Probing the interaction of arsenobetaine with blood plasma constituents in vitro: an SEC-ICP-AES study
Katie L.
Pei
and
Jürgen
Gailer
*
Department of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, AB, T2N 1 N4, Canada. E-mail: jgailer@ucalgary.ca; Fax: +1-403-289-9488; Tel: +1-403-210-8899
First published on 23rd July 2009
Abstract
Arsenobetaine, which is frequently ingested by humans via the consumption of seafood, is rapidly excreted unchanged in urine, but not much is known about its transport in the mammalian bloodstream. To assess whether this transport involves binding to plasma proteins, rabbit and human plasma were spiked with arsenobetaine and the mixture was analyzed (after 5 min and again after 6 h) by size-exclusion chromatography (SEC) coupled on-line to an inductively coupled plasma atomic emission spectrometer (ICP-AES). Simultaneous monitoring of the emission lines of As, Cu, Fe and Zn in the column effluent allowed us to determine the elution of arsenobetaine relative to that of the major Cu, Fe and Zn-containing metalloproteins. Over the investigated time period, a single As peak eluted near the inclusion volume on two different SEC columns with fractionation ranges of 600–10 KDa and 7000–100 Da. These results indicate that arsenobetaine did not bind to plasma proteins and that SEC-ICP-AES is a useful tool to rapidly probe toxicologically and pharmacologically-relevant interactions between organometalloid compounds and mammalian blood plasma constituents in vitro.
Introduction
Arsenobetaine [(CH3)3As+CH2COO−] has been identified as the predominant organoarsenic compound in a large variety of marine animals,1–3 some terrestrial mushrooms,4 and—quite unexpectedly—in terrestrial birds in an area of naturally and anthropogenically elevated concentrations of inorganic arsenic.5 Systematic studies into the accumulation of individual inorganic and organoarsenic compounds by blue mussels (Mytilus edulis) have revealed that arsenobetaine is efficiently accumulated, but not metabolized in the mussel tissues.6 In view of the fact that blue mussels as well as other marine organisms are frequently ingested by humans, it is not surprising that the mammalian toxicity of arsenobetaine has been extensively assessed. Several studies which involved the administration of varying doses of this compound to animals conclusively established it to be essentially non-toxic,7–12 non-mutagenic11,13 and non-embryotoxic.14 Investigations specifically aimed at detecting its cytotoxicity, morphological neoplastic transformation, intracellular retention and general metabolic behavior at the cellular level (using BALB/3T3 Cl A 31-1-1 cells) revealed that it does not appreciably interact with intracellular components and that its carcinogenic potential is negligible.9 Interestingly, however, in vitro studies with cultured murine bone marrow cells revealed a significant dose-dependent effect on the cell viability when μM concentrations of arsenobetaine were added to the growth medium during a 72 h incubation [this effect was not seen with the structurally related compound glycinebetaine, (CH3)3N+CH2COO−)].15,16 An early study into the excretion of orally administered arsenobetaine by humans revealed a rapid excretion of the unchanged compound in urine,17 but in a later study an unusually high urinary excretion of arsenobetaine was observed in some human control subjects who had not consumed As-rich food for 3 days prior to the experiment.18 Thus, even though it is now generally accepted that the ingestion of arsenobetaine-containing food items does not pose a human health hazard and may even exert a beneficial biological effect,16 some ambiguities about the metabolism of this organoarsenic compound in humans remain.18Although arsenobetaine has proven useful for studies into the mammalian metabolism of arsenite,19 not much is known about the mechanism by which arsenobetaine—after it has been absorbed from the gastrointestinal tract—is transported in the mammalian bloodstream for its subsequent excretionvia the kidneys. Since numerous medicinal drugs which are approved for administration to humans, (e.g.aspirin, warfarin and diazepam) bind to human serum albumin in blood plasma,20arsenobetaine could similarly bind to this or another plasma protein. Even though the latter question has not been investigated experimentally, it would require a liquid chromatographic separation method that is capable of separating the major plasma proteins using a physiologically relevant buffer and a detector that can simultaneously detect arsenobetaine and the separated plasma proteins. If plasma were then to be spiked with arsenobetainein vitro and analyzed, the elution of arsenobetaine relative to the separated plasma proteins would indicate if binding to plasma proteins had occurred. Importantly, this approach would reveal if binding occurred under near physiological conditions.
To this end, we have recently developed an instrumental analytical method for the separation of the major Cu, Fe, and Zn-containing metalloproteins that are contained in rabbit plasma using size-exclusion chromatography (SEC) and the simultaneous on-line detection of the associated metals by a state-of-the-art charge injection device (CID)-based inductively coupled plasma atomic emission spectrometer (ICP-AES).21 Since the latter instrument can simultaneously detect several metals and/or metalloids, this SEC-ICP-AES method should be suited to probe the in vitro binding of pharmacologically interesting metalloid compounds, such as arsenobetaine,16 to plasma proteins in blood plasma. Even though the related instrumental analytical technique, SEC-ICP-MS, has also been successfully used to study the binding of Pt-based anti-cancer drugs to plasma proteins, the majority of these studies employed Tris–HCl buffers for the separation of the proteins and monitored only the element of interest (e.g. Pt) by ICP-MS.22,23 Conversely, the SEC-ICP-AES method that was employed in the present study employs a phosphate buffered saline (PBS) mobile phase for the separation of plasma proteins and simultaneously detects Cu, Fe, and Zn in addition to the element of interest. This allows one to measure the elution of the element of interest relative to the major Cu, Fe, and Zn-containing metalloproteins (the latter are collectively referred to as the plasma metalloproteome in the context of this paper).
Our main focus was to evaluate our recently developed SEC-ICP-AES method for its capability to probe the in vitro binding of the organometalloid compound arsenobetaine to plasma constituents in blood plasma. We therefore added arsenobetaine to fresh rabbit and human plasma and analyzed the resulting mixture after 5 min and again after 6 h by SEC-ICP-AES. Simultaneous monitoring of the emission lines of As, Cu, Fe, and Zn in the SEC column effluent was intended to provide insight into the relative elution of arsenobetaine compared to the major plasma proteins and therefore its overall binding behavior. Our deliberate decision to study the binding of arsenobetaine to plasma proteins in vitro represents the first practical application of our systematically developed SEC-ICP-AES method and will simplify the interpretation of the chromatographic results since this organoarsenic compound is not metabolized by mammals.17 Accordingly, only a single As-peak is expected to elute in the SEC column effluent, whereas choosing an organometalloid compound that is biotransformed in plasma would likely result in the detection of multiple peaks and therefore unnecessarily complicate the interpretation of the results.
Experimental
Chemicals
Phosphate-buffered saline buffer (PBS) tablets were purchased from Sigma-Aldrich (St. Louis, MO, USA) and PBS of pH 7.4 (10 mM phosphate, 2.7 mM KCl and 137 mM NaCl) was prepared by dissolving PBS tablets in the appropriate volume of water (followed by pH adjustment with dilute HCl if necessary) and its filtration through 0.45 μm nylon-filter membranes (Mandel Scienctific, Guelph, ON, Canada). A mixture of protein standards which contained thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.35 kDa) was obtained from Bio-Rad Laboratories (Hercules, CA, USA) to calibrate the Superdex 200 SEC column. Another mixture of peptide and amino acid standards was prepared by dissolving 33 mg of oxidized glutathione (GSSG), 47 mg of reduced glutathione (GSH), and 75 mg glycine in 5.0 ml water from a Simplicity waterpurification system (Millipore, Billerica, MA, USA) to calibrate the Superdex Peptide SEC column. Arsenobetaine bromide was synthesized according to a previously published procedure and its recrystallization from ethanol yielded white crystals with a melting point of 225 °C (literature value 227 °C).24SEC-ICP-AES system
The SEC-ICP-AES system consisted of a Smartline 1000 HPLC pump (Knauer, Berlin, Germany), a Rheodyne 9010 PEEK injection valve (Rheodyne, Rhonert Park, CA, USA) equipped with a 0.5 mL PEEK injection loop and a pre-packed Superdex™ 200 10/300 GL Tricorn™ high performance size-exclusion chromatography column (30.0 × 1.0 cm I.D., separates globular proteins between ∼600 kDa and ∼10 kDa; GE Healthcare, Piscataway, NJ, USA) or a Superdex™ Peptide HR 10/30 size-exclusion chromatography column (30.0 × 1.0 cm I.D., separates globular proteins between 7 kDa and 100 Da; GE Healthcare, Piscataway, NJ, USA). The exit of each SEC column was connected to the Meinhard concentric glass tube nebulizer of the ICP-AES with FEP Teflon tubing (30 cm, I.D. 0.5 mm). PBS-buffer served as the mobile phase and a flow rate of 1.0 mL/min was employed (column temperature: 22 °C). Simultaneous multielement-specific detection of C (193.091 nm), S (180.731 nm), P (213.618 nm), As (189.042 nm), Cu (324.754 nm), Fe (259.837 nm), and Zn (213.856 nm) in the column effluent was achieved with a Prodigy, high-dispersion, radial-view ICP-AES (Teledyne Leeman Labs, Hudson, NH, USA) at an Ar gas-flow rate of 19 L min−1, an RF power of 1.3 kW and a nebulizer gas pressure of 35 psi. The detector technology utilized in the Prodigy (a charge injection device or CID) allows the simultaneous measurement of the peak and the background emissions to generate the net emission intensity. This capability is critical in experiments where the background emission intensity changes (e.g. when a major protein peak reaches the ICP-AES) so the operator is not mislead into believing that an analytically significant event has occurred when in fact it has not. This advantage, together with the ability of ICP-AES to handle salt-containing solutions, makes the Prodigy well suited for the LC analysis of metal or metalloid-containing compounds with salt-containing mobile phases. Time scans were performed using the time-resolved analysis (TRA) mode (Salsa software version 3.0) and a data acquisition rate of 1 data point per 2 s. The raw data were imported into Sigmaplot 11 and smoothed using the bisquare algorithm. According to the void volume of the Superdex 200 column of 7.95 mL (477 s) determined by the injection of blue dextran (and C-specific detection), a 7.0 min delay was implemented between injection and the beginning of data acquisition and a 1000 s acquisition window was used. With regard to the Superdex Peptide column and the experimentally determined void volume of 7.22 mL (433 s, blue dextran), a 6.0 min delay and a similar acquisition window was used.SEC-ICP-AES analysis of arsenobetaine-spiked rabbit and human plasma
The Animal Care Committee of the University of Calgary approved the procedure to collect blood from New Zealand white rabbits (Protocol Approval #BI 2008-05). Similarly, the collection of blood from humans was approved by the Calgary Conjoint Health Research Ethics Board (Approval No. E-21198) of the University of Calgary. Male New Zealand white rabbits were purchased from Casey Vandermeer (Edmonton, AB, Canada), fed ad libitum on a “high fiber” diet (Lab Diet 5321, Canadian Lab Diets, Leduc, AB, Canada) and fasted 4.5 h before blood collection. Rabbit blood (∼14 mL) was collected from the marginal ear vein with 20-gauge stainless blood collection needles (211 monoject, Sherwood Medical, St. Louis, MO, USA) into 2 heparinized trace metal testing blood collection tubes (Monoject, Tyco Healthcare Group, Mansfield, MA, USA). Blood was also collected from healthy male humans (12 h-fasted) into heparinized blood collection tubes. Thereafter, rabbit and human blood was centrifuged at 1100 g (4 °C) for 10 min and the supernatant plasma was removed with a micropipette. Plasma (1.3 mL) was then spiked with 5 μL of an aqueous solution of arsenobetaine (0.468 M). Five min after mixing, 0.5 mL of the spiked (rabbit or human) plasma (which contained 67 μg of As in form of arsenobetaine) was injected onto the SEC-ICP-AES system (Superdex 200 SEC column) and another aliquot (0.5 mL) was analyzed after 6 h standing at room temperature (since no changes with regard to the relative elution of arsenobetaine compared to the plasma metalloproteins were observed over this time period, no additional time point was investigated). These experiments were carried out in duplicate. Based on the obtained As, Cu, Fe and Zn-specific chromatograms, the total peak area of all detected peaks per element were determined. These data were intended to reveal potential losses of metals caused by the degradation of plasma proteins (e.g. to the container wall) over the 6 h time period. The retention time of albumin was delineated from the most intense S-peak in the simultaneously monitored S-specific chromatogram. A recovery experiment was conducted in which the peak area of the As-peak that was obtained from the spiked plasma (on the Superdex 200 column) was compared to that obtained after the injection of arsenobetaine in PBS-buffer only. The results revealed that virtually all arsenobetaine that was added to plasma eluted from the SEC column (recovery >98%). Finally, arsenobetaine-spiked human plasma was also analyzed by SEC-ICP-AES on a size-calibrated Superdex Peptide SEC column at the same time points that were used for the rabbit plasma experiments (the retention time of arsenobetaine was identical to that of an arsenobetaine standard and different from that of dimethylarsinic acid, methylarsonic acid and arsenite standards).Results and discussion
Owing to the exposure of the general population to a variety of environmentally abundant inorganic and organic metal and metalloid compounds via the diet,25 efficient instrumental analytical methods have been developed over the last ∼20 years to quantify these potentially toxic compounds in various food items to better assess their overall toxicity.25,26Arsenobetaine represents an essentially non-toxic compound which is present in a large variety of marine organisms that are used for human consumption and therefore probably represents the organoarsenic compound to which humans are most frequently exposed. Interestingly, a remarkably potent biological effect of arsenobetaine on the cell viability of mammalian bone marrow cells in cell culture experiments was discovered in 2001.16 This finding, which essentially elevates arsenobetaine to be regarded as a model compound for a metal-based drug combined with the fact that this compound is not metabolized in humans,17,27 prompted us to study its transport in the mammalian bloodstream. In particular, we were interested in investigating the potential in vitro binding of arsenobetaine to plasma proteins, such as albumin , by applying a recently developed instrumental analytical method.21 This method is based on the combination of a relatively crude SEC-based fractionation of blood plasma proteins using PBS-buffer as the mobile phase and the highly specific simultaneous detection of the major Cu, Fe and Zn-containing plasma metalloproteins by ICP-AES.21 Following this approach, the complexity that is usually associated with the analysis of a mixture as convoluted as plasma (it contains >3700 proteins), could be dramatically reduced and ∼12 Cu, Fe and Zn-containing metalloproteins were detected.21 Since ICP-AES inherently allows one to simultaneously detect other metals/metalloids in addition to the aforementioned ones, we applied this SEC-ICP-AES method to monitor the elution of arsenobetaine (which had been added to plasma) relative to the endogenous Cu, Fe and Zn-containing plasma metalloproteins.After the addition of arsenobetaine to fresh rabbit plasma, the obtained mixture (67 μg As/0.5 mL plasma) was analyzed after 5 min and again after 6 h. The corresponding As, Cu, Fe, and Zn-specific SEC-ICP-AES chromatograms are depicted in Fig. 1A and B. With regard to the result for the 5 min time-point (Fig. 1A), the total number of Cu, Fe and Zn-peaks that were detected as well as their respective retention times and their relative intensity (to each other) were comparable to our earlier study.21 Notably, however, the Cu-peaks that were previously shown to correspond to coagulation factor V and small molecular weight Cu compounds (see single headed arrows in Fig. 1A) were less intense in the present study, which can be explained by the relatively large standard deviation of the peak intensity/peak area that we reported for these peaks (we noticed during our previous study that the analysis of rabbit plasma often results in very small peaks for these two Cu-metalloproteins).21 Importantly, however, only a single As peak was detected, which—owing to the lack of metabolism of arsenobetaine by mammalian organisms17—likely corresponds to arsenobetaine. The fact that the latter peak essentially eluted in the inclusion volume of the employed SEC column (indicated by the molecular weight marker for vitamin B12 on top of Fig. 1), implies that arsenobetaine did not bind to plasma proteins >10 kDa. The simultaneous Cu, Fe, and Zn-specific chromatogram that was obtained for the 6 h time-point was slightly different from that of the 5 min time-point (Fig. 1A and B), which is expected since some plasma proteins will inevitably undergo proteolytic degradation. Nevertheless, the peak areas that were obtained for all Fe and Zn-peaks detected at the 6 h time-point were very similar to those for the 5 min time-point, whereas ∼20% of Cu was lost over the investigated time period (Table 1). The latter result is in accord with our previous findings and may involve the binding of Cu (which was liberated from labile Cu-metalloproteins, such as coagulation factor V) to the container walls.21 The retention time and the peak area of the peak corresponding to arsenobetaine, however, remained essentially unchanged over the investigated time period (see dotted line in Fig. 1A and B and Table 1).
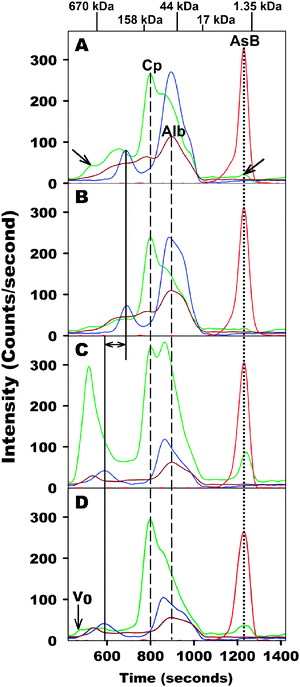 | ||
| Fig. 1 Simultaneous As, Cu, Fe and Zn-specific chromatogram of arsenobetaine-spiked rabbit plasma (A: after 5 min; B: after 6 h) and human plasma (C: after 5 min; D: after 6 h) on a Superdex 200 10/300 GL (30 × 1.0 cm I.D., 13 μm particle size) SEC column at 22 °C using a phosphate buffered saline buffer (PBS, pH 7.4) as the mobile phase (rabbit and human plasma were injected onto the same column). Abbreviations: Cp = ceruloplasmin, Alb = albumin , AsB=arsenobetaine. Flow-rate 1.0 mL/min, injection volume 500 μL, detectorICP-AES at 189.042 nm (As), 324.754 nm (Cu), 259.837 nm (Fe) and 213.856 nm (Zn). The retention times of the molecular weight markers are depicted on top of the figure. For explanation of other denotations (lines and arrows) see text. | ||
| Emission line | Total area counts | |||
|---|---|---|---|---|
| Rabbit (t0) | Rabbit (t6Hr) | Human (t0) | Human (t6Hr) | |
| As 189.042 nm | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 200 |
20100 |
19100 |
19100 |
| Cu 324.754 nm | 68100 |
53800 |
113400 |
61700 |
| Fe 259.837 nm | 45100 |
43200 |
24600 |
24300 |
| Zn 213.856 nm | 34400 |
34200 |
20600 |
20900 |
To corroborate these findings with fresh human plasma, the latter was spiked with arsenobetaine and the obtained mixture (67 μg As/0.5 mL plasma) was analyzed by SEC-ICP-AES as described above. The resulting As, Cu, Fe, and Zn-specific SEC-ICP-AES chromatograms are depicted in Fig. 1C and D. To a first approximation, the same number of Cu, Fe and Zn-peaks were detected in human plasma as in rabbit plasma. In addition, the retention times of the plasma Cu-metalloprotein ceruloplasmin and albumin -bound Zn were identical in both mammalian species (see vertical dashed lines in Fig. 1A–D). In human plasma, however, the retention times of ferritin, transferrin, and α2-macroglobulin were significantly shorter than those obtained for rabbit plasma (see double headed arrow in Fig. 1C corresponding to plasma ferritin; ferritin Δretention time = 104 s; transferrin Δretention time = 26 s; α2-macroglobulin Δretention time=109 s). These results imply different hydrodynamic radii of these metalloproteins in the respective mammalian organisms which could be caused by either a different hydrodynamic radius of the metalloproteins itself, or the aggregation of these metalloproteins with other plasma proteins during the chromatographic separation process. Whereas the comparatively small difference in the retention times for transferrin may be rationalized based on subtle differences in the hydrodynamic radius of the protein itself,28 the marked differences in the retention times for ferritin and α2-macroglobulin (in rabbit and human plasma) are more difficult to explain, especially since the specific cellular origin of mammalian plasma ferritins is currently unknown.28 Importantly, and in accord with the results obtained for rabbit plasma (Fig. 1A and B), however, a single As peak which co-eluted with the small molecular weight Cu-peak was detected at both time points (Fig. 1C and D). Similar to the results obtained for the analysis of rabbit plasma, small changes in the Cu, Fe and Zn-specific chromatogram were detected over the investigated time period (Fig. 1C and D). In addition, the peak areas that were obtained for all Fe and Zn-peaks at the 6 h time-point were very similar to those obtained after the 5 min time-point, whereas a considerably larger amount of Cu, namely ∼45%, was lost over the investigated time period (Table 1). Like the results obtained for rabbit plasma, the retention time and peak area of the As peak corresponding to arsenobetaine remained essentially unchanged over the investigated time period (see dotted line in Fig. 1C and D, Table 1).
These results indicate that arsenobetaine does not bind to plasma proteins >10 kDa in rabbit and human plasma. In order to investigate whether arsenobetaine may bind to plasma proteins <10 kDa, arsenobetaine-spiked human plasma (0.5 mL) was analyzed by SEC-ICP-AES on a Superdex Peptide SEC column, which offers the appropriate fractionation range (7 kDa–100 kDa), after 5 min and again after 6 h. The corresponding As, Cu, Fe, and Zn-specific chromatograms are depicted in Fig. 2. As expected, all Cu, Fe and Zn-containing plasma metalloproteins eluted close to the void volume and essentially in one broad peak. In addition, the area counts obtained for the peaks corresponding to these elements were comparable to the values reported in Table 1. Most importantly, arsenobetaine eluted close to the molecular weight standard GSH over the entire time period, which indicates that it did not bind to plasma constituents >300 Da (Fig. 2A and B).
 | ||
| Fig. 2 Simultaneous As, Cu, Fe and Zn-specific chromatogram of arsenobetaine-spiked human plasma (A: after 5 min; B: after 6 h) on a Superdex Peptide 10/300 GL (30 × 1.0 cm I.D., 13 μm particle size) SEC column at 22 °C using a phosphate buffered saline buffer (PBS, pH 7.4) as the mobile phase. AsB = arsenobetaine. Flow-rate 1.0 mL min−1, injection volume 500 μL, detectorICP-AES at 189.042 nm (As), 324.754 nm (Cu), 259.837 nm (Fe) and 213.856 nm (Zn). The retention times of the molecular weight markers are depicted in the upper right corner of A. | ||
Taken together, these findings indicate the absence of an arsenobetaine transport protein in human plasma. In the context of studies which demonstrated a rapid clearance of non-transferrin bound iron from human plasma,29 our results help to explain why ingested arsenobetaine is rapidly excreted from humans in urine.17 From a bioinorganic perspective, the absence of an arsenobetaine transport protein is somewhat unexpected in view of the fact that the most abundant mammalian plasma protein, serum albumin , displays prolific ligand-binding properties for a large variety of metals [e.g. Au(I), Hg(II), Cu(II), Ni(II)], metal-based drugs (e.g. auranofin, cisplatin),30 medicinal drugs (e.g.aspirin, warfarin, bilirubin, diazepam, ibuprofen) and amino acids (e.g.L-tryptophan).20 In order to put the results into a wider context, however, it is instructive to look at the structurally analogous zwitterionglycinebetaine[(CH3)3N+CH2COO−](the nitrogen analog of arsenobetaine), which is naturally present in human blood plasma at concentrations of 20–60 μmol L−1.31 With regard to the function of glycinebetaine in mammalian organisms, this compound has been demonstrated to serve as an osmolyte (to maintain normal cell volume),32 a methyl-donor and as a chaperone to protect proteins against denaturation.33 Since glycinebetaine must presumably be freely available in plasma (i.e. not bind to plasma proteins) in order to serve its role as an osmolyte and as a chemical chaperone, it is only reasonable to expect that the structurally analogous arsenobetaine should behave similarly, which—according to our results—it does (Fig. 1, Fig. 2). Furthermore, the fact that glycinebetaine has been identified as one of the major osmolytes in the renal medulla of kidneys in mammals,32 may explain the unusually high urinary excretion of arsenobetaine from human volunteers that had consumed an As-free diet for 3 days prior to the collection and analysis of urine.18 Based on our data, these results cannot be caused by the slow clearance of arsenobetaine from blood owing its binding to a plasma protein, but could be explained by a likely co-localization of glycinebetaine and arsenobetaine in the kidney and an experimental diet-induced release of the latter into the urine. Even though this explanation is somewhat speculative, it appears scientifically feasible.
According to our investigations into the binding of the biologically active “model metallodrug” arsenobetaine to plasma proteins in native plasma in vitro (Fig. 1 and 2), SEC-ICP-AES is identified as a useful tool to probe metallodrug–plasma protein interactions in a similar fashion. These interactions are of considerable pharmacological importance because the efficacy of new metal or metalloid-based drugs critically depends on their binding to plasma proteins. In fact, many promising new drugs have been rendered ineffective because of their unusually high affinity for albumin 20 and the binding of a promising new drug to albumin has therefore been referred to as the “second step in rational drug design”.20 In addition, interactions between metal-based drugs and plasma proteins are also important because the antitumor activity of a drug is usually modified when the drug is complexed by a protein.23 Finally, interactions between metal-based drugs and protein targets within a given proteome are much less studied than metallodrug–DNA interactions, yet they hold the potential to identify new targets for drug therapy.34 Considering the increased recent interest in such metallodrug–plasma protein interactions,22,34,35 appropriate analytical techniques are likely to become increasingly important.22,35 Even though a related analytical technique, namely SEC-ICP-MS, has also been successfully applied to study the binding of Pt-containing anti-cancer drugs to plasma proteins using 30 mM Tris-HCl buffer of pH 7.2 (to separate the proteins),23 the SEC-ICP-AES method that was employed in the present study uses a PBS-buffer to separate the plasma proteins. This should, in principle, avoid interactions that could possibly occur between plasma metalloproteins and the major Tris-buffer constituent tris(hydroxymethyl)aminomethane during the separation process itself when SEC-ICP-MS is used. In fact, tris(hydroxymethyl)aminomethane is known to form complexes with free metal ions36 and this mobile phase constituent could therefore possibly abstract metal ions from a metalloprotein which could result in ambiguous results when the binding of a metallodrug to plasma proteins is studied. Furthermore, the SEC-ICP-AES approach is based on the simultaneous monitoring of the emission lines of Cu, Fe and Zn in addition to the element of interest which offers the additional capability to visualize a potential dose-dependent effect of the compound of interest on the plasma Cu, Fe and Zn-metalloproteome (which could, for example, result in the disappearance of a specific metalloprotein peak and the corresponding elution of a new metal peak in the inclusion volume).21 Studies are currently underway to explore this potential capability of SEC-ICP-AES.
Conclusion
The addition of arsenobetaine to rabbit and human plasma followed by the analysis of the resulting mixture by SEC-ICP-AES revealed that arsenobetaine does not bind to plasma proteins >300 Da. These investigations represent the first practical application of our newly developed SEC-ICP-AES method and indicate that—at least from an analytical point of view—SEC-ICP-AES must be considered as an important new addition to the relatively few instrumental analytical methods that allow one to directly determine metallodrug–plasma protein interactions in undiluted plasma.22 Based on the conceptually simple approach of adding the metal or metalloid-containing compound of interest to plasma in vitro and to rapidly analyse the obtained mixture by SEC-ICP-AES, the binding of established metal-based anti-cancer drugs22 and novel inorganic medicinal drugs (that have already been demonstrated to exert the desired biological activity at the cellular level)37,38 to plasma constituents can now be studied. Taking into account the speed that SEC-ICP-AES offers in gathering relevant data about the in vitro binding of a metallodrug to plasma proteins in native plasma (results can be obtained in <25 min), this hyphenated analytical technique is destined to play an important role in probing toxicologically and pharmacologically relevant interactions between metal-based drugs and plasma constituents in the near future.Acknowledgements
This research was funded by the Natural Sciences and Engineering Research Council (NSERC) of Canada. The staff of the Animal Health Unit (LESARC) at the University of Calgary are gratefully acknowledged for the maintenance and the drawing of blood from the rabbits. Shawn A. Manley and Ted S. Sorensen are gratefully acknowledged for providing constructive feedback on the final draft of the manuscript.References
- K. A. Francesconi and J. S. Edmonds, Oceanogr. Mar. Biol. Annu. Rev., 1993, 31, 111–151 Search PubMed.
- E. H. Larsen, G. Pritzl and S. H. Hansen, J. Anal. At. Spectrom., 1993, 8, 1075–1084 RSC.
- Y. Shibata, M. Morita and K. Fuwa, Adv. Biophys., 1992, 28, 31–80 CrossRef CAS.
- A. R. Byrne, Z. Slejkovec, T. Stijve, L. Fay, W. Gössler, J. Gailer and K. J. Irgolic, Appl. Organomet. Chem., 1995, 9, 305–313 CrossRef CAS.
- I. Koch, V. Mace and K. J. Reimer, Environ. Toxicol. Chem., 2005, 24, 1468–1474 CrossRef CAS.
- J. Gailer, K. A. Francesconi, J. S. Edmonds and K. J. Irgolic, Appl. Organomet. Chem., 1995, 9, 341–355 CrossRef CAS.
- J. R. Cannon, J. B. Saunders and R. F. Toia, Sci. Total Environ., 1983, 31, 181–185 CrossRef CAS.
- T. Kaise and S. Fukui, Appl. Organomet. Chem., 1992, 6, 155–160 CrossRef CAS.
- E. Sabbioni, M. Fischbach, G. Pozzi, R. Pietra, M. Gallorini and J. L. Piette, Carcinogenesis, 1991, 12, 1287–1291 CAS.
- T. Kaise, S. Watanabe and K. Itoh, Chemosphere, 1985, 14, 1327–1332 CrossRef CAS.
- Y. Oya-Ohta, T. Kaise and T. Ochi, Mutat. Res., 1996, 357, 123–129 CrossRef.
- T. Kaise, T. Ochi, Y. Oya-Ohta, K. Hanaoka, T. Sakurai, T. Saitoh and C. Matsubara, Appl. Organomet. Chem., 1998, 12, 137–143 CrossRef CAS.
- W. M. F. Jongen, J. M. Cardinaals and P. M. J. Bos, Food Chem. Toxicol., 1985, 23, 669–673 CrossRef CAS.
- T. R. Irvin and K. J. Irgolic, Appl. Organomet. Chem., 1988, 2, 509–514 CrossRef CAS.
- T. Sakurai, Appl. Organomet. Chem., 2002, 16, 401–405 CrossRef CAS.
- T. Sakurai and K. Fujiwara, Br. J. Pharmacol., 2001, 132, 143–150 CrossRef CAS.
- Z. Slejkovec, A. R. Byrne and M. Dermelj, Acta Chim. Slov., 1994, 41, 83–85 CAS.
- V. W.-M. Lai, Y. Sun, E. Ting, W. R. Cullen and K. J. Reimer, Toxicol. Appl. Pharmacol., 2004, 198, 297–306 CrossRef CAS.
- A. J. Percy and J. Gailer, Bioinorg. Chem. Appl., 2008 Search PubMed (2008), Article ID 539082, 8 pages.
- D. C. Carter and J. X. Ho, Adv. Prot. Sci., 1994, 45, 153–203 Search PubMed.
- S. A. Manley, S. Byrns, A. W. Lyon, P. Brown and J. Gailer, J. Biol. Inorg. Chem., 2009, 14, 61–74 CrossRef CAS.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS.
- J. Szpunar, A. Makarov, T. Pieper, B. K. Keppler and R. Lobinski, Anal. Chim. Acta, 1999, 387, 135–144 CrossRef CAS.
- J. Gailer and K. J. Irgolic, J. Chromatogr. A, 1996, 730, 219–229 CrossRef CAS.
- M. Leermakers, W. Baeyens, M. De Gieter, B. Smedts, C. Meert, H. C. De Bisschop, R. Morabito and P. Quevauviller, Trends Anal. Chem., 2006, 25, 1–10 CrossRef CAS.
- A. J. Percy, M. Korbas, G. N. George and J. Gailer, J. Chromatogr. A, 2007, 1156, 331–339 CrossRef CAS.
- K. A. Francesconi, Environ. Chem., 2005, 2, 141–145 CrossRef CAS.
- A. M. Koorts and M. Viljoen, Arch. Physiol. Biochem., 2007, 113, 30–54 CrossRef CAS.
- D. M. De Silva, C. C. Askwith and J. Kaplan, Physiol. Rev., 1996, 76, 31–47 CAS.
- T. Yotsuyanagi, N. Ohta, T. Futo, S. Ito, D. Chen and K. Ikeda, Chem. Pharm. Bull., 1991, 39, 3003–3007 CAS.
- M. Lever, P. C. B. Sizeland, L. M. Bason, C. M. Hayman and S. T. Chambers, Biochim. Biophys. Acta, 1994, 1200, 259–264 CrossRef CAS.
- A. Miyai, A. Yamauchi, T. Moriyama, T. Kaneko, M. Takenaka, T. Sugiura, H. Kitamura, A. Ando, M. Tohyama, S. Shimada, E. Imai and T. Kamada, Kidney Int., 1996, 50, 819–827 CrossRef CAS.
- B. C. Schwahn, D. Hafner, T. Hohlfeld, N. Balkenhol, M. D. Laryea and U. Wendel, Br. J. Clin. Pharmacol., 2003, 55, 6–13 CrossRef CAS.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS.
- A. R. Timerbaev, C. G. Hartinger and B. K. Keppler, Trends Anal. Chem., 2006, 25, 868–875 CrossRef CAS.
- B. E. Fischer, U. K. Haering, R. Trobolet and H. Sigel, Eur. J. Biochem., 1979, 94, 523–530 CrossRef CAS.
- T. W. Hambley, Science, 2007, 318, 1392–1393 CrossRef CAS.
- T. W. Hambley, Dalton Trans., 2007, 4929–4937 RSC.
| This journal is © The Royal Society of Chemistry 2009 |