSi-Giso), and an aryl coupled bifunctional guanidine, {ArN(H)C(NPri2)NC6H2Pri2-2,6-}2 (PrisoH)2, have been prepared by other routes. All compounds have been crystallographically characterised and shown to exist in a number of isomeric forms in the solid state. These appear to be largely retained in solution. The deprotonation of GisoH with BunLi in either hexane or THF led to crystallographically characterised dimeric and monomeric complexes respectively, viz. [Li{Li(κ2-N,N′-Giso)2}] and [Li(THF)(η1-N,η3-Ar-Giso)]. Deprotonation of PrisoH and Pip(GisoH)2 with K[N(SiMe3)2] gave the unsolvated polymer, [{K(η1-N,η6-Ar-Priso)}∞], and the solvated complex, [{K(THF)2}{Pip(Giso)2}{K(THF)3}], respectively.
Introduction
The coordination chemistry of anionic amidinate ([RNC(R)NR]−, R = H, alkyl, aryl etc.) and guanidinate ([RNC(NR2)NR]−) ligands has been extensively studied, giving rise to numerous complexes incorporating metals from across the periodic table.1 In these, the ligands have displayed an impressive array of coordination modes which depend upon the nature and bulk of the substituents (R), and the metal involved. This structural diversity is one of the main factors that have led to such complexes finding many applications in catalysis,2–4 materials science5 and synthesis,1 to name but a few.Recent developments in this area have concentrated on the use of very bulky amidinates to stabilise low nuclearity s- and p-block metal complexes which show significant potential as, for example, lactide polymerisation catalysts.2 Of most note here is the Piso− ligand, [ArNC(But)NAr]−, which incorporates sterically demanding 2,6-diisopropylphenyl (Ar) substituents at its N-centres and a tert-butyl group on the backbone carbon. The spatial profile and ligating abilities of this ligand have been likened to those of β-diketiminates, the most commonly utilised examples of which also possess N–Ar substituents, e.g. [(ArNCR)2CH]− (R = Me or But).6 Although complexes of β-diketiminates are widely used in catalytic processes, they are perhaps more notable for their capacity to kinetically stabilise complexes containing low oxidation state metal centres. A salient illustration of this is the synthesis and structural characterisation of the homologous series of monomeric, N,N′-chelated group 13 metal(I) complexes, [:M{(ArNCMe)2CH}] (M = Al, Ga, In or Tl),7 which have shown remarkable further chemistry.
In contrast to β-diketiminates, bulky amidinates (e.g. Piso−) had rarely been employed in the preparation of low oxidation state metal complexes. In 2005, we began to address this paucity with the preparation of the group 13 metal(I) complexes [:M(Piso)] (M = In or Tl).8 However, unlike their β-diketiminate counterparts, [:M{(ArNCMe)2CH}], the Piso− ligand in these complexes is localised and chelates the metal centre in an η1-N,η3-arene-fashion. In addition, the analogous GaI and AlI complexes could not be stabilised. These results suggested that related, but bulkier ligands would need to be accessed to enforce N,N′-chelation and allow stabilisation of lighter group 13 metal(I) centres. To this end, the very large guanidinate ligand, [ArNC(NCy2)NAr]− (Giso−; Cy = cyclohexyl), was developed and used in the syntheses of the remarkably stable monomeric four-membered heterocycles, [:M(κ2-N,N′-Giso)] (M = Ga or In; N.B. the Al(I) heterocycle has not yet been accessed),9 the coordination chemistry of which was later explored.10 In addition to the increased steric bulk of Giso− over Piso−, the greater stabilising ability of the guanidinate can be attributed to the fact that it is a more N-electron rich donor than the amidinate, a result of it possessing a zwitterionic resonance form containing two negatively charged N-donor centres, viz. [Cy2N+C(N−Ar)2].
Over the last three years we have extended our application of Giso−, and a range of other Ar-substituted guanidinate and phosphaguanidinate ([ArNC(PR2)NAr]−)11 ligands, to the stabilisation of heterocyclic complexes containing low oxidation state metal centres from all blocks of the periodic table (e.g. Mg(I),12Ge(I),13As(I),14 various d-block metal(I)15 and f-block metal(II)16 species) with considerable success. Moreover, we have used these ligands in the synthesis of a variety of gallyl–metal complexes, including examples exhibiting unprecedented Ga–Zn17 and Ga–Sn18 bonds. In all these studies, the ligands have been prepared by the deprotonation of neutral guanidines or phosphaguanidines with alkali metal reagents. Although some preliminary details of the synthesis of the neutral ligand precursors have been previously been described by us,9–16 it seemed that a full report of the preparation and characterisation of these compounds would aid other researchers seeking to harness their unique properties for their own purposes. The value of this is highlighted by the fact that prior to our involvement in this field, only one guanidine bearing 2,6-diisopropylphenyl substituents at its N-centres, viz.ArNC{N(H)Ar}2, had appeared in the literature.19 Here, we report on the synthesis, structures and properties of eight N–Ar substituted guanidines and phosphaguanidines, and some of their alkali metal derivatives.
Results and discussion
(i) Synthesis of bulky guanidines and phosphaguanidines
A number of synthetic routes are known for the preparation of guanidines.1 One of the most versatile of these involves the addition of metallated amides to carbodiimides (RNCNR), followed by aqueous work-up. Here, this route has been employed to synthesise the guanidines GisoH (1), PrisoH (2), PipisoH (3), as well as the bifunctional guanidine, Pip(GisoH)2 (4), in high to quantitative yields (Scheme 1). In all preparations, THF was used as the solvent and the initial addition reactions were carried out at either ambient temperature and/or under reflux conditions.
 | ||
| Scheme 1
Reagents and conditions: (i) ArNCNAr, THF, 20 °C or reflux; (ii) H2O. | ||
It appears that this route does have steric and electronic limitations, as the attempted addition of some amides to the carbodiimide (ArNCNAr) were not successful. For example, lithiated cis-2,6-dimethylpiperidine adds to the carbodiimide to give compound 3, whereas lithiated 2,2,6,6-tetramethylpiperidine does not react with ArNCNAr in THF at reflux. Moreover, M[N(SiMe3)2] (M = Li, Na or K) do not react with ArNCNAr under similar conditions, though these reagents are known to add to smaller carbodiimides at room temperature.20
Although considerably less sterically demanding than some of the amide precursors mentioned above, lithium carbazolyl did not react with ArNCNAr in THF at reflux, and only carbazole and the carbodiimide were recovered after work-up. This lack of reactivity probably derives from the lower nucleophilicity of the aromatic carbozyl anion, relative to the bulkier amides used in the preparation of 1–3.
Interest in the coordination chemistry of phosphaguanidinates, [RNC(PR′2)NR]−, has recently begun to escalate.1d,21 One of the main reasons behind this is that the phosphino group of these ligands is pyramidal, unlike the planar amino substituent of guanidinates. Therefore, the zwitterionic resonance form of these ligands, [R′2P+C(N−R)2], does not play a significant role in their chemistry. As a result, phosphaguanidinates are coordinatively versatile, and in many of their complexes the phosphino group acts a P-lone pair donor.1d,21 Despite this emerging importance, there had been no reports of N–Ar substituted phosphaguanidinates or phosphaguanidines in the literature. We have reversed this situation with the synthesis of CyP-GisoH, 5, and PhP-GisoH, 6, via the addition of the relevant lithium phosphide to ArNCNAr (Scheme 1). Aqueous work-ups of these compounds were performed under an inert atmosphere to prevent oxidation of the phosphorus atom. However, we have found that the products can be handled in moist air as solids or in solution without significant oxidation occurring, as judged by 31P NMR spectroscopy. It is noteworthy that the addition of phosphines to smaller carbodiimides to form phosphaguanidines, in the presence of catalytic amounts of s-block amide or alkyl bases, has recently been reported.22,23
Although the addition of metal amides to ArNCNAr is a versatile route to bulky guanidine compounds, its limitations centre on the bulk of the reacting amide complex (as mentioned above). Because of this, a different approach was used to synthesise the exceedingly bulky guanidine, ArSi–GisoH 7 (Scheme 2). This involved lithiation of the known guanidine, ArNC{N(H)Ar}2, the product of which was subsequently quenched with Me3SiCl in THF at reflux to give 7 in good yield.
 | ||
| Scheme 2 Reagents and conditions: (i) BunLi, THF; (ii) Me3SiCl, THF reflux. | ||
One further bifunctional guanidine has been prepared in this study via a route not involving carbodiimide addition. Though this synthesis was originally not intended, it is moderately yielding, reproducible and thus is included here. In an attempt to form a Mn(II) complex of Priso−, K[Priso] was reacted with commercially available MnI2 in THF. This, instead led to the isolation of the aryl-coupled guanidine, (PrisoH)28, in a 30% yield (Scheme 3) without aqueous work-up. When the reaction was repeated with a pure sample of [MnI2(THF)3], compound 8 was not obtained. Presumably, the commercially sourced MnI2 initially employed, was contaminated with significant amounts of higher oxidation state manganese species. It is believed that the reaction of the impure MnI2 with K[Priso] led to the oxidative coupling of two Priso− anions through aryl para-positions on each. This seems reasonable in light of the fact that we have recently shown that Priso− can coordinate the Rh(COD) fragment (COD = 1,5-cyclooctadiene) solely through one aryl substituent in a η5-cyclohexadienyl fashion.15 A Mn(>II)-Priso complex in which the ligand exhibits this cyclohexadienyl binding mode can easily be envisaged as an intermediate in the oxidative coupling that gave 8. The possibility that 8 was alternatively formed via the oxidative coupling of two Priso− anions by a diiodine contaminant in the impure sample of MnI2 was examined and discounted.
![Reagents and conditions: (i) K[N(SiMe3)2], THF; (ii) MnI2, THF.](/image/article/2009/NJ/b809120j/b809120j-s3.gif) | ||
| Scheme 3 Reagents and conditions: (i) K[N(SiMe3)2], THF; (ii) MnI2, THF. | ||
(ii) Structural and spectroscopic properties of prepared compounds
The crystal structures of all compounds 1–8 have been determined (see Fig. 1–6 for the molecular structures of 1, 4–8; those of 2 and 3 can be found in ESI†). The compounds display solid state structures comparable to those of previously characterised guanidines and phosphaguanidines.1,21 Each of the guanidines, 1–3, possesses a close to planar backbone amino (–NR2) fragment which in no case is co-planar with the CN3 core of the molecule. Therefore, any interaction of the amino N-lone pair within the π-system of the largely localised guanidine CN3 backbone must be limited. It is noteworthy that the –NR2 fragments of 4 are significantly more distorted from planar than those of the monofunctional guanidines. Similarly, the two phosphaguanidines display distorted pyramidal phosphorus centres, the lone pairs of which are directional and therefore cannot be involved with the π-system of their localised CN2P cores. The bond lengths and angles within these core fragments (see Table 1) are consistent with these descriptions. | ||
| Fig. 1 Molecular structure of 1 (25% thermal ellipsoids are shown; hydrogen atoms, except H(1), omitted for sake of clarity). | ||
 | ||
| Fig. 2 Molecular structure of 4 (25% thermal ellipsoids are shown; hydrogen atoms, except H(1), omitted for sake of clarity). Symmetry operation:′−x + 1, −y + 1, −z. | ||
 | ||
| Fig. 3 Molecular structure of 5 (25% thermal ellipsoids are shown; hydrogen atoms, except H(2), omitted for sake of clarity). | ||
 | ||
| Fig. 4 Molecular structure of 6 (25% thermal ellipsoids are shown; hydrogen atoms, except H(1), omitted for sake of clarity). | ||
 | ||
| Fig. 5 Molecular structure of 7 (25% thermal ellipsoids are shown; hydrogen atoms, except H(3), omitted for sake of clarity). Selected bond lengths (Å) and angles (°): Si(1)–N(1) 1.7762(16), N(1)–C(1) 1.410(2), C(1)–N(2) 1.285(2), C(1)–N(3) 1.383(2); N(2)–C(1)–N(3) 130.87(17), N(2)–C(1)–N(1) 116.38(16), N(3)–C(1)–N(1) 112.73(16), C(1)–N(1)–C(5) 119.99(15), C(1)–N(1)–Si(1) 119.94(12), C(5)–N(1)–Si(1) 119.81(12). | ||
 | ||
| Fig. 6 Molecular structure of 8 (25% thermal ellipsoids are shown; hydrogen atoms, except H(1) and H(6), omitted for sake of clarity). Selected bond lengths (Å) and angles (°): N(1)–C(1) 1.391(3), C(1)–N(3) 1.290(4), C(1)–N(2) 1.378(4), N(4)–C(44) 1.290(3), N(5)–C(44) 1.383(4), N(6)–C(44) 1.387(4); N(3)–C(1)–N(2) 121.1(2), N(3)–C(1)–N(1) 122.2(3), N(2)–C(1)–N(1) 116.7(3), C(1)–N(2)–C(17) 120.2(2), C(1)–N(2)–C(14) 119.8(2), C(17)–N(2)–C(14) 115.5(2), C(44)–N(5)–C(45) 119.5(2), C(44)–N(5)–C(48) 119.9(2), C(45)–N(5)–C(48) 116.0(2), N(4)–C(44)–N(5) 120.8(3), N(4)–C(44)–N(6) 122.0(3), N(5)–C(44)–N(6) 117.3(2). | ||
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| ArNC |
1.290(2) | 1.2911(16) | 1.285(2) | 1.287(2) | 1.2909(19) | 1.311(2) |
| ArN–C | 1.384(3) | 1.3910(16) | 1.394(2) | 1.373(2) | 1.375(2) | 1.346(2) |
| C–ER2 | 1.388(2) | 1.3807(16) | 1.385(2) | 1.398(2) | 1.8708(17) | 1.8798(18) |
|
ArN–CN |
121.26(17) | 122.02(11) | 124.10(17) | 124.67(15) | 123.26(14) | 121.51(16) |
| R2E–CN |
121.66(18) | 120.57(11) | 119.91(16) | 119.84(15) | 121.76(11) | 119.98(13) |
| R2E–C–N | 117.08(17) | 117.42(10) | 115.99(16) | 115.48(14) | 114.96(11) | 118.51(13) |
| ∑ angles about E | 353.3 | 357.0 | 353.5 | 341.9 | 302.3 | 304.4 |
Several different isomeric forms of the compounds have been identified in this study. To allow comparisons with related amidines, the backbone unit (R2N or R2P) has been defined as the lower priority in determining the stereo-configuration of the compounds (see refs. 1d and 1e for a description of the four isomeric and tautomeric forms of amidines, viz. Z-anti, Z-syn, E-anti and E-syn). The guanidines, 1–3 (see Fig. 1 for the structure of 1), and the phosphaguanidine, 5 (Fig. 3), exist in the Z-anti-form which is common for guanidines but not for uncoordinated phosphaguanidines which normally occur in the solid state in their E-syn-form.1d,21 Indeed, this is the isomer adopted by the phosphaguanidine, 6, in the solid state (Fig. 4). In contrast, the extremely bulky guanidine, 7 (Fig. 5), crystallises in the rarely observed Z-syn-form, probably because of steric buttressing of its aryl groups by the larger N(Ar)SiMe3 substituent. It is of note that the Z-syn-isomer of amidines with very bulky backbone C-substituents have been previously reported, e.g.(tript)C{N(H)R}(NR) (tript = 9-triptycenyl, R = Cy or Pri).24 Both the bifunctional amidines, 4 and 8 (Fig. 2 and 6, respectively), exist in the solid state as Z-anti-,Z-anti-isomers, as has been previously documented for bifunctional amidines.1
Often, amidines and guanidines will be present in solution in more than one of their four possible isomeric forms. This can lead to complicated NMR spectra for such compounds. However, the guanidines and phosphaguanidines, 1–6, display relatively simple 1H and 13C{1H} NMR spectra, which are suggestive of only one, or predominantly one, isomer occurring in solution. These spectra imply that each compound has two chemically inequivalent Ar substituents, and that both alkyl or aryl groups on the backbone –ER2 (E = N or P) groups are equivalent. If the compounds retain their solid state isomeric forms in solution, which seems likely, the latter observation requires their –ER2groups to partially rotate on the NMR timescale, thus leading to the compounds possessing averaged mirror planes incorporating their ECN2 fragments.
Although the isomeric forms adopted by the guanidines, 1–4, in solution cannot be certain without two-dimensional NMR experiments, some insight into the solution conformations of the phosphaguanidines, 5 and 6, can be gained from their 1H NMR spectra. That for 5 shows only one isomer, the NH resonance of which exists as a doublet (3JPH = 14.1 Hz; 31P{1H} NMR: δ−2.9 ppm). The spectrum of 6 reveals the compound to exist as two isomers in solution in an approximately 90 : 10 ratio. The NH resonance of the major isomer (31P{1H} NMR: δ−18.5 ppm) is a singlet, while that for the minor isomer (31P{1H} NMR: δ−13.3 ppm) is a doublet (3JPH = 18.2 Hz). In an excellent paper on phosphaguanidinate solution behaviour, Coles et al. have shown that isomer interconversion can readily occur by one or more of a number of possible pathways.21e Importantly, they also showed that the closely related phosphaguanidine, Cy2PC{N(H)Pri}(NPri), is present in solution in both its E-syn- (major) and Z-anti- (minor) forms (14 : 1 ratio at 298 K). The NH resonance of the E-syn-form shows no coupling to the P-centre, while that of the minor Z-anti-isomer does (3JPH = 14.5 Hz). Accordingly, we conclude that compound 5 exists solely as its Z-anti-form in solution (as in the solid state), whereas the major solution state isomeric form of 6 is E-syn (as in the solid state), and the minor form is Z-anti.
Many of the signals in the 1H NMR spectrum of ArSi-GisoH, 7, are very broad and suggest one or more dynamic processes are occurring in solution. Despite efforts, the spectrum could not be resolved, and thus we could not shed light on the nature of the dynamic behaviour. One possibility, however, is that it involves a restricted rotation of the Ar and/or SiMe3 groups about the N–C or N–Si bonds of 7. In this respect, it should be noted that similar solution dynamic behaviour has been observed for the closely related compound, ArNC{N(H)Ar}2, an exhaustive variable-temperature NMR study of which showed this behaviour to be derived from restricted rotation of its three Ar groups.19 Another possibility for 7 is that there is a fluxional interconversion between two or more isomers of the compound, which is occurring at close to the NMR timescale. This seems less likely, however, when the imposing sterics of the compound are taken into account.
Little information could be gained from the solution NMR spectra of the bifunctional guanidine, 8. These are very complicated and point towards more than one isomer existing in solution. For example, several overlapping N–H resonances were seen in its 1H NMR spectrum, where only one would be expected if it retained its solid state Z-anti-, Z-anti-isomeric form in solution. As a result, the spectra proved difficult to assign.
(iii) Metallation of bulky guanidines and phosphaguanidines
The guanidines and phosphaguanidines prepared here (with the exception of ArSi-Giso 7), can be easily deprotonated by standard metallation procedures. The reactions of these ligands with one equivalent of BunLi or K[N(SiMe3)2] proceed rapidly and near quantitatively in common solvents such as hexane, toluene, THF or diethyl ether at ambient temperature or below. The solvent and metal involved in the reaction can have a striking bearing on the nuclearity of the formed complex, and the conformation adopted by the guanidinate or phosphaguanidinate ligand. This is important as it can influence the product obtained from, for example, further salt metathesis reactions of these alkali metal complexes with other metal halides. In this study, we have structurally and spectroscopically characterised four lithium or potassium salts of the ligands prepared above.The lithiation of GisoH, 1, with BunLi in hexane led to the solvent free dimeric complex, 9, whilst in THF the monomeric solvated complex, 10, was formed (Scheme 4). In contrast, metallation of PrisoH, 2, with K[N(SiMe3)2] in toluene afforded the polymeric, solvent free complex, 11, whereas metallation of Pip(GisoH)2 with the same reagent in THF gave the solvated complex, 12 (Scheme 4). The NMR spectroscopic data for 9–11 are more symmetrical than their solid state structures (vide infra) would suggest and imply that fluxional processes are occurring in solution that are rapid on the NMR timescale. This is not uncommon for alkali-metal amidinates and guanidinates,1 and therefore no efforts were made to investigate these dynamic behaviours by variable temperature NMR studies. Once crystallised from the reaction mixture, compound 12 has negligible solubility in normal deuterated solvents (including D8-THF) and therefore no meaningful NMR spectroscopic data could be obtained for this compound.
![Reagents and conditions: (i) BunLi, hexane (Cy = cyclohexyl); (ii) BunLi, THF; (iii) K[N(SiMe3)2], toluene; (iv) K[N(SiMe3)2], THF.](/image/article/2009/NJ/b809120j/b809120j-s4.gif) | ||
| Scheme 4 Reagents and conditions: (i) BunLi, hexane (Cy = cyclohexyl); (ii) BunLi, THF; (iii) K[N(SiMe3)2], toluene; (iv) K[N(SiMe3)2], THF. | ||
The molecular structure of 9 is depicted in Fig. 7 and shows it to be dimeric with two different lithium coordination environments. Li(1) is coordinated by two chelating Giso− ligands that have largely localised N(1)–C(1)–N(2) fragments. The Li(1)–N bond lengths of 2.072(2) Å (to N(2) and N(2)′) and 2.240(5) Å (to N(1) and N(1)′), although different, lie within the normal range for amidinate and guanidinate N–Li interactions.25 The two more distant N-atoms (N(1) and N(1)′) also coordinate the bent two-coordinate Li(2) centre with short interactions (1.954(4) Å). The coordination sphere of the both Li atoms is completed by agostic interactions to ligand hydrogen atoms; Li(1) has two such interactions (both ca. 2.23 Å), whereas Li(2) has four (from ca. 2.03 Å to ca. 2.27 Å). When these close contacts are taken into account, both Li-centres can be thought of as having heavily distorted octahedral geometries. A survey of the Cambridge Crystallographic Database revealed two similar dimeric lithium amidinates, [Li{κ2-N,N′-(SiMe3)NC(R)N(SiMe3)}2{Li(OEt2)}] (R = C6H5CF3-4 or C6H5F-2),26 though the non-chelated Li centre of both is further coordinated by an ether molecule.
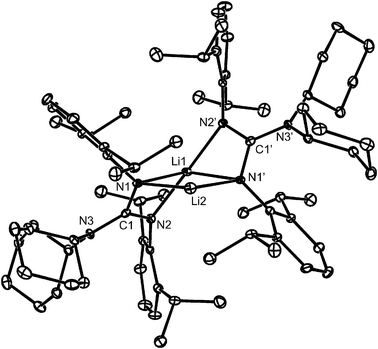 | ||
| Fig. 7 Molecular structure of 9 (25% thermal ellipsoids are shown; hydrogen atoms omitted for sake of clarity). Selected bond lengths (Å) and angles (°): N(1)–C(1) 1.394(3), N(2)–C(1) 1.323(3), N(3)–C(1) 1.409(3), N(1)–Li(2) 1.954(4), N(1)–Li(1) 2.240(5), N(2)–Li(1) 2.072(2); N(2)–C(1)–N(1) 114.3(3), N(2)–Li(1)–N(1) 63.78(12), N(2)′–Li(1)–N(1) 119.6(2), N(1)′–Li(2)–N(1) 121.7(4). Symmetry operation:′−x, y, −z + 1/2. | ||
The molecular structure of monomeric 10 is shown in Fig. 8. The localised guanidinate ligand is acting as an amide that coordinates the Li atom in an η1-fashion through N(2). In addition, there is an approximately η3-interaction of the Li-centre with the Ar-substituent of N(1). The coordination sphere on the Li(1) is completed by one THF molecule. A similar coordination mode (but minus the coordinated THF) has been reported for the thallium(I) complex, [Tl(η1-N,η3-Ar-Giso)].8
 | ||
| Fig. 8 Molecular structure of 10 (25% thermal ellipsoids are shown; hydrogen atoms omitted for sake of clarity). Selected bond lengths (Å) and angles (°): N(1)–C(1) 1.3149(16), C(1)–N(2) 1.3587(16), C(1)–N(3) 1.4092(16), Li(1)–N(2) 1.943(3), Li(1)–C(2) 2.290(3), Li(1)–C(3) 2.458(3), Li(1)–C(7) 2.591(3), O(1)–Li(1) 1.889(3); N(1)–C(1)–N(2) 121.56(11), N(1)–C(1)–N(3) 117.46(11), N(2)–C(1)–N(3) 120.98(11), C(1)–N(3)–C(32) 117.05(10), C(1)–N(3)–C(26) 120.82(10), C(32)–N(3)–C(26) 115.51(10), C(1)–N(2)–Li(1) 117.59(11). | ||
Like the structure of 10, the guanidinate moieties of the potassium complexes, 11 and 12 (Fig. 9 and 10, respectively), adopt the Z-anti-configuration but with more localised coordinated NCN fragments. In addition, the arene-K interactions in both are close to η6-, as opposed to the η3-Ar-Li coordination seen in 10. In 11, this leads to a one-dimensional polymeric structure in which one Ar-group of each ligand bridges two K-centres. Compound 12 is monomeric, and in addition to arene and N-attachments, one K-centre is coordinated by two THF molecules, while the other is ligated by three. All the distances to the K-centres in both complexes are in the normal range.25
 | ||
| Fig. 9 Molecular structure of 11 (25% thermal ellipsoids are shown; hydrogen atoms omitted for sake of clarity). Selected bond lengths (Å) and angles (°): K(1)–N(1) 2.755(3), K(1)–Ar centroid 3.077(1), K(1)′–Ar centroid 2.945(1), C(1)–N(2) 1.329(5), C(1)–N(3) 1.402(5), N(1)–C(1) 1.340(5); N(2)–C(1)–N(1) 121.7(3), N(2)–C(1)–N(3) 115.1(3), N(1)–C(1)–N(3) 123.2(3), C(1)–N(3)–C(26) 122.0(3), C(1)–N(3)–C(29) 121.5(3), C(26)–N(3)–C(29) 114.7(3), C(1)–N(1)–K(1) 128.2(2). Symmetry operation:′x− 1/2, −y + 1/2, −z. | ||
 | ||
| Fig. 10 Molecular structure of 12 (25% thermal ellipsoids are shown; hydrogen atoms and isopropyl groups omitted for sake of clarity). Selected bond lengths (Å) and angles (°): K(1)–O(1) 2.681(3), K(1)–O(2) 2.698(3), K(1)–O(5) 2.780(3), K(1)–N(1) 2.823(2), K(2)–O(3) 2.646(3), K(2)–O(4) 2.710(3), K(2)–N(5) 2.735(3), K(1)–Ar centroid 3.007(1), K(2)–Ar centroid 2.915(1), N(1)–C(1) 1.328(4), N(2)–C(1) 1.321(4), N(3)–C(1) 1.437(4), N(4)–C(30) 1.426(4), N(5)–C(30) 1.336(4), N(6)–C(30) 1.322(4); N(2)–C(1)–N(1) 124.6(3), N(2)–C(1)–N(3) 114.8(3), N(1)–C(1)–N(3) 120.6(2), C(1)–N(1)–K(1) 123.88(18), C(30)–N(5)–K(2) 129.42(18), N(6)–C(30)–N(5) 123.6(3), N(6)–C(30)–N(4) 115.2(2), N(5)–C(30)–N(4) 121.2(2). | ||
Conclusion
In conclusion, the synthesis and characterisation of a variety of guanidine, bifunctional guanidine and phosphaguanidine compounds, all bearing 2,6-diisopropylphenyl N-substituents, have been described. In the solid state, the Z-anti-isomeric form is observed for all guanidines, except in one extremely bulky example, ArSi-GisoH 7. The sterics of this necessitate it occurring as the rarely observed Z-syn-isomer. Of the phosphaguanidinates, the bulkier example, CyP-GisoH 5, crystallises in the Z-anti-form, while PhP-GisoH, 6, adopts the E-syn-conformation. In solution, most of the described compounds appear to retain their stereochemistry, though in some cases isomer mixtures were observed. Several of the prepared compounds have been deprotonated with alkali metal reagents and the resulting salts crystallographically characterised. In the case of the deprotonation of GisoH 1 with BunLi, the nuclearity and guanidinate coordination mode displayed by the formed complexes are dependent upon the reaction solvent employed. We are currently systematically exploring the use of bulky guanidinates and phosphaguanidinates, prepared from the neutral compounds 1–8, for the stabilisation of low oxidation metallacycles incorporating metals from all blocks of the periodic table.Experimental
General considerations
All manipulations were performed under an inert atmosphere (dinitrogen or argon) using Schlenk or glove box techniques. Aqueous organic work-ups were carried out in air, except those for the phosphaguanidines, 5 and 6. Melting points were determined in sealed capillaries under a dinitrogen atmosphere, except those for the guanidines, 1–4, which were determined in open capillaries. Reaction solvents were dried over potassium or Na/K alloy prior to use, except dichloromethane and chloroform which were used as received. Mass spectra were recorded at the EPSRC National Mass Spectrometric Service, Swansea University. Microanalyses were obtained from either Medac Ltd or Campbell Microanalytical, Ottago. IR spectra were recorded using a Nicolet 510 FT-IR spectrometer as Nujol mulls between NaCl plates. 1H and 13C{1H} NMR spectra were recorded on either Bruker DXP400, Bruker DPX300, Jeol Eclipse 300 or Bruker WM250 spectrometers and were referenced to the resonances of the solvent used. 31P{1H} NMR spectra were recorded on a Jeol Eclipse 300 spectrometer and were referenced to external 85% H3PO4. Cy2NH, Pri2NH, cis-2,6-dimethylpiperidine and piperazine were obtained commercially, dried over molecular sieves, and distilled under dinitrogen prior to use. K[N(SiMe3)2] was prepared by treating (SiMe3)2NH with KH in toluene at 20 °C. ArNCNAr27 and ArNC{N(H)Ar}219 were synthesised according to literature procedures. All other reagents were obtained from commercial sources and used as received.
Preparation of GisoH 1
BunLi (5.33 cm3 of a 1.6 M solution in hexanes, 8.52 mmol) was added to a solution of Cy2NH (1.58 g, 1.73 cm3, 8.69 mmol) in THF (40 cm3) at 20 °C over 5 min and the resultant solution stirred for 1 h. ArNCNAr (3.00 g, 8.27 mmol) was then added, the suspension stirred for 15 min, followed by heating at reflux for 1.5 h (or alternatively stirred at room temperature for 4 h). All volatiles were removed under reduced pressure and diethyl ether (40 cm3) and H2O (10 cm3) added to the residue. The mixture was stirred for 30 min to give two clear solution phases. The organic phase was separated and the aqueous layer was extracted with CH2Cl2 (3 × 30 cm3). The combined organic phases were dried (MgSO4), filtered, and volatiles evaporated from the filtrate under vacuum. The oily residue solidified upon standing to give 1 as colourless crystals (yield 4.40 g, 98%). The product can be recrystallised from hot hexane (yield 80%); mp 140–141 °C. 1H NMR (300 MHz, 298 K, CDCl3): δ 0.91 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.08–0.90 (m, 8 H, CH2), 1.21 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.36 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.38 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.47–1.70 (m, 8 H, CH2), 2.05 (m, 4 H, CH2CHN), 2.97 (tt, J = 11.7, 3.3 Hz, 2 H, CHN), 3.22 (sept, J = 6.8 Hz, 2 H, CH(CH3)2), 3.32 (sept, J = 6.8 Hz, 2 H, CH(CH3)2), 4.95 (s, 1 H, NH), 6.89–7.17 (m, 6 H, ArH); 1H NMR (250 MHz, 298 K, C6D6): δ 0.96 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.12–1.32 (m, 6 H, CH2), 1.44 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.50–1.68 (m, 2 H, CH2), 1.54 (d, J = 6.8 Hz, 12 H, CH(CH3)2), 1.75–1.92 (m, 8 H, CH2), 2.21–2.44 (m, 4 H, CH2), 3.23 (tt, J = 11.7, 3.3 Hz, 2 H, CHN), 3.60 (sept, J = 6.8 Hz, 4 H, CH(CH3)2), 5.32 (s, 1 H, NH), 7.06–7.44 (m, 6 H, ArH); 13C{1H} NMR (75.5 MHz, 298 K, CDCl3): δ 21.6 (CH(CH3)2), 22.5 (CH(CH3)2), 24.9 (CH2), 26.0 (CH(CH3)2), 26.1 (CH(CH3)2), 27.1 (CH(CH3)2), 28.6 (CH(CH3)2), 29.0 (CH2), 32.6 (CH2), 58.0 (HCN), 121.6, 122.8, 123.5, 126.9, 135.9, 140.0, 145.5, 145.6, (ArC), 148.0 (CN3), 13C{1H} NMR (75.5 MHz, 298 K, C6D6): δ 21.7 (CH(CH3)2), 22.3 (CH(CH3)2), 25.2 (CH2), 26.2 (CH(CH3)2), 27.3 (CH(CH3)2), 28.7 (CH(CH3)2), 29.3 (CH(CH3)2), 32.9 (CH2), 39.8 (CH2), 58.2 (HCN), 122.6, 123.3, 123.7, 127.1, 136.1, 139.9, 145.5, 145.6 (ArC), 148.5 (CN3); IR (Nujol): ν/cm−1 = 3384 (m), 1614 (s), 1583 (s), 1259 (m), 1163 (m), 1110 (m), 1072 (m), 986 (m), 954 (w), 894 (m), 799 (m), 761 (m), 700 (w); MS/APCI: m/z (%) = 544.7 (MH+, 100).
Preparation of PrisoH 2
A procedure analogous to that used to prepare 1 was employed, but using Pri2NH (colourless crystals: crude yield 99%; ca. 90% after recrystallisation); mp 144–145 °C; 1H NMR (250 MHz, 298 K, CDCl3): δ 0.89 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.12 (overlapping d, J = 6.8 Hz, 18 H, CH(CH3)2), 1.20 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.37 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 3.17 (sept, J = 6.8 Hz, 2 H, CH(CH3)2), 3.25 (sept, J = 6.8 Hz, 2 H, CH(CH3)2), 3.49 (sept, J = 6.8 Hz, 2 H, CH(CH3)2), 4.80 (s, 1 H, NH), 6.80–7.18 (m, 6 H, ArH); 1H NMR (400 MHz, 298 K, C6D6): δ 0.98 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.36 (overlapping d, J = 6.8 Hz, 18 H, CH(CH3)2), 1.51 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.54 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 3.68 (sept, J = 6.8 Hz, 6 H, CH(CH3)2), 5.25 (s, 1 H, NH), 7.09–7.39 (m, 6 H, ArH), 13C{1H} NMR (75.5 MHz, 298 K, CDCl3): δ 21.6 (CH(CH3)2), 21.9 (NCH(CH3)2), 22.7 (CH(CH3)2), 24.7 (CH(CH3)2), 25.7 (CH(CH3)2), 28.2 (CH(CH3)2), 28.8 (CH(CH3)2), 47.7 (HCN), 121.7, 122.7, 123.6, 126.9, 135.4, 139.8, 145.3, 145.8 (ArC), 148.3 (CN3); 13C{1H} NMR (100.6 MHz, 298 K, C6D6): δ 22.2 (CH(CH3)2), 22.5 (NCH(CH3)2), 23.1 (CH(CH3)2), 25.5 (CH(CH3)2), 26.0 (CH(CH3)2), 28.8 (CH(CH3)2), 29.7 (CH(CH3)2), 48.3 (HCN), 123.2, 123.8, 124.3, 127.7, 136.1, 140.3, 145.9, 146.2 (ArC), 149.1 (CN3); IR (Nujol): ν/cm−1 = 3364 (m), 1608 (s), 1580 (s), 1303 (m), 1245 (m), 1184 (m), 1154 (m), 1109 (m), 1046 (m), 1002 (m), 932 (m), 828 (m), 798 (m), 767 (m), 714 (m); MS/APCI: m/z (%) = 464.4 (MH+, 100).Preparation of PipisoH 3
A procedure analogous to that used to prepare 1 was employed, but using cis-2,6-dimethylpiperidine (colourless crystals: crude yield 98%; ca. 88% after recrystallisation); mp 128–130 °C. 1H NMR (400 MHz, 298 K, CDCl3): δ 0.89 (d, J≈ 6.1 Hz, 6 H, NCH(CH3)), 1.11 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.16 (d, J = 6.8 Hz, 12 H, CH(CH3)2), 1.18–1.76 (m, 6 H, CH2), 1.26 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 3.12 (sept, J = 6.8 Hz, 2 H, CH(CH3)2), 3.16 (sept, J = 6.8 Hz, 2 H, CH(CH3)2), 3.70 (mc, 2 H, NCH(CH3)), 4.85 (s, 1 H, NH), 6.88–7.18 (m, 6 H, ArH); 1H NMR (400 MHz, 298 K, C6D6): δ 1.01 (d, J≈ 6.0 Hz, 6 H, NCH(CH3)), 1.28–1.73 (m, 6 H, CH2), 1.42 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.49 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.54 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 1.56 (d, J = 6.8 Hz, 6 H, CH(CH3)2), 3.55 (sept, J = 6.8 Hz, 4 H, CH(CH3)2), 4.11 (mc, 2 H, NCH(CH3)), 5.34 (s, 1 H, NH), 7.11–7.39 (m, 6 H, ArH); 13C{1H} NMR (75.5 MHz, 298 K, CDCl3): δ 14.4 (CH2), 20.8 (NCH(CH3)), 21.5 (CH(CH3)2), 22.9 (CH(CH3)2), 24.1 (CH(CH3)2), 25.6 (CH(CH3)2), 28.2 (CH(CH3)2), 28.9 (CH(CH3)2), 30.0 (CH2), 48.3 (HCN), 121.8, 122.6, 123.5, 126.7, 135.1, 139.4, 145.3, 145.6 (ArC), 149.8 (CN3); 13C{1H} NMR (100.6 MHz, 298 K, C6D6): δ 15.0 (CH2), 21.4 (NCH(CH3)), 22.2 (CH(CH3)2), 23.4 (CH(CH3)2), 25.0 (CH(CH3)2), 25.9 (CH(CH3)2), 28.9 (CH(CH3)2), 29.8 (CH(CH3)2), 30.7 (CH2), 49.0 (HCN), 123.3, 123.7, 124.3, 127.6, 135.9, 140.0, 145.8, 146.1 (ArC), 150.7 (CN3); IR (Nujol): ν/cm−1 = 3378 (m), 1616 (s), 1579 (s), 1303 (m), 1258 (m), 1183 (m), 1145 (m), 1169 (m), 1079 (m), 1023 (m), 934 (m), 803 (m), 765 (m), 755 (m); MS/APCI: m/z (%) = 476.4 (MH+, 100).Preparation of Pip(GisoH)24
BunLi (5.00 cm3 of a 1.6 M solution in hexanes, 8.00 mmol) was added to a solution of piperazine (0.339 g, 3.94 mmol) in THF (40 cm3) at 20 °C over 5 min and the resultant solution stirred for 1 h. ArNCNAr (2.93 g, 8.08 mmol) was then added and the mixture stirred for 30 min, before being heated at reflux for 2 h. After cooling to ambient temperature, water (ca. 3 cm3) was added and volatiles removed under reduced pressure. More water (ca. 30 cm3) and CH2Cl2 (60 cm3) were then added to the residue and the mixture vigorously stirred until two clear solution phases were formed. The organic phase was separated and the aqueous layer was extracted with CH2Cl2 (3 × 30 cm3). The combined organic phases were dried (MgSO4), filtered and volatiles removed from the filtrate under reduced pressure. The residue was recrystallised from CHCl3 at –30 °C to give 4 as colourless crystals (yield: 1.88 g, 75%); mp 196–198 °C; 1H NMR (400 MHz, 298 K, CDCl3): δ 0.89 (br d, J = 6.8 Hz, 12 H, CH(CH3)2), 1.00 (d, J = 6.8 Hz, 12 H, CH(CH3)2), 1.15 (br d, J = 6.8 Hz, 12 H, CH(CH3)2), 1.23 (d, J = 6.8 Hz, 12 H, CH(CH3)2), 2.89 (br s, 8 H NCH2), 3.04 (mc of overlapping sept., J = 6.8 Hz, 8 H, CH(CH3)2), 4.93 (s, 2 H, NH), 6.90–7.16 (m, 12 H, ArH); 13C{1H} NMR (100.6 MHz, 298 K, CDCl3): δ 22.8 (CH(CH3)2), 23.3 (CH(CH3)2), 24.5 (CH(CH3)2), 25.7 (CH(CH3)2), 28.8 (CH(CH3)2), 28.9 (CH(CH3)2), 47.3 (NCH2), 123.1, 123.4, 124.3, 127.3, 134.1, 140.1, 144.4, 145.2 (ArC), 150.8 (N3C); IR (Nujol): ν/cm−1 = 3391 (m), 1623 (s), 1585 (m), 1261 (m), 1196 (m), 1145 (m), 1109 (m), 1041 (m), 988 (m), 935 (m), 840 (m), 799 (m), 759 (m); MS/APCI: m/z (%) = 811.4 (MH+, 100).
Preparation of CyP-GisoH 5
BunLi (4.00 cm3 of a 1.6 M solution in hexanes, 6.40 mmol) was added to a solution of Cy2PH (1.27 g, 6.40 mmol) in THF (20 cm3) at 0 °C over 5 min. The resultant solution was stirred for 1 h at room temperature. A solution of ArNCNAr (2.54 g, 6.28 mmol) in THF (15 cm3) was then added to the mixture which was subsequently heated at reflux for 1.5 h. After cooling, degassed water (1 cm3) was added, the mixture vigorously stirred for 1 h, and all volatiles removed under reduced pressure. The residue was extracted with warm hexane (2 × 50 cm3). The extract was dried over MgSO4, then filtered and concentrated to ca. 15 cm3. Slow cooling of the filtrate to −30 °C overnight yielded colourless crystals of 5 (yield: 2.85 g, 81%); mp 150–152 °C. 1H NMR (400 MHz, 298 K, CDCl3): δ 0.81 (d, J = 6.7 Hz, 6 H, CH(CH3)2), 1.09 (d, J = 6.7 Hz, 6 H, CH(CH3)2), 1.10–1.25 (m, 8 H, CH2), 1.23 (d, J = 6.7 Hz, 6 H, CH(CH3)2), 1.25 (d, J = 6.7 Hz, 6 H, CH(CH3)2), 1.58–2.04 (m, 14 H, CHP and CH2), 3.00 (sept., J = 6.7 Hz, 2 H, CH(CH3)2), 3.18 (sept., J = 6.7 Hz, 2 H, CH(CH3)2), 5.44 (d, JPH = 14.1 Hz, 1 H, NH), 6.92–7.18 (m, 6 H, ArH); 13C{1H} NMR (100.6 MHz, 298 K, CDCl3): δ 22.3 (CH2), 22.4 (CH2), 25.2 (CH(CH3)2), 26.1 (CH(CH3)2), 27.0 (CH(CH3)2), 27.8 (CH(CH3)2), 27.9 (CH(CH3)2), 28.0 (CH(CH3)2), 28.1 (CH2), 28.8 (CH2), 29.1 (CH2), 29.1 (CH2), 32.2 (d, J = 20 Hz, CH2), 33.7 (d, J = 13.2 Hz, CH2), 123.3, 123.4, 123.5, 128.4, 133.9, 139.0, 145.2, 147.2 (ArC), 160.1 (d, J = 13.1 Hz, backbone PCN2); 31P{1H} NMR (121 MHz, 298 K, C6D6): δ–2.9 (s); IR (Nujol): ν/cm−1 = 3354 (NH), 1620 (m), 1592 (m), 1568 (s), 1324 (m), 1259 (s), 1173 (m), 1109 (m), 1043 (m), 934 (m), 884 (m), 852 (m), 799 (s), 756 (s); MS/EI: m/z (%) = 560.4 (M+, 4), 517.4 (M+− C3H7, 100). Accurate mass (EI), m/z: calc. for M+: 560.4254, found: 560.4251.
Preparation of PhP-GisoH 6
BunLi (2.80 cm3 of a 1.6 M solution in hexanes, 4.52 mmol) was added to a solution of Ph2PH (0.85 g, 4.56 mmol) in THF (10 cm3) at −70 °C over 5 min then warmed to room temperature and stirred for 2 h. To the resultant red solution was added ArNCNAr (1.61 g, 4.43 mmol) in THF (10 cm3) at −70 °C. The mixture was subsequently heated under reflux for 1.5 h. It was then cooled to room temperature and ca. 0.3 cm3 degassed H2O was added with stirring. Volatiles were removed in vacuo and the residue extracted into diethyl ether (80 cm3) and filtered. The filtrate was concentrated and stored at –30 °C to give colourless blocks of 6 (yield: 1.66 g, 68%); mp 160–162 °C. 1H NMR (400 MHz, 298 K, CDCl3): δ 0.83 (4 × overlapping d, J = 6.8 Hz, 24 H, CH3), 2.78 (sept, J = 6.8 Hz, 2 H, CH), 3.15 (sept, J = 6.8 Hz, 2 H, CH), 5.60 (s, 1 H, NH), 6.85–7.51 (m, 16 H, Ar–H); 13C{1H} NMR (100.6 MHz, 298 K, CDCl3): δ 21.9, 22.1, 24.4, 25.4 (CH(CH3)2), 28.5, 28.8 (CH(CH3)2), 122.2, 122.8, 123.0, 123.1, 127.6, 128.0, 128.4, 129.1, 129.7, 137.4, 138.7, 145.9, 146.1, 146.4 (ArC), 155.6 (J = 16.1 Hz, PCN2); 31P{1H} NMR (121 MHz, 298 K, CDCl3,): δ−18.5; MS/APCI, m/z (%): 549 (M+, 100); IR (Nujol): ν/cm−1 = 1607 (s), 1579 (s), 1434 (m), 1258 (s), 1185 (m), 1099 (m), 742 (m), 693 (m); C37H45N2P requires: C 80.99%, H 8.27%, N 5.10%, found: C 80.84%, H 8.38%, N 5.25%.
Preparation of ArSi-GisoH 7
BunLi (1.64 cm3 of a 1.6 M solution in hexanes, 2.62 mmol) was added to a solution of ArNC{N(H)Ar}2 (1.35 g, 2.50 mmol) in THF (15 cm3) at room temperature over 5 min. The solution was then stirred for 1 h. Me3SiCl (0.36 g, 2.85 mmol) was added at room temperature and the mixture subsequently heated at reflux for 2.5 h. All volatiles were removed under reduced pressure and the residue was extracted into warm hexane (60 cm3). The solution was concentrated under reduced pressure to ca. 12 cm3 and cooled to 4 °C to obtain colourless crystals of 7 (yield 0.96 g, 63%); mp 257–258 °C. 1H NMR (400 MHz, 298 K, CDCl3): δ−0.3 (v br s, 9 H, Si(CH3)3), 0.92–0.64 (m, 12 H, CH(CH3)2), 1.14 (d, J = 6.8 Hz, 12 H, CH(CH3)2), 1.17 (d, J = 6.8 Hz, 12 H, CH(CH3)2), 2.81 (sept, J = 6.8 Hz, 2 H, CH(CH3)2), 3.54–3.14 (m, 4 H, CH(CH3)2), 5.59 (s, 1 H, NH), 7.28–6.54 (m, 9 H, ArH); 13C{1H} NMR (75.5 MHz, 298 K, CDCl3): only resonances of one aryl substituent are resolved. Others, as well as those for the SiMe3 group, are too broad to be detected. δ 23.2, 26.0, 28.2, (CH(CH3)2 and CH(CH3)2), 122.4, 126.6, 134.2, 145.5 (ArC), 146.8 (CN3); IR (Nujol): ν/cm−1= 3378 (m), 1618 (s), 1578 (s), 1378 (m), 1246 (m), 1223 (m), 1107 (m), 1007 (m), 970 (m), 843 (m), 822 (m), 752 (m); MS/EI: m/z (%) = 611 (M+, 14), 596 (M+− CH3, 5), 568 (M+− C3H7, 15), 539 (M+− SiMe3, 15), 496 (M+− SiMe3− C3H6, 18).Preparation of (PrisoH)28
A solution of K[N(SiMe3)2] (0.65 g, 3.24 mmol) in THF (10 cm3) was added to PrisoH 2 (1.50 g, 3.24 mmol) in THF (10 cm3) at 20 °C and the mixture stirred for 1 h. A solution of impure MnI2 (Aldrich Chemical Company, 1.00 g, 3.24 mmol) in THF (20 cm3) was then added at −78 °C and the reaction mixture slowly warmed to room temperature overnight. All volatiles were removed in vacuo and the residue was extracted with hexane (40 cm3). Filtration, concentration and slow cooling overnight to −30 °C yielded colourless crystals of 8 (yield 0.44 g, 30%); mp 223–225 °C. 1H NMR (400 MHz, 298 K, C6D6): δ 0.81–1.01 (m of overlapping br d, 18 H, CH(CH3)2), 1.05–1.47 (m of overlapping broad d, 54 H, CH(CH3)2), 3.11–3.49 (m of overlapping br sept, 12 H, CH(CH3)2), 4.74–4.91 (m, 2 H, NH), 6.88–7.29 (m, 10 H, ArH); 13C{1H} NMR (100.6 MHz, 298 K, C6D6): δ 21.83, 21.87, 22.70, 22.75, 24.73, 25.69, 28.12, 28.28, 28.81, 28.94, 31.63 (CH(CH3)2 and CH(CH3)2), 47.60 (CHN), 121.32, 121.41, 121.89, 122.68, 123.55, 123.61, 139.69, 139.78, 140.12, 145.59, 145.78 (ArC), 148.24, 148.33 (CN3), NB: more than one isomer present. Only major resonances reported with tentative assignments; IR (Nujol): ν/cm−1 = 3364 (m), 1611 (s), 1589 (s), 1376 (s), 1342 (m), 1261 (m), 1184 (m), 1111 (m), 999 (m), 870 (m), 802 (m), 761 (m), 715 (m); MS/EI: m/z (%) = 924.7 (M+, 14), 881.7 (M+–C3H7, 52). Accurate mass (EI), m/z: calc. for M+: 924.7691, found: 924.7688; CHN: C62H96N6 requires: C 80.46%, H 10.45%, N 9.08%; found: C 79.77%, H 10.99%, N 9.31%.Preparation of [Li{Li(Giso)2}] 9
BunLi (0.70 cm3 of a 1.6 M solution in hexanes, 1.12 mmol) was added over 5 min to a solution of GisoH 1 (0.58 g, 1.07 mmol) in hexane (20 cm3) at 20 °C. The resultant solution was stirred for 30 min then concentrated under reduced pressure to ca. 8 cm3. It was then stored at 4 °C overnight to afford colourless crystals of 9 (yield: 0.42 g, 71%); mp 190–192 °C (melts with slow decomposition); 1H NMR (300 MHz, 298 K, C6D6): δ 0.85–1.17 (m, 16 H, CH2), 1.18 (d, J = 6.8 Hz, 24 H, CH(CH3)2), 1.49 (d, J = 6.8 Hz, 24 H, CH(CH3)2), 1.78–1.48 (m, 16 H, CH2), 2.03 (m, 8 H, CH2CHN), 3.35 (br t, J≈ 11 Hz, 4 H, CHN), 3.57 (br sept, J = 6.8 Hz, 8 H, CH(CH3)2), 7.26–6.94 (m, 12 H, ArH); 13C{1H} NMR (75.5 MHz, 298 K, C6D6): δ 23.7, 25.0, 26.9, 27.9, 28.7, (CH2), CH(CH3), CH(CH3)), 35.1 (CH2), 58.6 (HCN), 120.8 (ArC), 124.0 (ArC), 141.3 (ArC), 150.2 (br, ArC), 160.6 (v br, CN3); 7Li NMR (155.5 MHz, 298 K, C6D6): δ 2.6 (s); IR (Nujol): ν/cm−1 = 1612 (s), 1583 (s), 1236 (s), 1156 (m), 1110 (m), 1027 (m), 933 (m), 895 (m), 792 (m), 748 (m); MS/EI: m/z (%) = 543.7 (GisoH+, 5), 500 (GisoH+− C3H6, 62).Preparation of [Li(THF)(Giso)] 10
BunLi (2.00 cm3 of a 1.6 M solution in hexanes, 3.20 mmol) was added over 5 min to a solution of GisoH 1 (1.71 g, 3.14 mmol) in THF (20 cm3) at 0 °C. The solution was then stirred for 1 h and volatiles removed under reduced pressure. Hexane (15 cm3) was added to the residue and the resultant solution concentrated to ca. 6 cm3. This was filtered and cooled to −30 °C to yield large colourless crystals of 10. Concentration of the supernatant solution at room temperature yielded another crop of 10 (yield 1.52 g; 78%); mp 208–210 °C. 1H NMR (400 MHz, 298 K, C6D6): δ 0.97 (mc, 4 H, THF-CH2), 1.27 (br mc, 12 H, CH(CH3)2), 1.30–1.45 (m, 6 H, CH2), 1.63 (d, 3JHH = 6.8 Hz, 12 H, CH(CH3)2), 1.71 (mc, 2 H, CH2), 1.93 (mc, 4 H, CH2), 2.06 (mc, 4 H, CH2), 2.52 (mc, 4 H, CH2), 2.63 (mc, 4 H, THF-OCH2), 3.38 (mc, 2 H, NCH), 3.85 (sept, J = 6.8 Hz, 4 H, CH(CH3)2), 6.98 (t, J = 7.5 Hz, 2 H, p-ArH), 7.26 (d, J = 7.5 Hz, 4 H, m-ArH); 13C{1H} NMR (100.6 MHz, 298 K, C6D6): δ 22.8 (CH(CH3)2), 23.4 (CH2), 25.3 (CH2), 26.3 (CH2, THF), 27.1 (CH(CH3)2), 32.3 (CH2), 56.5 (NCH), 66.7 (OCH2), 118.8, 122.2, 140.1, 149.7 (ArC), 156.6 (backbone, CN3); 7Li NMR (116.8 MHz, 298 K, C6D6): δ 1.64 (s); IR (Nujol): ν/cm−1 = 3378 (m), 1618 (s), 1578 (m), 1378 (m), 1246 (s), 1228 (m), 1198 (m), 1107 (m), 1008 (m), 970 (m), 843 (m), 822 (m), 752 (m); MS/EI: m/z (%) = 543.7 (GisoH+, 100).Preparation of [{K(Priso)}∞] 11
Toluene (30 cm3) was added to a mixture of PrisoH 2 (1.02 g, 2.20 mmol) and K[N(SiMe3)2] (0.45 g, 2.26 mmol) and the resultant suspension stirred vigorously for 4 h at room temperature. All volatiles were removed under reduced pressure and the residue washed with hexane (15 cm3). Recrystallisation from a toluene solution at −30 °C yielded colourless crystals of 11 (yield 0.95 g, 86%); mp >300 °C. 1H NMR (400 MHz, 298 K, C6D6): δ 1.08 (d, J = 6.8 Hz, 12 H, CH(CH3)2), 1.43 (d, J = 6.8 Hz, 12 H, CH(CH3)2), 1.51 (d, J = 6.8 Hz, 12 H, CH(CH3)2), 3.41 (sept, J = 6.8 Hz, 2 H, CH(CH3)2), 3.68 (two overlapping sept, J = 6.8 Hz, 4 H, CH(CH3)2), 6.76–7.18 (m, 6 H, ArH); 13C{1H} NMR (100.6 MHz, 298 K, C6D6): δ 22.7 (NCH(CH3)2), 24.0 (CH(CH3)2), 24.2 (CH(CH3)2), 27.4 (CH(CH3)2), 47.0 (HCN), 117.3, 122.5, 141.1, 145.5 (ArC), 153.2 (CN3); IR (Nujol): ν/cm−1 = 1613 (s), 1584 (m), 1261 (m), 1152 (m), 1098 (m), 933 (m), 779 (m); MS/EI: m/z (%) = 501.3 (M+, 3), 420.3 (M+− K − C3H6, 100). Accurate mass (EI), m/z: calc. for M+: 501.3480, found: 501.3484.Preparation of [{K(THF)2}{Pip(Giso)2}{K(THF)3}] 12
A solution of K[N(SiMe3)2] (0.336 g, 1.68 mmol) in THF (15 cm3) was added to a solution of Pip(GisoH)24 (0.65 g, 0.801 mmol) in THF (25 cm3) at 20 °C. The resultant mixture was stirred for 1 h, concentrated to ca. 15 cm3 and then cooled to −30 °C to afford colourless crystals of 12 (yield 0.46 g, 52%); mp >300 °C; IR (Nujol): ν/cm−1 = 1620 (s), 1584 (m), 1238 (m), 1195 (m), 1050 (m), 987 (m), 840 (m), 799 (m), 758 (m), 736 (m); MS/APCI: m/z (%) = 811.4 (Pip(GisoH)2H+, 100). N.B. The very low solubility of 12 in common deuterated solvents precluded the acquisition of meaningful NMR data.X-Ray crystallography
Crystals of 1–12 suitable for X-ray structural determination were mounted in silicone oil. Crystallographic measurements were made using a Nonius Kappa CCD diffractometer. The structures were solved by direct methods and refined on F2 by full-matrix least squares (SHELX97)28 using all unique data. Hydrogen atoms have been included in calculated positions (riding model) for all structures, with the exception of the methyl hydrogens of C(21) and C(24) in the structure of 11 which were not included in the refinement. Two crystallographically independent molecules were refined in the asymmetric units of the crystal structures of 3 and 7. No significant geometric differences were found between the two molecules in each structure and therefore only the metrical parameters for one molecule from each structure are reported here. The Flack parameter for the crystal structure of compound 11 is 0.01(6). Crystal data, details of data collections and refinement are given in Table 2.| Compound | 1 | 2 | 3 | 4·2CHCl3 | 5 | 6 |
|---|---|---|---|---|---|---|
| Empirical formula | C37H57N3 | C31H49N3 | C32H49N3 | C56H80Cl6N6 | C37H57N2P | C37H45N2P |
| M r | 543.86 | 463.73 | 475.74 | 1049.96 | 560.82 | 548.72 |
| T/K | 123(2) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/n | Pbca | P21/c | P21/c | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 12.265(3) | 18.397(4) | 19.236(4) | 13.042(3) | 10.960(2) | 10.847(2) |
| b/Å | 17.424(4) | 15.542(3) | 16.327(3) | 12.030(2) | 26.459(5) | 10.942(2) |
| c/Å | 15.775(3) | 20.168(4) | 19.536(4) | 18.989(4) | 12.934(3) | 14.156(3) |
| α/° | 90 | 90 | 90 | 90 | 90 | 96.42(3) |
| β/° | 90.43(3) | 90 | 106.01(3) | 91.97(3) | 112.61(3) | 101.60(3) |
| γ/° | 90 | 90 | 90 | 90 | 90 | 102.50(3) |
| V/Å3 | 3371.2(12) | 5767(2) | 5898(2) | 2977.7(10) | 3462.6(12) | 1585.4(6) |
| Z | 4 | 8 | 8 | 2 | 4 | 2 |
| D c/Mg m−3 | 1.072 | 1.068 | 1.072 | 1.171 | 1.076 | 1.149 |
| μ(Mo-Kα)/mm−1 | 0.062 | 0.062 | 0.062 | 0.328 | 0.105 | 0.114 |
| F(000) | 1200 | 2048 | 2096 | 1120 | 1232 | 592 |
| No. reflections collected | 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 325 325 |
19664 |
21080 |
10581 |
14712 |
10001 |
| No. independent reflns | 7339 | 5343 | 11498 | 5520 | 7517 | 5420 |
| R int | 0.1167 | 0.0382 | 0.0500 | 0.0272 | 0.0299 | 0.0306 |
| Final R1 (I > 2σ(I)) | 0.0629 | 0.0435 | 0.0603 | 0.0465 | 0.0479 | 0.0464 |
| Final wR2 (all data) | 0.1584 | 0.1069 | 0.1527 | 0.1147 | 0.1227 | 0.1159 |
| Compound | 7 | 8·hexane | 9 | 10 | 11 | 12·2THF |
|---|---|---|---|---|---|---|
| Empirical formula | C40H61N3Si | C68H110N6 | C74H112Li2N6 | C41H64LiN3O | C31H48KN3 | C82H132K2N6O7 |
| M r | 612.01 | 1011.62 | 1099.58 | 621.89 | 501.82 | 1392.14 |
| T/K | 123(2) | 150(2) | 123(2) | 150(2) | 123(2) | 150(2) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21/c | P21/c | C2/c | P21/c | P212121 | P21/n |
| a/Å | 35.510(7) | 15.467(3) | 20.707(4) | 18.807(4) | 11.462(2) | 24.500(5) |
| b/Å | 9.9877(2) | 22.956(5) | 12.031(2) | 11.783(2) | 11.997(2) | 16.473(3) |
| c/Å | 21.966(4) | 19.774(4) | 26.802(5) | 18.773(4) | 21.319(4) | 20.494(4) |
| α/° | 90 | 90 | 90 | 90 | 90 | 90 |
| β/° | 104.59(3) | 111.87(3) | 104.33(3) | 113.20(3) | 90 | 93.48(3) |
| γ/° | 90 | 90 | 90 | 90 | 90 | 90 |
| V/Å3 | 7456(3) | 6516(2) | 6469(2) | 3823.8(13) | 2931.6(10) | 8256(3) |
| Z | 8 | 4 | 4 | 4 | 4 | 4 |
| D c/Mg m−3 | 1.090 | 1.031 | 1.129 | 1.080 | 1.137 | 1.120 |
| μ(Mo-Kα)/mm−1 | 0.093 | 0.059 | 0.064 | 0.063 | 0.204 | 0.168 |
| F(000) | 2688 | 2240 | 2416 | 1368 | 1096 | 3040 |
| No. reflections collected | 58137 |
16169 |
18157 |
24208 |
22486 |
27961 |
| No. independent reflns | 16069 |
11404 |
5651 | 8282 | 5060 | 14497 |
| R int | (0.0810) | (0.0423) | (0.1428) | (0.0304) | (0.1036) | (0.0408) |
| Final R1 (I > 2σ(I)) | 0.0546 | 0.0755 | 0.0669 | 0.0501 | 0.0737 | 0.0713 |
| Final wR2 (all data) | 0.1380 | 0.1828 | 0.1301 | 0.1236 | 0.1371 | 0.1858 |
Acknowledgements
We gratefully acknowledge financial support from the Australian Research Council (fellowships for C. J. and A. S.), the Leverhulme Trust (fellowship for A. S.), the Erasmus scheme of the European Union (travel grant for K. L.), the Royal Society (fellowship for G. J.), and the US Air Force Asian Office of Aerospace Research and Development. Thanks also go to the EPSRC Mass Spectrometry Service, Swansea.References
- For general references on the structure and reactivity of amidinate and guanidinate complexes, see: (a) J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219 CrossRef CAS; (b) F. T. Edelmann, Coord. Chem. Rev., 1994, 137, 403 CrossRef; (c) P. J. Bailey and S. Price, Coord. Chem. Rev., 2001, 214, 91 CrossRef CAS; (d) M. P. Coles, Dalton Trans., 2006, 985 RSC; (e) P. C. Junk and M. L. Cole, Chem. Commun., 2007, 1579 RSC , and references therein.
- N. Nimitsiriwar, V. C. Gibson, E. L. Marshall, A. J. P. White, S. H. Dale and M. R. J. Elsegood, Dalton Trans., 2007, 4464 RSC.
- S. R. Foley, Y. Zhou, G. P. A. Yap and D. S. Richeson, Inorg. Chem., 2000, 39, 924 CrossRef CAS.
- S. Dagorne, I. A. Guzei, M. P. Coles and R. F. Jordan, J. Am. Chem. Soc., 2000, 122, 274 CrossRef CAS.
- J. Barker, N. C. Blacker, P. R. Phillips, N. W. Alcock, W. Errington and M. G. H. Wallbridge, J. Chem. Soc., Dalton Trans., 1996, 431 RSC.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031 CrossRef CAS.
- (a) C. Cui, H. W. Roesky, H. -G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274 CrossRef CAS; (b) N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991 RSC; (c) M. S. Hill and P. B. Hitchcock, Chem. Commun., 2004, 1818 RSC; (d) M. S. Hill, P. B. Hitchcock and R. Pongtavornpinyo, Dalton Trans., 2005, 273 RSC.
- C. Jones, P. C. Junk, J. A. Platts, D. Rathmann and A. Stasch, Dalton Trans., 2005, 2497 RSC.
- C. Jones, P. C. Junk, J. A. Platts and A. Stasch, J. Am. Chem. Soc., 2006, 128, 2206 CrossRef CAS.
- S. P. Green, C. Jones and A. Stasch, Inorg. Chem., 2007, 46, 11 CrossRef CAS.
- G. Jin, C. Jones, P. C. Junk, A. Stasch and W. D. Woodul, New J. Chem., 2008, 32, 835 RSC.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754 CrossRef CAS.
- S. P. Green, C. Jones, P. C. Junk, K.-A. Lippert and A. Stasch, Chem. Commun., 2006, 3978 RSC.
- S. P. Green, C. Jones, G. Jin and A. Stasch, Inorg. Chem., 2007, 46, 8 CrossRef CAS.
- C. Jones, D. P. Mills and A. Stasch, Dalton Trans., 2008, 4799 RSC.
- D. Heitmann, C. Jones, P. C. Junk, K. -A. Lippert and A. Stasch, Dalton Trans., 2007, 187 RSC.
- C. Jones, R. P. Rose and A. Stasch, Dalton Trans., 2007, 2997 RSC.
- S. P. Green, C. Jones, K. -A. Lippert, D. P. Mills and A. Stasch, Inorg. Chem., 2006, 45, 7242 CrossRef CAS.
- R. E. Boeré, R. T. Boeré, T. Masuda and G. Wolmershäuser, Can. J. Chem., 2000, 78, 1613 CrossRef CAS.
- Z. Lu, G. P. A. Yap and D. S. Richeson, Inorg. Chem., 1999, 38, 5788 CrossRef CAS.
- See for example (a) M. P. Coles and P. B. Hitchcock, Chem. Commun., 2002, 2794 RSC; (b) J. Grundy, M. P. Coles and P. B. Hitchcock, Dalton Trans., 2003, 2573 RSC; (c) N. E. Mansfield, M. P. Coles and P. B. Hitchcock, Dalton Trans., 2005, 2833 RSC; (d) N. E. Mansfield, M. P. Coles and P. B. Hitchcock, Dalton Trans., 2006, 2052 RSC; (e) N. E. Mansfield, J. Grundy, M. P. Coles, A. G. Avent and P. B. Hitchcock, J. Am. Chem. Soc., 2006, 128, 13879 CrossRef CAS.
- W. -X. Zhang, M. Nishiura and Z. Hou, Chem. Commun., 2006, 3812 RSC.
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2008, 27, 497 CrossRef CAS.
- R. J. Baker and C. Jones, J. Organomet. Chem., 2006, 691, 65 CrossRef CAS.
- As determined from a survey of the Cambridge Crystallographic Database, May, 2008.
- C. Knapp, E. Lork, P. G. Watson and R. Mews, Inorg. Chem., 2002, 41, 2014 CrossRef CAS.
- K. Ogawa and M. Akazawa, Jpn. Pat. Appl., JP 91-208987910517, 1993.
- G. M. Sheldrick, SHELX-97, University of Göttingen, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP diagrams for 2 and 3. Crystallographic data (excluding structure factors) for the structures of 1–12. CCDC reference numbers 704662 (1), 704663 (2), 704664 (3), 704665 (4·2CHCl3), 704666 (5), 704667 (6), 704668 (7), 699384 (8·hexane), 704669 (9), 704670 (10), 704671 (11), 704672 (12·2THF). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b809120j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |