Advances in developing tris(8-quinolinolato)gallium(III) as an anticancer drug: critical appraisal and prospects
Andrei R.
Timerbaev
*
Vernadsky Institute of Geochemistry and Analytical Chemistry, Russian Academy of Sciences, Kosygin St. 19, 119991, Moscow, Russia. E-mail: andrei.timerbaev@univie.ac.at
First published on 9th April 2009
Abstract
Gallium-based anticancer chemotherapeutics are appreciably progressing in clinical studies. A steady interest of drug developers and clinicians in gallium compounds is due to a proven ability of gallium cations to inhibit tumour growth, on the one hand, and enhanced bioavailability and moderate toxicity provided by the conversion of gallium into chelate complexes, on the other. One of the complexes suitable for a more convenient oral administration is tris(8-quinolinolato)gallium(III) (KP46). Nominated from a range of gallium complexes for the clinical stage of development, KP46 has finished phase I trials with the outcome of promising tolerability and evidence of clinical activity in renal cell carcinoma. Therefore, there is obviously a need to codify and critically evaluate the continuing advances in the emergence of KP46 as a lead-drug candidate. Additionally, many questions remain unanswered regarding the relevant biological reactivity, modes of delivery and action and potential cell target(s) of KP46. The timely publication of the present review is also an attempt to shed light on these pertinent drug assets and to accelerate research activities towards further clinical development of KP46.
 Andrei R. Timerbaev Andrei R. Timerbaev | Andrei R. Timerbaev received his MS in Chemistry from Moscow State University (1975) and PhD in analytical chemistry from Vernadsky Institute of Geochemistry and Analytical Chemistry, Moscow (1985). He was a research scientist at this institute from 1975 to 1985 and joined it again as a head scientist in 1996. From 1986, he spent 6 years at the Department of Analytical Chemistry of Mendeleev University of Chemical Technology, Moscow, ending as a senior researcher, and habilitated in 1991, earning a Doctor of Science degree in chemistry. In 1991, he became a visiting research professor at Johannes-Kepler University, Linz, Austria. He has also been part-time professor in analytical chemistry at the Mendeleev University (2001–2005) and research professor at the Institute of Inorganic Chemistry, University of Vienna (2003–2008). |
Introduction
Among the incredible number and variety of roles that metals play in contemporary medicine, cancer treatment is appreciated as arguably the most prominent application of metal-based drugs. After the success of cisplatin and its analogues in chemotherapy, ruthenium and gallium are second in importance, the anticancer potential of whose compounds has been assessed in an enormous number of studies.1–7 These efforts have led, and are leading, to vast improvements in molecular design, to improved anticancer behaviour and to the better understanding of the basic underlying chemistry and biochemical mechanisms of cytotoxicity. A number of therapeutic agents have been advanced to entering clinical trials but the rate of failure continues to be overwhelming, with the depressing outcome of no non-platinum anticancer metallo-drugs ever being approved. The main reason for this is the fact that a definite rationale of desirable therapeutic efficacy and pharmacological profiles is lacking for drug-like metal compounds. This situation is complicated by the often non-parallel research activities of inorganic and biological chemists. Furthermore, the expanding literature tends to be fragmented into scholarly specialized discourse. To address these challenges, the summarizing of available published accounts on a particular compound appears to be a solution that would provide drug developers with an integrated guide on how to progress a drug’s development in the most effective manner. This was a major driving force behind the publication of the present review related to the title gallium(III) complex, which is, before long, expected to be scheduled for phase II clinical examination.A state-of-the-art picture of gallium compounds in cancer therapy
The systematic investigation of gallium compounds for their applicability as tumour-inhibiting agents commenced in the early 1970s. As highlighted in a series of reviews,2,6–12 the past decades of rigorous research both in academic and clinical laboratories revealed certain attractive features of gallium cytotoxics compared to classical platinum-based or developmental ruthenium(III) chemotherapeutic drugs. It has been proven that gallium(III) exerts a strong antiproliferative action, and such an activity has much to do with its interference with iron cellular metabolism. A weaker inclination to hydrolysis and redox transformations under physiological conditions makes Ga3+ ions more bioavailable than, for instance, Ru3+. Coordination characteristics similar to iron(III) facilitate binding to human serum proteins which ensures gallium-based agents a lesser toxicity than that of platinum drugs. Unfavourable pharmacokinetic and toxicological properties typical to gallium salts can be largely overcome by using gallium complexes with organic ligands (one of which is the target KP46).The mode of action of gallium compounds can be represented by a scheme shown in Fig. 1, the application of which is well proven at least for gallium salts. Briefly, the cellular acquisition of gallium mainly occurs by transferrin-mediated uptake followed by accumulation in endosomes. After transport into the cytosol, gallium(III) binds to and inhibits the functioning of ribonucleotide reductase (RR), an enzyme recognized as the most significant intracellular target for antiproliferative activity of gallium. This impairs the reduction of ribonucleotides to deoxyribonucleotides, DNA replication, activates cell cycle arrest and results finally in apoptosis through the mitochondrial pathway (see Fig. 1 and also ref. 6 for more detail). Apart from the apoptotic activity described, gallium (in the form of nitrate salt) is effective against cancer-related hypercalcemia. Note that this acute complication is frequently observed in advanced cancer forms.
 | ||
| Fig. 1 Schematic representation of the mode of action of gallium. Abbreviations: Tf = transferrin; NDP = nucleoside diphosphate; dNDP = desoxynucleoside diphosphate; BAX = a proapoptosic protein. Reproduced from ref. 6, with permission. | ||
Of a range of gallium compounds tested, four drug candidates have been transferred to clinical trials. Gallium nitrate showed notable efficacy in lymphomas and bladder cancer and in the treatment of hypercalcemia; however, its intravenous administration was accompanied by severe side effects (nephrotoxicity, optical neuropathy). After clinical evaluation was first abandoned for this reason, low-dose gallium nitrate is currently under re-evaluation in patients with non-Hodgkin’s lymphoma. Oral administration of gallium chloride attempting to render prolonged exposure of low steady-state gallium concentrations, was basically ineffective because intestinal absorption was too low to obtain therapeutically active drug doses. Significant and fairly stable gallium blood levels, no dose-limiting toxicities and oral formulations that could be further optimised were found in phase I studies of tris(3-hydroxy-2-methyl-4H-pyran-4-onato)gallium(III) (gallium maltolate) (Fig. 2). This gallium complex was designed to be similar to the iron(III)–maltol complex, which is known to provide iron in a bioavailable, readily absorbable form. The current status of preclinical and early clinical development of another oral gallium-based drug, KP46 (see Fig. 2), will be given in the following sections.
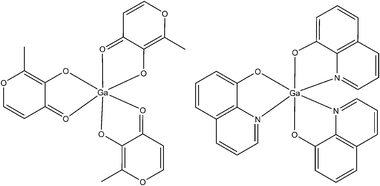 | ||
| Fig. 2 Structural formulas of tris(3-hydroxy-2-methyl-4H-pyran-4-onato)gallium(III) (left) and tris(8-quinolinolato)gallium(III), KP46 (right). | ||
Drug-relevant chemical properties of KP46
A successful drug development program requires careful consideration of various chemical and biochemical assays conducted before, or in parallel with, in vitro tumour inhibiting studies. The characterisation of a metal-based drug includes tests of solubility, stability, lipophilicity (log P), reactivity towards serum transport proteins, etc., the core properties that determine drugs’ absorption, distribution and transport in the body. It should be emphasised here that the concepts of developing anticancer gallium complexes were essentially derived from experience with gallium salts. Surprisingly, while both gallium nitrate and chloride are well characterised in terms of desirable drug-like properties, some tests seem to be incomplete in the case of KP46.(a) Solubility
The only systematic study on the solubility of KP46 (among other cytotoxic gallium complexes)13 reports an aqueous solubility of 3.6 × 10−5 M. It is unlikely that this threshold could be higher in the slightly basic environment of the small intestine. This assumption implies that, unless there exists some (enzymatic) mechanism of metal release from the complexed form, KP46, after being absorbed in the intestine, would enter the bloodstream holding gallium at a concentration of essentially 10−5 M.(b) Stability
As a matter of fact, hydrolysis is an attribute of the majority of metal ions and their compounds. To emphasise, it is hydrolytic degradation that places clinical limitations on gallium salts. Therefore, the assessment of hydrolysis survival rates is a basic requirement in the systematic metallo-drug discovery process. Due to the chelating ability of 8-quinolinol, KP46 possesses extremely high thermodynamic stability: logβ3 = 40.7.14 Its kinetic stability, commonly assessed in terms of the water-exchange rate constant of a metal ion, i.e. 4.0 × 102 for Ga3+,15 can be considered, however, as intermediate. For instance, KP46 was found to dissociate in the stationary phase of silica gel used in HPLC separation,16 thus behaving like a labile compound. Nonetheless, the integrity of KP46 should not be much affected in the intestinal juice environment. Recently, the drug stability to biotransformation via hydrolysis was demonstrated by the monitoring of hydrolysis kinetics in water and physiological buffer solutions.13 In either solution the gallium complex retained its chemical stability at a level of 50% for at least several hours. Furthermore, calculations based on a simple blood plasma model17 suggest that KP46 may persist even in the presence of physiological transferrin concentrations. In this way, its route of distribution differs substantially from that of gallium maltolate, from which gallium is liberated during, or before, passage across the intestinal epithelium to become nearly all bound to transferrin in blood18 (see also Fig. 1). On the other hand, the release of gallium may be facilitated under the slightly acidic environment of tumour tissue. Further studies are needed to examine these hypotheses and to assess which chemical properties of KP46 translate better into the observed pharmacological behaviour.(c) Lipophilicity
Efficient penetration through biomembranes is a prerequisite for an oral drug on the way from the intestine to the bloodstream and then to the tumour cell. The ability of a drug candidate to cross these barriers until it binds to the biomolecular target and induces the desired response is most often described by the n-octanol–water partition coefficient, log P. A single log P value reported for KP46 is 0.88.13 Although only characteristic of the intact complex (not its metabolites, e.g. protein adducts), this figure witnesses an appropriate hydrophilic/lipophilic balance of KP46 that should afford adequate membrane permeability after oral administration. It is worthwhile noting that the same conclusion was drawn in the pioneering drug-discovery study of KP46, dealing purposely with in vivo bioavailaibilty testing.19 It is known, however, that in the case of metal chelates, partition coefficients determined by the shake-flask method may have limited use. In this regard, recent attempts to approximate the lipophilicity of KP46 (within a set of other cytotoxic gallium complexes) within the framework of the quantitative structure–activity relationship principle using log P calculated by molecular computation20,21 should be mentioned.(d) Reactivity towards biological transporters
Such interactions would by all means affect the bioavailability and the pharmacokinetic profile of the drug. However, only some first data on KP46 interaction with serum transport proteins can be found in recent literature. The binding rate constants of KP46 to albumin and transferrin measured by capillary electrophoresis were 3.3 × 10−5 and 5.0 × 10−5 s−1, respectively.13 The fact that transferrin binding is kinetically more favourable has a certain mode-of-delivery implication as gallium is known to be taken up into cells mainly by the transferrin receptor-mediated route. Moreover, the maximum development of both protein-binding reactions observed at ≥4 h is consistent with the results of clinical studies revealing that the peak plasma levels are reached within 5–7 h after the intake of KP46.22The aforementioned kinetic data can be supported by unambiguous confirmation of protein-adduct formation under equilibrium conditions using a metal-specific detection technique, inductively coupled plasma-mass spectrometry (Fig. 3).23 Another specific method that holds promise for following changes in the coordination sphere of Ga in the presence of potential binding proteins, is X-ray absorption spectroscopy.24 Nonetheless, characterisation of the affinity of KP46 towards individual proteins in terms of association constants is, so far, missing. Consequently, the role and proper functions of protein-binding for KP46 remain unclear. One can reasonably assume that the delivery of gallium from blood to tumour cells is altered by binding of its oral drug to proteins much less than in the case of its intravenous administration as a simple salt. Besides, other transporters, e.g. multidrug resistance (MDR1), may likely contribute in pharmacokinetics and oral bioavailability of KP46.
 | ||
| Fig. 3 Speciation of gallium induced by the interaction of KP46 with human serum proteins. Drug–protein mixtures were incubated for 3 days and subjected to capillary electrophoretic analysis (electropherograms show a gallium-specific signal of the respective species). | ||
Cytotoxicity investigations
According to the number of literature accounts, examination of antiproliferative activity in cancer cells has received the most consideration in the preclinical development of KP46. The following subsections underscore the results of in vitro testing obtained by applying KP46 as a single agent and in combination with platinum drugs.(a) Single-agent activity
Considerable apoptosis-inducing effects in rat Walker carcinosarcoma 256 cells were found in the first related report,25 and this brought KP46 recognition as an antihypercalcemic agent superior to gallium nitrate. However, one should not overestimate this distinction as the doses tested in this and the following study,26i.e. 10−4–10−3 M, are manifold higher than those available after the oral administration of KP46 (see above). Using the malignant A549 human lung adenocarcinoma cell line it was demonstrated that KP46 is 10 times more potent an inhibitor of tumour cell proliferationin vitro than gallium chloride.27 The inhibitory effect of KP46 appears to be only dose-dependent (but not time-dependent), with IC50 reached at 2.5 μM concentration after 48 h of exposure. A high cytotoxic potency of KP46 has been further confirmed in clonogenic cells obtained from gastrointestinal (including colorectal) and urogenital (mainly kidney) tumours using a human tumour cloning assay.28 Sufficient drug response was obtained at a concentration of 5 μM which is a factor of 2–4 lower than the target steady-state plasma Ga concentrations for therapeutic approaches employing oral administration. In a more systematic treatment,29 the drug was investigated for its antimelanoma potency in a variety of human melanoma cell lines. As can be seen from the data in Table 1, the IC50 values fall in a relatively narrow low-micromolar range. When compared to a cell panel representing other solid tumours, melanoma cell models can be categorized as, on average, more sensitive to KP46 than other cell lines.Furthermore, the activity of KP46 was assessed in terms of uni- and multicellular resistance in cell culture models of the parent A549 cell line and its resistant subline.30 In contrast to several approved chemotherapeutic agents (including cisplatin), cell killing ability of the gallium drug is fully retained in cells grown as spheroids (resembling in vivo metastatic modules) as compared to monolayer cultures. However, one should take into account that without proper corrections for a strong adsorption of KP46 onto culture (polystyrene) plates typically used cell line experiments, there is always the risk of producing artefacts and poor data repeatability.31
(b) Drug combination effect
A major objective of discovering KP46 was its application for combination therapy with cisplatin18 which represents one of the most successful ways to treat cancer. However, such an option has been demonstrated in vitro only after almost 10 years by Jakupec et al.32 who investigated the interactions of KP46 with the platinum drugs, cisplatin, carboplatin and oxaliplatin, in human cancer cells. Comparison of the drug combination effects with single-agent activities revealed additive and synergistic antiproliferative efficiency both in ovarian (41M, CH1) and colon carcinoma cells (SW480). Importantly, the synergy is more pronounced in less platinum-sensitive cells but is largely independent of the schedule of drug exposure.33 While the molecular mechanism underlying the drug interactions observed remains to be clarified, the established effects may provide a basis for the design of clinical trials using combination chemotherapy protocols.34Cellular uptake and an early look into the mechanism of action
The intention of designing KP46 as an antineoplastic drug was to enhance the cellular effects of gallium salts, particularly inhibiting DNA synthesis due to interaction with RR (see Fig. 1), provided that their toxicological and pharmacokinetic disadvantages are overcome. In response to this rationale, a higher cellular uptake exhibited by KP46 in comparison to gallium chloride has recently been proved in rat glioblastoma cells.35,36 A superior bioavailability may explain not only a greater cytotoxicity but also a considerable decrease of intracellulardeoxyribonucleosidetriphosphate (dNTP) levels (assayed by HPLC) induced by KP46. In the authors’ opinion, the observed depletion of cellular dNTP pools is consistent with the hypothesis that KP46 inhibits the activity of RR. Additional support for this assumption is provided by data on the induction of a cell cycle arrest with increased S-phase fraction, in which KP46 acts with a much higher potency than the gallium salt, and on the initiation of apoptosis, both discernible in the same cell line . It should be emphasized that the above findings are only a first attempt to elucidate the mechanism of action of KP46.In vivo preclinical studies
Therapeutic experiments in animals are an essential link between testing in vitro cytotoxic activity and clinical studies of an investigational drug. Indeed, there are prominent examples where compounds with moderate or even low activity in cancer cell-line screens showed promising anticancer activity or vice versa, when a high systemic cytotoxic potency of a drug candidate was offset by its low therapeutic index. Therefore, it is of paramount importance to assess the applicability and efficacy of a drug in animal studies that might increase the chances for clinical success.For KP46, the therapeutic range has already been evaluated at an early stage of its preclinical evaluation. The acute toxicity after oral administration to healthy mice was found by Collery et al.37 to be moderate, though somewhat higher than that of simple gallium salts. In comparison to gallium chloride, higher serum and tissue gallium concentrations were achieved with a smaller number of administrations of equimolar drug doses, with the highest concentrations being determined by two independent methods in bone, liver and spleen, followed by kidneys, lungs and testes19,37,38 (see Fig. 4 for an example). The dose of 62.5 mg kg−1 d−1 (ca. 1/40 of LD50) was well tolerated without deaths or renal, hepatic and haematological toxicities, and as a no observed-adverse-effect level for KP46-induced toxicity it was recommended for the evaluation of drug therapeutic potential.37 The preclinical toxicology of KP46 was later reinvestigated in mice and rats.39 The maximally tolerated single dose was defined as 900 mg kg−1, and LD50 were interpolated to 1724 and 1352 mg kg−1, respectively. Only high-dosed rats experienced severe toxicities, males being more susceptible to the gallium drug. The toxic effects in high-dosed mice were generally less pronounced and similar in both sexes.
 | ||
| Fig. 4 Dose-dependent concentration of gallium in different mice tissues after oral administration of KP46 for two weeks. Reproduced from ref. 37, with permission. | ||
In vivo tumour models offer more reliable predictions of the clinical situation. The antitumour and antihypercalcemic effects of KP46 were tested by the group of Keppler and Ziegler25,26,38,40 in several experimental series with Walker carcinosarcoma 256 cells transplanted in rats. A significant decrease in tumour weight and serum calcium level was observed with a dose of 24 mg kg−1, which in addition showed no side effects on the kidney or liver.26 This striking finding allowed the authors to anticipate that KP46 could serve as a potent and safe drug for hypercalcemia of malignancy, deserving further investigation in the clinical setting.
Clinical studies
A phase I dose-escalation trial was conducted in 2004 in order to evaluate the drug’s safety, toxicity profile and pharmacokinetics .22,39 Of several pharmaceutical gallium compositions suitable for oral administration,41 a solid (tablet) formulation has been chosen for clinical application. The content of the active substance was varied in the range of 10–30% (w/w), and sufficient stability of KP46 in different tablet batches stored over two years was validated by using two independent analytical techniques.42 Seven patients with advanced malignant solid tumours (located in the kidney, ovary, stomach and parotid gland) received KP46 in doses from 30 to 480 mg m−2 daily over two weeks without dose-limiting toxicities observed.22 The fact that the drug was well tolerated up to the highest dose level investigated still does not allow for giving an explicit dose recommendation for further clinical studies. In this regard, the production of higher dose tablets appears to be needed.Preliminary evidence of efficacy was seen in three of four patients with renal cell carcinoma. It is noteworthy that a long-lasting stable disease (49 weeks) was achieved in a patient receiving a low dose of 30 mg m−2 day−1. The summary of the pharmacokinetic parameters is presented in Table 2. It can be seen that gallium pharmacokinetics are characterized by a long terminal half-life, a high total clearance and a large apparent volume of distribution. However, the data showing that gallium has linear pharmacokinetics are not yet available. Another critical comment is that being assessed by atomic absorption spectrometry technique, the Ga concentrations are unrelated to a specific metal species (i.e., metal speciation) occurring in plasma.
| Parameter | Patient (dose, mg m−2) | |
|---|---|---|
| 1 (30) | 7 (480) | |
| Maximum plasma concentration/mg l−1 | 15.3 | 62.7 |
| t 1/2/h | 53.5 | 121.5 |
| Area under the curve (AUC)/μg h ml−1 | 0.33 | 11.22 |
| Total clearance (CL/F)/l h−1 | 25.1 | 11.1 |
| Apparent volume of distribution (V/F)/l | 388 | 1952 |
Conclusion/outlook
The KP46drug is one of the classic examples of successful metal-based therapeutics originated from university research and/or small companies. Indeed, despite the fact that a large pharmaceutical company has not been involved in its discovery and evaluation, the keen efforts of a rather small group of anticancer drug developers resulted in the advancement of a viable, orally applicable gallium drug. Designing the 8-quinolinol ligands around the gallium scaffold led to a less “shotgun” cytotoxic agent than gallium nitrate or chloride. Due to the well-defined toxicological and pharmacokinetic advantages, use of KP46 not only enables higher and well tolerable tissue gallium concentrations to be established, but inhibitory effects on cell growth proliferation in vitro and in vivo (with IC50 values typically in the low micromolar range) superior to gallium salts are also induced. Remarkably, this compound exhibits both antitumour and antihypercalcemic efficiency in preclinical tumour models. Proceeding through first clinical trials in an orally administered formulation revealed the preliminary evidence for efficacy in patients with renal cell cancer. These encouraging results highlight the potential usefulness of KP46 for the treatment of tumours and a prompt necessity for phase II testing, in which it could qualify for combinations with other drugs (e.g., cisplatin) for increased effect.However, to make the clinical development of KP46 more straightforward, further endeavours towards establishing the drug mechanism should be undertaken. This seems to be no trivial task, even though there was a target definition in the drug design, namely exerting antitumour activity by interaction with RR followed by impairment of the catalytic activity of the enzyme. Perhaps the role of proteins should not be overlooked since binding to serum proteins is implicated in the transport mechanism for most metallo-drugs, and there are also certain indications that KP46 binds to albumin and transferrin under simulated extracellular conditions. In this regard, it is interesting to see whether the protein adducts play a (major) role in drug delivery and if so, how the release of gallium functionality in the cell interior takes place. Likewise, mapping drug–protein interactions appears to be important as a way of indentifying areas in the cellular proteomic profile which KP46 could target. Also notably, no biologically relevant compounds other than some proteins (e.g., DNA model compounds or DNA itself) were tested with respect to their affinity towards KP46. Therefore, for the time being, the only assumption on the mode of action for KP46 that the author can assert is that it probably involves transport through the body and accumulation in tumour cells mostly in the intact, complexed form, subsequent release of gallium ions (or other reactive species to be identified), reaction with RR, followed by blocking of DNA synthesis and induction of apoptosisvia the mitochondrial pathway. One can anticipate increased research activities in order to bring this sketch close to reality. Gallium has a radioisotope (68Ga) suitable for synthesizing the drug complex and hence positron emission tomography application may facilitate this task, as pointed out an anonymous referee.
Acknowledgements
The author wishes to thank Dr M. A. Jakupec of the University of Vienna for his assistance in conducting the comprehensive literature search and L. S. Foteeva (Vernadsky Institute) for creating the image for the Table of Contents entry.References
- M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511 CrossRef CAS.
- T. Pieper, K. Borsky and B. K. Keppler, Top. Biol. Inorg. Chem., 1999, 1, 171 Search PubMed.
- M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209 CrossRef CAS.
- M. Galanski, V. B. Arion, M. A. Jakupec and B. K. Keppler, Curr. Pharm. Des., 2003, 9, 2078 CrossRef CAS.
- W. H. Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 4003 CrossRef CAS.
- M. J. Hannon, Pure Appl. Chem., 2007, 79, 2243 CrossRef CAS.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183 RSC.
- P. Collery, B. Keppler, C. Madoulet and B. Desoize, Crit. Rev. Oncol. Hematol., 2002, 42, 283 CrossRef.
- C. R. Chitambar, Curr. Opinion Oncol., 2004, 16, 547 Search PubMed.
- M. A. Jakupec and B. K. Keppler, Metal Ions Biol. Syst., 2004, 42, 425 CAS.
- M. A. Jakupec and B. K. Keppler, Curr. Top. Med. Chem., 2004, 4, 1575 CrossRef CAS.
- L. R. Bernstein, in Metallotherapeutic Drugs and Metal-Based Diagnostic Agents, ed. M. Gielen and E. R. T. Tiekink, Wiley, New York, 2005, 259 Search PubMed.
- A. V. Rudnev, L. S. Foteeva, C. Kowol, R. Berger, M. A. Jakupec, V. B. Arion, A. R. Timerbaev and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 1819 CrossRef CAS.
- A. P. Savostin, Russ. J. Inorg. Chem., 1965, 10, 2765.
- L. Helm and A. E. Merbach, Chem. Rev., 2005, 105, 1923 CrossRef CAS.
- A. R. Timerbaev, A. Yu. Malykhin, O. P. Semenova, O. M. Petrukhin, T. A. Bol’shova and E. M. Basova, Dokl. Akad. Nauk SSSR, 1988, 301, 1151 CAS.
- D. J. Clevette and C. Orvig, Polyhedron, 1990, 9, 151 CrossRef CAS.
- L. R. Bernstein, T. Tanner, C. Godfrey and B. Noll, Metal-Based Drugs, 2000, 7, 33 Search PubMed.
- P. Collery, H. Millart, C. Pechery, F. Kratz and B. K. Keppler, in Metal Ions in Biology and Medicine, ed. J. Anastassopoulou, P. Collery, J. C. Etienne and T. Theophanides, Libbey Eurotext, Paris, 1992, vol. 2, 173 Search PubMed.
- A. R. Timerbaev, O. O. Vasylenko, L. S. Foteeva, A. V. Rudnev, O. Semenova and B. K. Keppler, J. Sep. Sci., 2007, 30, 399 CrossRef CAS.
- S. Oszwałdowski and A. R. Timerbaev, Electrophoresis, 2008, 29, 827 CrossRef CAS.
- R.-D. Hofheinz, C. Dittrich, M. A. Jakupec, A. Drescher, U. Jaehde, M. Gneist, N. Graf von Keyserlingk, B. K. Keppler and A. Hochhaus, Int. J. Clin. Pharmacol. Ther., 2005, 43, 590 Search PubMed.
- J. K. Abramski, L. S. Foteeva, K. Pawlak, A. R. Timerbaev, B. K. Keppler and M. Jarosz, Proc. 19th Int. Sym. Pharm. Biomed. Anal., Gdansk, Poland, 2008, 317 Search PubMed.
- A. Rompel, A. E. Mijovilovich, W. Meyer-Klaucke, R. Eichinger, M. A. Jakupec and B. K. Keppler, J. Biol. Inorg. Chem., 2007, 12, S7.
- B. Winter, T. Schilling, D. C. Gey, B. K. Keppler and R. Ziegler, Exp. Clin. Endocrinol. Diabetes, 1998, 106, S83 Search PubMed.
- M. Thiel, T. Schilling, D. C. Gey, R. Ziegler, P. Collery and B. K. Keppler, in Contributions to Oncology, ed. H. H. Fiebig and A. M. Burger, Karger, Basel, 1999, vol. 54, 439 Search PubMed.
- P. Collery, F. Lechenault, A. Cazabat, E. Juvin, L. Khassanova, A. Evangelou and B. K. Keppler, Anticancer Res., 2000, 20, 955 CAS.
- M. Fremuth, M. A. Jakupec, W. Hulla, R. Königsberg, C. Dittrich and B. K. Keppler, Proc. Symp. “Novel Approaches for the Discovery of Anticancer Agents”, Freiburg, Germany, 2003, 95 Search PubMed.
- S. M. Valiahdi, M. A. Jakupec, R. Marculescu and B. K. Keppler, in Metal Ions in Biology and Medicine, ed. M. Carmen Alpoim, P. Vasconcellos Morais, M. Amélia Santos, A. L. Cristóvão, J. A. Centeno and P. Collery, Libbey Eurotext, Paris, 2006, vol. 9, 282 Search PubMed.
- B. Desoize, P. Collery, M.-G. Akéli, J.-C. Etienne and B. Keppler, in Metal Ions in Biology and Medicine, ed. J. A. Centeno, P. Collery, G. Vernet, R. B. Finkelman, H. Gibb and J. C. Etienne, Libbey Eurotext, Paris, 2000, vol. 6, 573 Search PubMed.
- A. E. Egger, C. Rappel, M. A. Jakupec, C. G. Hartinger, P. Heffeter and B. K. Keppler, J. Anal. At. Spectrom., 2009, 24, 51 RSC.
- M. A. Jakupec, P. Collery and B. K. Keppler, Proc. 92nd Ann. Meet. Am. Assoc. Cancer Res., New Orleans, LA, 2001, vol. 42, 425 Search PubMed.
- M. A. Jakupec, P. Collery and B. K. Keppler, in Metal Ions in Biology and Medicine, ed. P. Collery, I. Maymard, T. Theophanides, L. Khassanova and T. Collery, Libbey Eurotext, Paris, 2008, vol. 10, 110 Search PubMed.
- B. K. Keppler, PCT Int. Appl., 2002, 31 pp, CODEN: PIXXD2, WO 2002074304, A2 20020926 Search PubMed.
- M. Pongratz, M. Fremuth, Z. Horvath, M. A. Jakupec, T. Szekeres and B. K. Keppler, Proc. Symp. “Novel Approaches for the Discovery of Anticancer Agents”, Freiburg, Germany, 2003, 113 Search PubMed.
- M. A. Jakupec, P. Heffeter, M. Pongratz, M. Fremuth, Z. Horvath, P. Unfried, N. Graf von Keyserlingk, W. Berger, T. Szekeres and B. K. Keppler, Clin. Cancer Res., 2005, 11, S9159.
- P. Collery, J. L. Domingo and B. K. Keppler, Anticancer Res., 1996, 16, 687 CAS.
- M. Thiel, T. Schilling, D. C. Gey, R. Ziegler, P. Collery and B. K. Keppler, Proc. 8th Int. Sym. Platinum Other Metal Coord. Comp. Cancer Chemother., 1999, P3 Search PubMed.
- P. Collery, M. A. Jakupec, B. Kynast and B. K. Keppler, in Metal Ions in Biology and Medicine, ed. M. Carmen Alpoim, P. Vasconcellos Morais, M. Amélia Santos, A. L. Cristóvão, J. A. Centeno and P. Collery, Libbey Eurotext, Paris, 2006, vol. 9, 521 Search PubMed.
- D. C. Gey, T. Schilling, B. K. Keppler, M. Thiel, H. Schmidt-Gayk, F. Raue and R. Ziegler, J. Cancer Res. Clin. Oncol., 1997, 123(Suppl. 1), 6.
- L. R. Bernstein, US Pat. Appl. Publ., 2007, 10 pp, CODEN: USXXCO, US 2007098815, A1 20070503 Search PubMed.
- L. S. Foteeva, N. V. Stolyarova, A. R. Timerbaev and B. K. Keppler, J. Pharm. Biomed. Anal., 2008, 48, 218 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |