Molecular probes for the in vivo imaging of cancer
Raphael
Alford
ab,
Mikako
Ogawa
a,
Peter L.
Choyke
a and
Hisataka
Kobayashi
*a
aMolecular Imaging Program, Center for Cancer Research, National Cancer Institute, NIH, Building 10, Room 1B40, MSC1088, Bethesda, Maryland, MD 20892-1088, USA. E-mail: kobayash@mail.nih.gov; Fax: +1 301-402-3191; Tel: +1 301-451-4220
bCase Western Reserve School of Medicine, Cleveland, Ohio, USA
First published on 19th August 2009
Abstract
Advancements in medical imaging have brought about unprecedented changes in the in vivo assessment of cancer. Positron emission tomography, single photon emission computed tomography, optical imaging, and magnetic resonance imaging are the primary tools being developed for oncologic imaging. These techniques may still be in their infancy, as recently developed chemical molecular probes for each modality have improved in vivo characterization of physiologic and molecular characteristics. Herein, we discuss advances in these imaging techniques, and focus on the major design strategies with which molecular probes are being developed.
Introduction
Successful cancer therapy hinges on early diagnosis and accurate staging. Cancer deaths are expected to rise from 7.4 million in 2004 to 23.4 million by 2030.1 In diagnosing and monitoring malignancies, physicians classically rely on changes in size and location, criteria which are late indicators of disease. Biopsies provide the most detailed information about a cancer, but monitoring with biopsies is both impractical and dangerous. Moreover, biopsy specimens are subject to sampling error as the entire tumor burden cannot be evaluated. Very often, a solid tumor large enough to visualize and biopsy is very heterogeneous, making needle sampling even more problematic. Noninvasive molecular imaging of a cancer before, during, and after therapy could increase our understanding of cancer biology, provide earlier diagnoses, and a means to monitor therapy, contributing to the realization of “personalized medicine”.The term molecular imaging was first applied to the imaging of gene structure and function. Driven by advances in molecular biology and imaging technology, molecular imaging has grown rapidly in the last 5 years and now extends into in vivo applications. Molecular imaging can include three types of imaging: physiologic, metabolic, and targeted. The imaging of physiologic processes, such as blood flow and vessel permeability, generally involves non-targeted imaging agents that are distributed according to their size, hydrophilicity, and surface charge. This is molecular imaging at its broadest definition. Metabolic imaging refers to the visualization of such processes as glucose uptake by cells or DNA proliferation. Targeted imaging, the strictest category of molecular imaging, involves labeling an imaging agent with a ligand that binds to a specific cell surface marker, such as folate or epithelial growth factor receptors.
The growth of molecular imaging depends on multidisciplinary cooperation between chemists, molecular biologists, physicists, and imaging specialists. Moreover, there are substantial legal, regulatory, and economic barriers. Today, the field of molecular imaging is developing both in the range of modalities and the diversity of molecular probes employed. The purpose of this review is to describe the most common molecular imaging modalities—nuclear imaging, optical, and magnetic resonance imaging (MRI )—and demonstrate current developments in molecular probes for in vivo oncologic imaging.
Imaging modalities
The three modalities at the forefront of in vivo molecular imaging research are nuclear imaging, optical imaging, and magnetic resonance imaging (MRI ). Each has its own strengths and weaknesses, varying in sensitivity, spatial resolution, temporal resolution, cost, and depth of tissue penetration.Nuclear imaging is the temporal/spatial detection of ionizing radiation from injected radionuclides. The primary methods through which in vivo oncologic imaging is accomplished are positron emission tomography (PET) and single photon emission computed tomographic (SPECT) imaging. In both modalities an imaging agent, or ‘tracer’, is created by labeling a compound with a radioisotope. PET utilizes positron emitters (11C, 18F are most common), detecting γ rays that result from positron/electron annihilation. SPECT directly images γ emitters (123I, 111In, and 99mTc are most common).
PET and SPECT are valuable molecular imaging modalities as both are capable of detecting minute amounts of radioactive tracer—e.g. PET is able to image a minimum of 10−11–10−12 molar of a molecular probe; SPECT, a minimum of 10−10–10−11 molar—while minimally perturbing the biological system. PET is currently more sensitive (∼2–3 orders of magnitude) than SPECT, has better resolution, and offers superior tracer quantification. This difference is due largely to the design of each device: PET electronically determines the radionucleotide locations through the detection of coincident rays, whereas SPECT requires the use of a collimator to reject radionucleotide emissions beyond a certain angle to determine the incidence of the ray. SPECT is less expensive, has a broader array of approved radionuclides, and can distinguish multiple emission energies simultaneously. It should be noted that these generalizations are not static, as advances in collimators, including multi-pinhole collimator and gamma camera design, have allowed small animal SPECT systems to surpass small animal PET systems in spatial resolution along with considerable improvement of sensitivity, especially when the small objects are imaged. The ability of SPECT to distinguish multiple emission energies holds the potential to distinguish co-administered tracers that differ in their targets and emission energies.2 Although this currently remains to have some difficulties on a longtudinal study because of the limited choices of relevant isotope combinations, multiple isotopes targeted against distinct markers may eventually allow for a single image of a cancer’s molecular expression profile. The development of PET and SPECT with computed tomography (CT) has enabled the relatively low spatial resolution of radionuclide images to be fused to a high resolution anatomical CT image, thus improving localization, quantification, and sensitivity.3,4
PET and SPECT do have limitations: they are more costly than other methods, expose the patient to ionizing radiation, and the temporal and spatial resolution (1–30 min, 4–10 mm) are inferior to other imaging modalities. The use of ionizing radiation limits the number of research studies that can be performed in a single patient. As a result the radioactivity dose is kept low (100–1000 MBq), and is further limited if the tracer’s biodistribution results in accumulation in particular organ(s).
Optical in vivo molecular imaging techniques refer to the imaging of light photons with charge-coupled device (CCD) cameras. These cameras allow for the imaging of light both within and outside of the visible range (ultraviolet (UV) and near infrared (NIR)). Fluorescence, light emitted when an electronically excited molecule transitions from its lowest singlet state to its ground state, is the source of contrast for most in vivo optical imaging.
In comparison to nuclear imaging and MRI methods, optical techniques are cost effective and widely available. The two most utilized optical techniques for in vivo imaging are bioluminescence (the imaging of enzymatically mediated light producing chemical reactions), and fluorescence intensity imaging (in which an injected or applied fluorophore is stimulated to fluoresce by an exogenous light source, also referred to as fluorescence reflectance imaging). Endogenous fluorescence for bioluminescence imaging is created by genetically manipulating the cell/tissue to produce a fluorescent protein whose catabolism creates light. Although these techniques have had an immeasurable impact on the understanding of cancer through laboratory models, the gene transfer techniques required for bioluminescence are not likely to be relevant for clinical imaging in humans in the near term since the proteins produced are cross species (e.g.firefly luciferase). The clinical translation of the genetically transfected fluorescent proteins (e.g. jelly fish or coral fluorescent proteins) is distant clinically for similar reasons.
Fluorescence intensity imaging (FII) is the primary optical technique for which there has been development of a broad array of molecular probes. FII is sensitive, offering the ability to image as little as 10−9–10−12 molar of probe. FII also has high temporal and spatial resolution (2–3 mm). Furthermore, a broad range of fluorophores are available, with emissions ranging from visible spectrum (390–650) to the near infrared (650–900, NIR). Unlike nuclear modalities, optical techniques do not involve any form of ionizing radiation.
The primary hurdles in the development of fluorescence imaging techniques have been twofold: poor methods of quantification, and high fluorescent light absorption and scattering secondary to endogenous tissue chromophores (hemoglobin, melanin, lipids, water, etc.). The recent advent of fluorescence molecular tomography (FMT) has demonstrated a capacity for quantification, but this technology is not yet widely available.5 The development and use of fluorophores in the NIR range has done much to overcome the limitations of depth penetration, as much less light at these longer wavelengths is absorbed by biological tissues.6 There is also much less inherent autofluorescence induced by excitation of NIR probes, improving tumor to background ratios. Even with the NIR fluorophores, the maximum imaging depth through tissue is ∼3–4 cm, much less than the detection depths of the other molecular imaging modalities (FMT is reported to image to a 10 cm depth5). It should be noted, however, with increasing depth not only is the amount of light reduced, but the scatter results in a significant degradation of resolution. Fluorescence intensity imaging techniques are most applicable to oncologic targets near tissue surfaces (breast cancer, lymph nodes, etc.), directly visible with an endoscope, or under open surgery (peritoneal tumors, colon cancers, bladder cancers, etc.).
The third technique, MRI , is a powerful imaging modality that provides excellent soft tissue contrast. When a material (such as tissue) is placed within a strong magnetic field (1.5–3 Tesla are common) the proton spins align to create a multitude of precessing dipoles oriented in parallel to the main magnetic field direction (the z axis). When a radiofrequency pulse of a certain frequency (known as the resonance or Larmor frequency) is applied, the targeted dipoles are displaced from the z axis onto the x–y plane. Once the RF pulse has been turned off, the dipoles relax to their normal, z-aligned state. T1 (longitudinal relaxation) is the time required for a certain amount of recovery of the dipole along the z axis, while T2 (transverse relaxation) is the time required for a certain amount of dephasing in the x–y plane. T1 and T2 weighting are used to create MR images which highlight different anatomical structures and fluids. In addition to T1 and T2, many other MR parameters can be manipulated to influence the MR image. Metals with magnetic moments (Gd3+, Mn2+, Fe3+) are effective contrast agents for the MRI . These agents can alter the T1 and T2 of tissue, permitting detection on an MR image.
Of the three molecular imaging modalities, MRI has the best spatial resolution (10–100 μm) and thus provides superior anatomical information. MRI furthermore has no depth limit and does not utilize any ionizing radiation. There are no known health impacts of magnetic field exposure.7 Of the three approaches however, MRI is the least sensitive, requiring 10−3–10−5 molar metals as a molecular probe for detection. MRI has an intermediate temporal resolution of seconds to minutes, and is costly, on par with the nuclear imaging technologies.
Molecular imaging probes
Nuclear imaging probes
The radiochemist is primarily concerned with isotope characteristics and pharmacokinetics when developing a new tracer. The recurring theme in isotope choice is half-life: it must be long enough to allow time for the radiotracer to be synthesized and administered to the patient, ideally matching the biologic half life of the end compound, but no longer than is absolutely needed. For instance, antibodies have long clearance times and therefore, longer lived radioisotopes such as 131I, 111In, 186Re, 67Ga, 177Lu, for SPECT, and 124I, 64Cu, 86Y for PET are preferred. On the other hand, small molecules that are cleared rapidly may require labeling with shorter lived isotopes such as 11C and 18F (Table 1).8–11 These isotopes require rapid synthesis of the compound containing them due to their short half-lives, and if the isotope is cyclotron based, this costly equipment may be required onsite. For example, since the halflife of 11C is 20 min, radiochemists have to finish the synthesis, including purification, within 60 min for a tracer to have practical value. In addition, to avoid the radiation, the reaction should be done in the “hot-cell”, which is a radio-protected closed box equipped with an automatic synthesis system. Simple and fast reactions are therefore required for short-half life radionuclides, and this is often challenging for radiochemists.| Isotope | Half-life | Energy use for imaging (keV) | Beta emission (± and %) |
|---|---|---|---|
| a IC: internal conversion beta. | |||
| (Single photon) | |||
| 125I | 60.1 d | 36 | −(Auger) |
| 131I | 8.02 d | 365 | β−, 606 keV (89.9%) |
| 177Lu | 6.73 d | 208 | β−, 498.3 keV (78.6%) |
| 186Re | 3.72 d | 137 | β−, 932 keV (21.5 %) 1069 keV (70.9%) |
| 67Ga | 3.26 d | 93, 185, 300 | — |
| 201Tl | 3.05 d | 69–80, 167 | |
| 111In | 2.80 d | 171, 245 | −(ICa ) |
| 123I | 13.13 h | 159 | — |
| 99mTc | 6.02 h | 141 | — |
| (PET) | |||
| 124I | 4.17 d | 511 | β+ 1535 keV (11.8 %), 2138 keV (11.0 %) |
| 86Y | 14.74 h | 511 | β+ 1254 keV (12.4%), 1578 keV (5.6%) |
| 75Br | 96.0 min | 511 | β+ 1740 keV (75.5%) |
| 68Ga | 68.0 min | 511 | β+ 1899 keV (87.7%) |
| 66Ga | 9.4 h | 511 | β+ 4153 keV (49.3%) |
| 64Cu | 12.7 h | 511 | β+ 653 keV(17.9%), β− 578.7 keV (39%) |
| 18F | 109.8 min | 511 | β+ 633 keV (96.7%) |
| 15O | 2.0 min | 511 | β+ 1732 keV (99.9%) |
| 13N | 10.0 min | 511 | β+ 1199 keV (99.8%) |
| 11C | 20.39 min | 511 | β+ 960 keV (99.8 %) |
Because radionuclides are “always on”, once injected, probes that are unbound create a high background signal, decreasing the effective tumor to background ratio. This “always on” characteristic also results in nonspecific signal if an isotope escapes from the targeting probe. The agent’s pharmacokinetics are therefore, critically important when designing a radiotracer. The ideal tracer will have excellent tissue penetration, bind the target with high affinity, yet the unbound agent will also be rapidly cleared from the system. Longer systemic circulation does provide greater target exposure, which is desirable if the agent is meant for therapeutic purposes. Thus, in the rational design of an imaging agent a balance must be struck between the competing demands of adequate binding and rapid excretion.
 | ||
| Fig. 1 (A) D-Glucose, (B) 18F-FDG, (C) an 18F-FDG PET image and (D) a PET/CT image of a patient with a paradudodenal mass secondary to malignant melanoma. Image also demonstrates high 18F-FDG uptake (shown in color scale) in normal brain, heart, and bladder (secondary to radiotracer excretion) overlapped on the underlying anatomical information simultaneously obtained by an X-ray CT scan (shown in the gray scale). Courtesy of Dr Baris Turkbey, Molecular Imaging Program, NCI/NIH. | ||
Despite the ubiquity of glucose metabolism throughout the body, 18F-FDG has been successful because healthy cells slow glycolysis in the presence of oxygen (known as the Pasteur effect) in favor of the more efficient aerobic respiration, whereas many types of malignant cells rely on an upregulated glycolytic anaerobic pathway (known as the Warburg effect)13,14 even when oxygen is present. Although the preference for a less efficient mechanism of energy production seems paradoxical, current theories suggest it may be an adaptation by the tumor to chronic hypoxia,15 or may be induced directly by oncogenes,13 either of which lead to an increased demand for the body’s limited resources and may allow cancerous cells to outcompete healthy ones for nutrients.14,16–19 With the exception of normal myocardium and brain, where glucose uptake is naturally high, the result is a buildup of 18F-FDG in hypermetabolic cells, thus identifying many neoplasms.
Clinically, the degree of uptake of 18F-FDG often reflects tumor aggressiveness,20,21 and can provide accurate estimations of metabolic activity and viable tumor.22 Moreover, significant decreases in metabolic activity can serve to delineate responders from nonresponders to therapy earlier than would otherwise be possible, although for improved clinical outcomes the metabolic response must be dramatic (>80%).23–28 Large reductions in 18F-FDG uptake after therapy correlate with improved survival and disease free survival in non small cell lung cancer, lymphoma, and colorectal cancer to name a few.20,21,29 The amount of reduction of activity needed to portend an improved outcome is still controversial, and varies by tumor type and treatment.
Since upregulated glycolysis is a non specific process, found in normal organs as well non-malignant disease processes, 18F-FDG PET can often fail to distinguish tumor from benign conditions. The high rates of glucose utilization are also not specific to malignancies—infectious (tuberculosis, sarcoidosis, pneumonia) and inflammatory processes, brown fat deposits, thyroid gland uptake, hyperplastic bone marrow, and gastrointestinal digestion are all non-malignant processes that commonly take up 18F-FDG, potentially leading to false positive results.30,31 Furthermore, not all cancers exhibit high 18F-FDG uptake—mucinous carcinomas, prostate cancers, neuroendocrine tumors, and bronchioloalveolar cell lung carcinomas are notorious for their lack of 18F-FDG uptake, especially with early stage disease.31,32
Attempts to overcome the limitations of 18F-FDG have focused on developing alternatives to glucose-based imaging. An example of such an alternative that is finding success is monitoring the uptake of radiolabeled thymidine, a nucleotide analog, to provide a reflection of cellular DNA proliferation. The most widely used thymidine analogue in in vivo imaging is 18F-3′-fluoro-3′-deoxy-L-thymidine (18F-FLT). 18F-FLT utilizes the same cellular transporters as thymidine to enter the cell.33,34 Once intracellular, thymidine is phosphorylated either by thymidine kinase 1 (TK1) or thymidine kinase 2 (TK2), whereas 18F-FLT has a high affinity for phosphorylation by TK1.35 While thymidine is rapidly incorporated into DNA, less than 1% of 18F-FLT-monophosphate is incorporated, and the majority of the molecular probe remains trapped in the cytoplasm.34 At high enough doses, 18F-FLT operates as a competitive inhibitor of TK1 (FLT was originally synthesized as a chemo- and HIV-therapeutic analogous to AZT, but caused bone marrow suppression and hepatoxicity at therapeutic doses). Similar to 18F-FDG, at tracer doses 18F-FLT has been proven safe.36
Although not yet approved for routine clinical use, 18F-FLT has emerged as the most promising of the thymidine analogues due to its specificity for TK1, and resistance to degradation. In cases where 18F-FLT and 18F-FDG have been directly compared, 18F-FDG typically demonstrates higher tumor uptake resulting in higher tumor to background ratio and a higher sensitivity. In contrast, 18F-FLT appears to have a higher specificity for cancer.37,3818F-FLT has furthermore overcome some of the limitations of 18F-FDG. In the brain for example, normal 18F-FDG uptake is high, masking uptake by metabolic tumors. 18F-FLT normally has poor uptake in the brain, and has been shown to be effective in the imaging of brain tumors. However, it is unclear whether the uptake of 18F-FLT in brain tumors is related to leakage through damaged blood brain barrier or to cellular proliferation, but 18F-FLT clearly improves brain tumor imaging as compared to 18F-FDG.39 It is hoped that proliferation imaging with 18F-FLT will enable the non-invasive grading of tumors and improve the ability to assess cancer response to therapy. 18F-FLT has shown some success in demonstrating cancer stage and grade over 18F-FDG,38,40–42 but not uniformly.37,43–4518F-FLT has also shown promising results in demonstrating a response of therapeutics in multiple cancer types.46–50
Pharmacokinetics are critical to radiolabled antibody design. At around 150 kDa in size, antibodies are much larger than the ∼60–70 kDa threshold for first pass renal filtration, resulting in slowed vascular clearance (days to weeks) and low tissue penetration. This slow clearance rate is deleterious to an imaging agent because it results in high background activity (Fig. 2). A number of engineered antibody fragments and derivatives have been developed to improve the pharmacokinetics , including CH2-domain-deleted antibodies (∼122 kDa),55 F(ab)2 (∼100 kDa),56 minibodies (∼75 kDa),53 diabodies (∼50 kDa),57 disulfide-stabilized and linear single chain variable fragments (scFv) (∼25 kDa),58 and affibodies (∼7 kDa).59,60 These engineered antibody fragments have been successful in preclinical studies. Another approach to improving pharmacokinetics is to employ a two step labeling and clearing (or pre-targeting) process. Pre-targeting involves the use of an initial targeting agent, which itself can be bound by secondarily injected agents. Common secondary agents are either quickly clearing radiotracers that bind the initial agent with high affinity61 or “chase” reagents that clear an unbound radiolabeled antibody circulating in the blood pool.62
 | ||
| Fig. 2 In vivo radio-immuno imaging of a mouse bearing a 3T3/HER2+ tumor, which overexpresses HER2 receptors. A 111In-labeled trastuzumab-CHX-A conjugate was injected into the mouse. A planar scintigram at 2 day post-injection allowed for the detection of the tumor by HER2 expression. | ||
Radiolabled antibodies are of particular interest because interchanging the radioisotope could transform a diagnostic probe to a targeted radiotherapeutic agent. In a therapeutic situation, slower antibody clearance is advantageous. Commonly used therapeutic isotopes 186Re, 177Lu, 90Y and 131I are γ and β− emitters; 213Bi and 211At are α emitters; and 125I and 111In are an auger and an internal conversion β− emitter. With the exception of 90Y, all of these isotopes are simultaneously imagable (Table 1). Choice of isotope depends on the intended target, as they vary in mean radiation range, and, of course, half-life.63 The therapeutic use of 90Y conjugated to ibritumomab tiuxetan (Zevalin), a monoclonal antibody to CD20, was clinically approved for the treatment of non-Hodgkins lymphoma in 2002.64,65111In-labeled ibritumomab tiuxetan is often administered separately or simultaneously for imaging and dosimetry. Although few have been approved for clinical use, over 400 radiolabeled therapeutic antibodies are in clinical trials.66
The prototype radiolobeled peptides are the radiolabeled-somatostatin analogues (∼1.5 kDa). Commonly labeled with 111In, 99mTc, or 68Ga, these analogues target somatostatin receptors that are commonly overexpressed on neuroendocrine tumors (pituitary adenomas, pheochromocytomas, paragangliomas, neuroblastomas, medullary thyroid cancers, and neuroendocrine tumors).71 There are 5 somatostatin receptor subtypes, and subtype 2 is the most widely over expressed in neuroendocrine tumors.67 Through binding of G-protein receptors, the analogue is internalized and builds up within the tumor to allow detection. Although their small size results in rapid systemic clearance, images of tumors millimetres in size can be obtained. Initially, somatostatin analogues suffered setbacks secondary to rapid peptide degradation following internalization, and loss of nuclide specificity. These limitations have been overcome through the development of synthetic peptides with high chemical stability.72 This is achieved by introducing non-natural or phosphorylated amino acids, amidating the C-terminus, cyclization, and PEGylation (linking peptide to polyethylene glycol chains).73 Such modifications typically prolong vascular clearance, and may alter receptor specificity; a balance must be struck between serum stability and clearance. The clinically approved 111In labeled DTPA-octreotide (OctreoScan) has proven to be a successful and versatile molecular imaging agent.72,74 Other hormone analogues in various stages of preclinical and clinical development include bombesin to target gastrin releasing peptide receptor, vasoactive intestinal peptide (VIP) to target VIP receptor, and RGD peptide to target αvβ3 integrin.75
Optical imaging probes
Optical imaging probes are still in the preclinical stages of development. Exogenous fluorescent dyes fall into two categories: the organic small molecule (such as fluorophore dyes), and nanosized particles (e.g.quantum dots). Broad arrays of fluorescent dyes are available, each with particular excitation and emission characteristics; NIR cyanine-based dyes are the most commonly used for in vivo fluorescence intensity imaging. Quantum dots are semiconductor nanocrystals that offer tunable narrow fluorescence emission, fluoresce more brightly, are more resistant to photobleaching, and have a broader excitation band than organic dyes.81 The broad excitation band allows multiple quantum dots to be excited by the same excitation source, a useful feature for the simultaneous imaging of multiple fluorophores. The toxicity profile of quantum dots, however, remains to be investigated since quantum dot cores are composed of inorganic metals such as Cadmium and Selenium.81 Through the conjugation of either category of dye to a high affinity ligand (antibody, antibody derivative, peptideetc.), molecular specificity can be added.Although fluorescent probes can be used in ways exactly analogous to the commonly used radionuclide probes, they are limited by the poor penetration of light in tissue. Instead, optical molecular imaging has focused on developing niche imaging applications, utilizing the advantages of optical imaging for surgical and endoscopic near field imaging. Galactosyl serum albumin –rhodamine green conjugate, a florescent molecular probe bound by ovarian tumors, has shown promise in mouse models of ovarian peritoneal metastases as an aid to intraoperative identification and resection82 (Fig. 3). Beyond this, optical molecular imaging techniques have focused on the development of its strengths: the ability to simultaneously distinguish between multiple probes, development of activatable probes, and improved characterization of fluorescence signal.
 | ||
| Fig. 3 Surface fluorescence imaging of peritoneally disseminated ovarian cancer with the injection of galactosyl serum albumin –rhodamine green in a mouse. (A) White light image, (B) green fluorescence image without unmixing, (C) rhodamine green spectral image after unmixing the autofluorescence. Sub-millimetre disseminated nodules are clearly identified with unmixed rhodamine green spectral image with minimal background signal. | ||
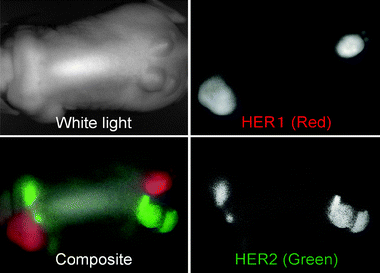 | ||
| Fig. 4 In vivo optical imaging of two tumor types, 3T3/HER2+ and A431, which express HER1 receptors. One of each type of tumor of was injected into the flanks and shoulders of athymic mice. A cocktail of cetuximab-conjugated Cy5.5 (HER1 directed shown in red), and trastuzumab-conjugated Cy7 samples (HER2+ shown in green) directed was injected into the mouse. Optical multispectral imaging allows for the differentiation of the two tumors by molecular expression pattern. | ||
Beyond the identification of multiple potential targets, there are obstacles to the broad application of multicolor imaging. First, totally overlapping fluorescence emissions can be difficult to delineate, even with spectral unmixing software. This currently requires the choice of fluorophores with fluorescence emissions distinctly spaced at least 20 nm apart, ultimately limiting the number of potential probes (currently up to 5, especially if one is attempting to work in the crowded NIR range alone). Second, spectral unmixing with the tunable crystal filter technology lowers the temporal resolution of imaging to about 5 s per frame, in a manner directly proportional to the number of emissions being identified. This has been problematic for the development of fluorescence guided surgery techniques as, until recently, video frame rate imaging of multiple colors has not been possible.87 However, both limitations could be minimized or overcome by further development of spectral imaging technology.
A recently developed novel approach to fluorophore activation utilizes the PeT mechanism.94 At physiologic pH (∼7.4), a nonprotonated N,N-dialkylated aniline is able to virtually eliminate fluorescence emission from the independent 2,6-dicarboxyethyl-1,3,5,6-tetramethyl boron-dipyrromethane (BODIPY) fluorophore through PeT. At lysosomal pH however (pH ∼ 5–6), protonation eliminates PeT, resulting in a 300 fold increase in photon emission.94 Conjugated to trastuzumab, this probe is able to localize HER-2+ cells, where it is internalized within target cells. Because fluorescence in this case is pH dependent, this probe demonstrates reversibility (or “deactivation”) if ejected from the tumor cell into the more neutral extracellular environment, or if the tumor cell becomes non-viable.94 This is potentially advantageous in comparison to extracellularproteaseactivation, where an activated agent has the potential to leak away from the activation site, whereas pH activated probes deactivate if they escape their target environment.
Translation of the potential of FLI from the field of microscopy to in vivo imaging is still occurring. In FLI microscopy, where autofluorescence, light absorption, and scattering are not an issue, much work has been done with fluorophores in the lower visible spectrum (∼500–600).95 To report an accurate lifetime value from within greater than 1 to 2 mm of a turbid media requires an accurate photon migration model, which remains a point of controversy.100,101 As in vivo FLI is still new, there has yet to be a widely validated equipment standard. Once established, the combination of FLI with FII will add several layers of information to molecular investigation of in vivo processes.
Magnetic resonance imaging probes
The two major categories of magnetic resonance probes are paramagnetic and superparamagnetic agents. Paramagnetic agents are composed of a metal ion possessing a permanent magnetic moment due to unpaired electrons (e.g. gadolinium (Gd3+) or manganese (Mn2+)) and a chelating ligand (e.g. diethylene triamine pentaacetic acid, DTPA). The chelate prevents the paramagnetic lanthanide ion from becoming toxic. Paramagnetic agents create magnetic moments that hasten the relaxation of water protons following a radiofrequency pulse. The result is shorter T1 and T2 relaxation times (the impact of paramagnetic agents is greater on T1), increasing signal in the presence of the agent. Superparamagnetic agents consist of an iron oxide core or a Fe/Mn composite metal core covered in a polymer matrix to prevent aggregation. Superparamagnetic agents form a significantly larger magnetic moment than the paramagnetic agents. These agents primarily shorten T2 relaxation times, although a newer generation of smaller superparamagnetic agents have been reported to affect T1 as well.103 As opposed to T1 agents, signal is locally decreased by the presence of a T2 agent. Both paramagnetic agents and superparamagnetic agents function primarily through a perfusion mediated process, distributing throughout the intravascular and interstitial space. Due to high vascularity and inefficient lymphatic drainage, they can successfully aid in oncologic imaging by localizing to tumors, and provide a means of following response to therapy. They both have also demonstrated usefulness in distinguishing benign from malignant tumors. Through conjugation to peptides and antibodies in manners similar to other modalities, targeted delivery of MRI contrast agents is achievable,104–107 but this approach has not been broadly adopted.Low sensitivity is the major obstacle in the development of targeted MRI contrast agents. With Gd3+ based agents for example, a local concentration of 0.5 mM is required for 50% contrast enhancement.108 Although this might be overcome by delivering more agent to the target tissue or improving the relaxivity of Gd3+, minimization of dose is desirable, especially with growing concern over contrast agent related toxicities such as nephrogenic systemic sclerosis.109 Additionally, receptors are not necessarily always overexpressed to the degree necessary to create this concentration of signal. As such, attempts to deliver higher magnetic payloads can be achieved through the conjugation of multiple magnetic nanoparticles to a targeted carrier molecule, such as a peptide, dendrimer, liposome, etc. Although this alters the pharmacokinetics of the probe as well as the magnetic effects of the metal nanoparticles, many of these probes have shown some success in preclinical studies.110 A number of activatable MRI contrast agents have been developed as well, usually either through an enzymatically cleavable blocker that shields water from the magnetic particle111 or via the paramagnetic chemical exchange saturation transfer technique (PARACEST).112 Briefly, PARACEST agents are designed to receive radiofrequency energy at a separate frequency from that directed at water protons. Prior to interrogating of water protons, PARACEST agents are pulsed and allowed to chemically transfer (or saturate) this energy to nearby water protons. Shortly thereafter, imaging attempts will demonstrate a decreased signal from saturated protons secondary to PARACEST agents. Such an agent would bring to MRI imaging the tandem imaging of multiple agents with multiple targets. Still, the success of targeted and activatable agents is remains limited by low sensitivity.
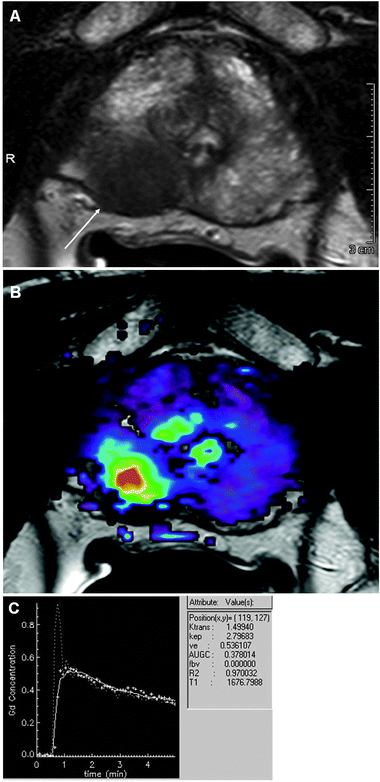 | ||
| Fig. 5 (A) Axial T2 image of prostate with a tumor (arrow). (B) DCE-MRI using low molecular weight contrast agent (Gd-DTPA). (C) Time intensity curve from site of tumor demonstrating rapid and strong rise, intense peak enhancement with subsequent wash-out, which is characteristic for highly angiogenic malignant tumors. Courtesy of Dr Baris Turkbey, Molecular Imaging Program, NCI/NIH. | ||
The ultrasmall superparamagentic iron oxide particles (USPIO) are one class of MRI contrast agent that has demonstrated a capacity for lymph node imaging. Once intravenously injected, they are phagocytosed by macrophages and accumulate in the reticulo-endothelial system of lymph nodes. On T2 weighted images the result is a localized decrease in signal within normal nodal tissue.123 As they are not able to uptake the USPIO agents, nodal tumors continue to produce signal. Multiple clinical studies have shown USPIO imaging to be a sensitive and specific technique.124,125 The limitations of this strategy include the possibility of a healthy lymph node hiding a micrometastasis,126 and a lymph node filled with signal producing lymphocytes due to an active inflammatory process.127 Also, this technique is only of limited value for the imaging of lymphatic vessels.
Dendrimers, polymer spheres that can be designed and synthesized to exact sizes, can be utilized to image both the lymphatics and lymph nodes (Fig. 6). Multiple Gd3+-DTPA molecules can be conjugated to the surface of a single nano-sized dendrimer leading to increased relaxivity by as much as ∼10-fold compared with clinically available Gd3+-DTPA chelates.128 The generation 6 (or G6) dendrimer has been found to be the optimal size (∼9 nm) for lymphatic imaging.128 Injected interstitially, the particles are efficiently taken up by the lymphatic system, providing T1 enhancement of lymphatic channels and nodes. If visualized dynamically, lymphatic flow can be investigated.
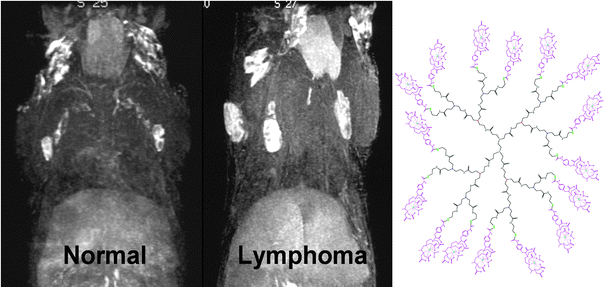 | ||
| Fig. 6 MR Lymphangiogram of a normal mouse (left) compared to a IL-15 transgenic, lymphoproliferative/lymphoma mouse (right), utilizing 0.005 mmol Gd per kg of generation 8 (G8) PAMAM dendrimer (13 nm) at 30 min post-injection. Lymph nodes and lymphatics are visualized through the uptake of the dendrimer agent. Enlarged lymph nodes involved with lymphoma lesions are clearly determined in the lymphoma mouse with MR Lymphangiogram. The chemical structure of generation 2 (G2) based PAMAM dendrimer contrast agent including 1B4M chelate is shown. The G8 contains repetitive extensions of this interior structure three more times. | ||
Monomolecular multimodality imaging agents (MOMIAs)
The combination of multiple imaging modalities holds the potential to take advantage of the strengths of each device and overcome individual limitations. Although this does not imply the need for a single probe that coregisters on multiple platforms over multiple distinct probes, anticipated toxicity and regulatory hurdles, plus the likely separate biodistributions of multiple probes for multimodality in vivo imaging has driven the development of MOMIAs. An optical-PET/SPECT or -MRI combination could localize targeted tumor tissue for presurgical planning, and provide optical enhancement during surgery. The G6 dendrimers described above conjugated dually with Gd3+ and a fluorophore130 provide one example of probes that could serve this purpose. MOMIAs also serve as an excellent validation tool. Recently, a fluorophore labeled liposome (a T1 MRI agent) was created in two batches; either targeted for the integrin αvβ3, overexpressed in the angiogenic blood vessels of many tumors, or in a non-targeted from. When applied separately to a mouse tumor model, both resulted in an in vivo signal increase on the T1-weighted image. Ex vivofluorescence microscopy however, revealed only the targeted agent in association with the endothelial cell surface, the non-targeted agent simply building up in the extravascular space.131,132 PET/MRI is another combination of modalities which has generated much interest recently, bringing the excellent soft tissue characterization of MRI , with multiple methods of soft tissue analysis, to the highly sensitive PET techniques without the burden of additional radiation. Multiple PET/MRI MOMIAs have been developed133,134 although this technology faces significant technical, financial, and regulatory burdens to overcome before it will be widely available.Conclusion
Functional and molecular imaging techniques are gradually being incorporated into every aspect of cancer management. SPECT, PET, optical, and MRI promise earlier and more accurate diagnoses, a functional and sometimes molecular understanding of the disease process, and a method of observing therapeutic efficacy. Agents/tracers/probes all aid in this regard, and can be designed to overcome an individual modality’s limitations. In some cases they may act as a therapeutic themselves. These techniques represent the next step in the evolution of in vivo oncologic imaging, reaching past the historical role of localizing and measuring cancer size.Disclosures
Raphael Alford’s research year was made possible through the Clinical Research Training Program, a public-private partnership supported jointly by the NIH and Pfizer Inc (via a grant to the Foundation for NIH from Pfizer Inc.).Acknowledgements
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.References
- The Global Burden of Disease: 2004 Update, http://www.who.int/healthinfo/global_burden_disease/GBD_report_2004update_full.pdf.
- M. D. Devous, Sr, J. L. Lowe and J. K. Payne, J. Nucl. Med., 1992, 33, 2030–2035 CAS.
- F. U. Chowdhury and A. F. Scarsbrook, Clinical Radiology, 2008, 63, 241–251 Search PubMed.
- O. Mawlawi and D. W. Townsend, Eur. J. Nucl. Med. Mol. Imaging, 2009, 36(Suppl 1), S15–29.
- V. Ntziachristos, Annu. Rev. Biomed. Eng., 2006, 8, 1–33 CrossRef CAS.
- R. Weissleder and V. Ntziachristos, Nat. Med., 2003, 9, 123–128 CrossRef CAS.
- J. F. Schenck, J. Magn. Reson. Imaging, 2000, 12, 2–19 CrossRef CAS.
- C. A. Boswell and M. W. Brechbiel, Nucl. Med. Biol., 2007, 34, 757–778 CrossRef CAS.
- P. McQuade, D. W. McCarthy and M. J. Welch, in Positron Emission Tomography, editon edn, 2005, pp. 237–250 Search PubMed.
- G. Saha, Basics of PET Imaging, Springer, New York, x edn, 2005, pp. 99–110 Search PubMed.
- S. Y. F. Chu, L. P. Ekstrom and R. B. Firestone, WWW Table of radioactive isotopes, http://nucleardata.nuclear.lu.se/nucleardata/toi/, accessed July, 2009.
- G. J. Kelloff, J. M. Hoffman, B. Johnson, H. I. Scher, B. A. Siegel, E. Y. Cheng, B. D. Cheson, J. O'Shaughnessy, K. Z. Guyton, D. A. Mankoff, L. Shankar, S. M. Larson, C. C. Sigman, R. L. Schilsky and D. C. Sullivan, Clin. Cancer Res., 2005, 11, 2785–2808 CrossRef CAS.
- T. Bui and C. B. Thompson, Cancer Cell, 2006, 9, 419–420 CrossRef CAS.
- J. W. Kim and C. V. Dang, Cancer Res., 2006, 66, 8927–8930 CrossRef CAS.
- R. A. Gatenby and R. J. Gillies, Nat. Rev. Cancer, 2008, 8, 56–61 CrossRef CAS.
- T. Pfeiffer, S. Schuster and S. Bonhoeffer, Science, 2001, 292, 504–507 CrossRef CAS.
- C. V. Dang and G. L. Semenza, Trends Biochem. Sci., 1999, 24, 68–72 CrossRef CAS.
- D. A. Tennant, R. V. Duran, H. Boulahbel and E. Gottlieb, Carcinogenesis, 2009, 30, 1269–1280 CrossRef CAS.
- G. L. Semenza, D. Artemov, A. Bedi, Z. Bhujwalla, K. Chiles, D. Feldser, E. Laughner, R. Ravi, J. Simons, P. Taghavi and H. Zhong, Novartis Found. Symp., 2001, 240, 251–260 Search PubMed ; discussion 260–254.
- K. Higashi, Y. Ueda, K. Ayabe, A. Sakurai, H. Seki, Y. Nambu, M. Oguchi, H. Shikata, S. Taki, H. Tonami, S. Katsuda and I. Yamamoto, Nucl. Med. Commun., 2000, 21, 707–714 CrossRef CAS.
- L.-F. de Geus-Oei, H. F. M. v. d. Heijden, Frans H. M. Corstens and Wim J. G. Oyen, Cancer, 2007, 110, 1654–1664 CrossRef.
- I. F. Ciernik, E. Dizendorf, B. G. Baumert, B. Reiner, C. Burger, J. B. Davis, U. M. Lütolf, H. C. Steinert and G. K. Von Schulthess, Int. J. Radiat. Oncol. Biol. Phys., 2003, 57, 853–863 CrossRef.
- D. A. Mankoff and L. K. Dunnwald, PET Clinics, 2006, 1, 71–81 CrossRef.
- C. Haioun, E. Itti, A. Rahmouni, P. Brice, J.-D. Rain, K. Belhadj, P. Gaulard, L. Garderet, E. Lepage, F. Reyes and M. Meignan, Blood, 2005, 106, 1376–1381 CrossRef CAS.
- N. G. Mikhaeel, M. Hutchings, P. A. Fields, M. J. O'Doherty and A. R. Timothy, Ann. Oncol., 2005, 16, 1514–1523 CrossRef CAS.
- M. E. Juweid, S. Stroobants, O. S. Hoekstra, F. M. Mottaghy, M. Dietlein, A. Guermazi, G. A. Wiseman, L. Kostakoglu, K. Scheidhauer, A. Buck, R. Naumann, K. Spaepen, R. J. Hicks, W. A. Weber, S. N. Reske, M. Schwaiger, L. H. Schwartz, J. M. Zijlstra, B. A. Siegel and B. D. Cheson, J. Clin. Oncol., 2007, 25, 571–578 CrossRef.
- G. L. Ceresoli, A. Chiti, P. A. Zucali, M. Rodari, R. F. Lutman, S. Salamina, M. Incarbone, M. Alloisio and A. Santoro, J. Clin. Oncol., 2006, 24, 4587–4593 CrossRef.
- T. J. Kim, H. Y. Kim, K. W. Lee and M. S. Kim, Radiographics, 2009, 29, 403–421 CrossRef.
- N. Avril, S. Sassen, B. Schmalfeldt, J. Naehrig, S. Rutke, W. A. Weber, M. Werner, H. Graeff, M. Schwaiger and W. Kuhn, J. Clin. Oncol., 2005, 23, 7445–7453 CrossRef.
- P. D. Shreve, Y. Anzai and R. L. Wahl, Radiographics, 1999, 19, 61–77 CAS ; quiz 150–151.
- E. Patz, Jr, V. Lowe, J. Hoffman, S. Paine, P. Burrowes, R. Coleman and P. Goodman, Radiology, 1993, 188, 487–490 Search PubMed.
- P. J. Effert, R. Bares, S. Handt, J. M. Wolff, U. Bull and G. Jakse, J. Urol., 1996, 155, 994–998 CrossRef CAS.
- X. B. Kong, Q. Y. Zhu, P. M. Vidal, K. A. Watanabe, B. Polsky, D. Armstrong, M. Ostrander, S. A. Lang, Jr, E. Muchmore and T. C. Chou, Antimicrob. Agents Chemother., 1992, 36, 808–818 CAS.
- J. R. Grierson, J. L. Schwartz, M. Muzi, R. Jordan and K. A. Krohn, Nucl. Med. Biol., 2004, 31, 829–837 CrossRef CAS.
- B. Munch-Petersen, L. Cloos, G. Tyrsted and S. Eriksson, J. Biol. Chem., 1991, 266, 9032–9038 CAS.
- J. R. Bading and A. F. Shields, J. Nucl. Med., 2008, 49(Suppl 2), 64S–80S CrossRef CAS.
- H. L. van Westreenen, D. C. Cobben, P. L. Jager, H. M. van Dullemen, J. Wesseling, P. H. Elsinga and J. T. Plukker, J. Nucl. Med., 2005, 46, 400–404 CAS.
- Y. Yamamoto, Y. Nishiyama, N. Kimura, S. Ishikawa, M. Okuda, S. Bandoh, N. Kanaji, M. Asakura and M. Ohkawa, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 236–245 CrossRef CAS.
- W. Chen, T. Cloughesy, N. Kamdar, N. Satyamurthy, M. Bergsneider, L. Liau, P. Mischel, J. Czernin, M. E. Phelps and D. H. S. Silverman, J. Nucl. Med., 2005, 46, 945–952 CAS.
- A. K. Buck, K. Herrmann, C. M. z. Buschenfelde, M. E. Juweid, M. Bischoff, G. Glatting, G. Weirich, P. Moller, H.-J. Wester, K. Scheidhauer, T. Dechow, C. Peschel, M. Schwaiger and S. N. Reske, Clin. Cancer Res., 2008, 14, 2970–2977 CrossRef CAS.
- A. K. Buck, M. Bommer, S. Stilgenbauer, M. Juweid, G. Glatting, H. Schirrmeister, T. Mattfeldt, D. Tepsic, D. Bunjes, F. M. Mottaghy, B. J. Krause, B. Neumaier, H. Dohner, P. Moller and S. N. Reske, Cancer Res., 2006, 66, 11055–11061 CrossRef CAS.
- S. J. Choi, J. S. Kim, J. H. Kim, S. J. Oh, J. G. Lee, C. J. Kim, Y. S. Ra, J. S. Yeo, J. S. Ryu and D. H. Moon, Eur. J. Nucl. Med. Mol. Imaging, 2005, 32, 653–659 CrossRef.
- A. K. Buck, M. Hetzel, H. Schirrmeister, G. Halter, P. Moller, C. Kratochwil, A. Wahl, G. Glatting, F. M. Mottaghy, T. Mattfeldt, B. Neumaier and S. N. Reske, Eur. J. Nucl. Med. Mol. Imaging, 2005, 32, 525–533 CrossRef.
- G. Halter, A. K. Buck, H. Schirrmeister, I. Wurziger, F. Liewald, G. Glatting, B. Neumaier, L. Sunder-Plassmann, S. N. Reske and M. Hetzel, J. Thorac. Cardiovasc. Surg., 2004, 127, 1093–1099 CrossRef CAS.
- D. C. Cobben, P. H. Elsinga, H. J. Hoekstra, A. J. Suurmeijer, W. Vaalburg, B. Maas, P. L. Jager and H. M. Groen, J. Nucl. Med., 2004, 45, 1677–1682 CAS.
- B. S. Pio, C. K. Park, R. Pietras, W. A. Hsueh, N. Satyamurthy, M. D. Pegram, J. Czernin, M. E. Phelps and D. H. Silverman, Mol. Imaging Biol., 2006, 8, 36–42 Search PubMed.
- L. Kenny, R. C. Coombes, D. M. Vigushin, A. Al-Nahhas, S. Shousha and E. O. Aboagye, Eur. J. Nucl. Med. Mol. Imaging, 2007, 34, 1339–1347 CrossRef.
- H. J. Sohn, Y. J. Yang, J. S. Ryu, S. J. Oh, K. C. Im, D. H. Moon, D. H. Lee, C. Suh, J. S. Lee and S. W. Kim, Clin. Cancer Res., 2008, 14, 7423–7429 CrossRef CAS.
- K. Herrmann, H. A. Wieder, A. K. Buck, M. Schoffel, B. J. Krause, F. Fend, T. Schuster, C. Meyer zum Buschenfelde, H. J. Wester, J. Duyster, C. Peschel, M. Schwaiger and T. Dechow, Clin. Cancer Res., 2007, 13, 3552–3558 CrossRef CAS.
- W. Chen, S. Delaloye, D. H. Silverman, C. Geist, J. Czernin, J. Sayre, N. Satyamurthy, W. Pope, A. Lai, M. E. Phelps and T. Cloughesy, J. Clin. Oncol., 2007, 25, 4714–4721 CrossRef CAS.
- V. Tolmachev, M. Friedman, M. Sandstrom, T. L. Eriksson, D. Rosik, M. Hodik, S. Stahl, F. Y. Frejd and A. Orlova, J. Nucl. Med., 2009, 50, 274–283 CrossRef.
- J. Y. C. Wong, D. Z. Chu, L. E. Williams, D. M. Yamauchi, D. N. Ikle, C. S. Kwok, A. Liu, S. Wilczynski, D. Colcher, P. J. Yazaki, J. E. Shively, A. M. Wu and A. A. Raubitschek, Clin. Cancer Res., 2004, 10, 5014–5021 CrossRef CAS.
- J. V. Leyton, T. Olafsen, E. J. Lepin, S. Hahm, K. B. Bauer, R. E. Reiter and A. M. Wu, Clin. Cancer Res., 2008, 14, 7488–7496 CrossRef CAS.
- A. M. E.-S. Steven M. Larson, Chaitanya R. Divgi, George Sgouros, Ronald D. Finn, Jorg Tschmelitsch, Antonio Picon, Marc Whitlow, Jeffrey Schlom, Jiaju Zhang and Alfred M. Cohen, Cancer, 1997, 80, 2458–2468 CrossRef.
- S. B. Larson, J. S. Day, S. Glaser, G. Braslawsky and A. McPherson, J. Mol. Biol., 2005, 348, 1177–1190 CrossRef CAS.
- P. M. Smith-Jones, D. B. Solit, T. Akhurst, F. Afroze, N. Rosen and S. M. Larson, Nat. Biotechnol., 2004, 22, 701–706 CrossRef CAS.
- T. Olafsen, C. W. Cheung, P. J. Yazaki, L. Li, G. Sundaresan, S. S. Gambhir, M. A. Sherman, L. E. Williams, J. E. Shively, A. A. Raubitschek and A. M. Wu, Protein Eng., Des. Sel., 2004, 17, 21–27 CrossRef CAS.
- H. Kobayashi, E. S. Han, I. S. Kim, N. Le, V. Rajagopal, R. J. Kreitman, I. Pastan, C. H. Paik and J. A. Carrasquillo, Nucl. Med. Biol., 1998, 25, 387–393 CrossRef CAS.
- A. Orlova, M. Magnusson, T. L. Eriksson, M. Nilsson, B. Larsson, I. Hoiden-Guthenberg, C. Widstrom, J. Carlsson, V. Tolmachev, S. Stahl and F. Y. Nilsson, Cancer Res., 2006, 66, 4339–4348 CrossRef CAS.
- A. M. Wu, J. Nucl. Med., 2009, 50, 2–5 CAS.
- D. M. Goldenberg, E. A. Rossi, R. M. Sharkey, W. J. McBride and C. H. Chang, J. Nucl. Med., 2008, 49, 158–163 CAS.
- H. Kobayashi, H. Sakahara, M. Hosono, Z. S. Yao, S. Toyama, K. Endo and J. Konishi, J. Nucl. Med., 1994, 35, 1677–1684 CAS.
- D. E. Milenic, E. D. Brady and M. W. Brechbiel, Nat. Rev. Drug Discovery, 2004, 3, 488–499 CrossRef CAS.
- G. A. Wiseman, C. A. White, T. E. Witzig, L. I. Gordon, C. Emmanouilides, A. Raubitschek, N. Janakiraman, J. Gutheil, R. J. Schilder, S. Spies, D. H. Silverman and A. J. Grillo-Lopez, Clin. Cancer Res., 1999, 5, 3281s–3286s CAS.
- A. J. Grillo-Lopez, Expert Rev. Anticancer Ther., 2002, 2, 485–493 Search PubMed.
- P. M. Deckert, Curr. Drug Targets, 2009, 10, 158–175 Search PubMed.
- J. C. Reubi, Endocr. Rev., 2003, 24, 389–427 CrossRef CAS.
- J. G. McAfee and R. D. Neumann, Nucl. Med. Biol., 1996, 23, 673–676 CrossRef CAS.
- R. Cescato, T. Maina, B. Nock, A. Nikolopoulou, D. Charalambidis, V. Piccand and J. C. Reubi, J. Nucl. Med., 2008, 49, 318–326 CrossRef CAS.
- T. Maina, B. A. Nock, H. Zhang, A. Nikolopoulou, B. Waser, J. C. Reubi and H. R. Maecke, J. Nucl. Med., 2005, 46, 823–830 CAS.
- V. Rufini, M. L. Calcagni and R. P. Baum, Semin. Nucl. Med., 2006, 36, 228–247 Search PubMed.
- M. de Jong, R. Valkema, F. Jamar, L. K. Kvols, D. J. Kwekkeboom, W. A. P. Breeman, W. H. Bakker, C. Smith, S. Pauwels and E. P. Krenning, Semin. Nucl. Med., 2002, 32, 133–140 CrossRef.
- D. Zwanziger and A. G. Beck-Sickinger, Curr. Pharm. Des., 2008, 14, 2385–2400 CrossRef CAS.
- D. J. Kwekkeboom and E. P. Krenning, Semin. Nucl. Med., 2002, 32, 84–91 CrossRef.
- I. Dijkgraaf, O. C. Boerman, W. J. Oyen, F. H. Corstens and M. Gotthardt, Anticancer Agents Med. Chem., 2007, 7, 543–551 CAS.
- V. Kersemans, B. Cornelissen, K. Kersemans, M. Bauwens, E. Achten, R. A. Dierckx, J. Mertens and G. Slegers, J. Nucl. Med., 2005, 46, 532–539 CAS.
- K. A. Krohn, J. M. Link and R. P. Mason, J. Nucl. Med., 2008, 49(Suppl 2), 129S–148S CrossRef CAS.
- T. Saga, M. Koizumi, T. Furukawa, K. Yoshikawa and Y. Fujibayashi, Cancer Sci., 2009, 100, 375–381 CrossRef CAS.
- F. G. Blankenberg, Curr. Pharm. Des., 2008, 14, 2974–2982 CrossRef CAS.
- Z. Levashova, M. V. Backer, G. Horng, D. Felsher, J. M. Backer and F. G. Blankenberg, Bioconjugate Chem., 2009, 20, 742–749 CrossRef CAS.
- L. A. Bentolila, Y. Ebenstein and S. Weiss, J. Nucl. Med., 2009, 50, 493–496 CrossRef CAS.
- Y. Hama, Y. Urano, Y. Koyama, P. L. Choyke and H. Kobayashi, J. Biomed. Opt., 2007, 12, 051501 CrossRef.
- W. F. Anderson and R. Matsuno, J. Natl. Cancer Inst., 2006, 98, 948–951 CrossRef CAS.
- H. Kobayashi, Y. Hama, Y. Koyama, T. Barrett, C. A. Regino, Y. Urano and P. L. Choyke, Nano Lett., 2007, 7, 1711–1716 CrossRef CAS.
- R. M. Levenson, D. T. Lynch, H. Kobayashi, J. M. Backer and M. V. Backer, ILAR J., 2008, 49, 78–88 Search PubMed.
- T. Barrett, Y. Koyama, Y. Hama, G. Ravizzini, I. S. Shin, B. S. Jang, C. H. Paik, Y. Urano, P. L. Choyke and H. Kobayashi, Clin. Cancer Res., 2007, 13, 6639–6648 CrossRef CAS.
- M. Longmire, N. Kosaka, M. Ogawa, P. L. Choyke and H. Kobayashi, Cancer Sci., 2009, 100, 1099–1104 CrossRef CAS.
- K. Marten, C. Bremer, K. Khazaie, M. Sameni, B. Sloane, C. H. Tung and R. Weissleder, Gastroenterology, 2002, 122, 406–414 CrossRef.
- P. Wunderbaldinger, K. Turetschek and C. Bremer, Eur. Radiol., 2003, 13, 2206–2211 CrossRef.
- C. Bremer, C. H. Tung and R. Weissleder, Acad. Radiol., 2002, 9(2), S314–315 CrossRef.
- U. Mahmood and R. Weissleder, Mol. Cancer Ther., 2003, 2, 489–496 CAS.
- M. Ogawa, C. A. Regino, P. L. Choyke and H. Kobayashi, Mol. Cancer Ther., 2009, 8, 232–239 CrossRef CAS.
- M. Ogawa, N. Kosaka, P. L. Choyke and H. Kobayashi, Cancer Res., 2009, 69, 1268–1272 CrossRef CAS.
- Y. Urano, D. Asanuma, Y. Hama, Y. Koyama, T. Barrett, M. Kamiya, T. Nagano, T. Watanabe, A. Hasegawa, P. L. Choyke and H. Kobayashi, Nat. Med., 2009, 15, 104–109 CrossRef CAS.
- E. B. van Munster and T. W. Gadella, Adv. Biochem. Eng. Biotechnol., 2005, 95, 143–175.
- R. M. Clegg, O. Holub and C. Gohlke, Methods Enzymol., 2003, 360, 509–542 CAS.
- K. Suhling, P. M. French and D. Phillips, Photochem. Photobiol. Sci., 2005, 4, 13–22 RSC.
- A. E. Cerussi, J. S. Maier, S. Fantini, M. A. Franceschini, W. W. Mantulin and E. Gratton, Appl. Opt., 1997, 36, 116–124 CrossRef CAS.
- E. Kuwana and E. M. Sevick-Muraca, Biophys. J., 2002, 83, 1165–1176 CrossRef CAS.
- K. Vishwanath, B. Pogue and M. A. Mycek, Phys. Med. Biol., 2002, 47, 3387–3405 CrossRef.
- M. Hassan, J. Riley, V. Chernomordik, P. Smith, R. Pursley, S. B. Lee, J. Capala and A. H. Gandjbakhche, Mol. Imaging, 2007, 6, 229–236 CAS.
- R. A. Sheth, R. Upadhyay, L. Stangenberg, R. Sheth, R. Weissleder and U. Mahmood, Gynecol. Oncol., 2009, 112, 616–622 CrossRef CAS.
- D. T. Kehagias, A. D. Gouliamos, V. Smyrniotis and L. J. Vlahos, J. Magn. Reson. Imaging, 2001, 14, 595–601 CrossRef CAS.
- C. Curtet, F. Maton, T. Havet, M. Slinkin, A. Mishra, J. F. Chatal and R. N. Muller, Invest. Radiol., 1998, 33, 752–761 CrossRef CAS.
- D. A. Sipkins, D. A. Cheresh, M. R. Kazemi, L. M. Nevin, M. D. Bednarski and K. C. Li, Nat. Med., 1998, 4, 623–626 CrossRef CAS.
- M. Kresse, S. Wagner, D. Pfefferer, R. Lawaczeck, V. Elste and W. Semmler, Magn. Reson. Med., 1998, 40, 236–242 CAS.
- S. Ozawa, Y. Imai, T. Suwa and M. Kitajima, Recent Results Cancer Res., 2000, 155, 73–87 Search PubMed.
- C. F. Geraldes and S. Laurent, Contrast Media Mol. Imaging, 2009, 4, 1–23 Search PubMed.
- H. S. Thomsen, P. Marckmann and V. B. Logager, Cancer Imaging, 2007, 7, 130–137 Search PubMed.
- T. L. Colpitts, M. Prorok and F. J. Castellino, Biochemistry, 1995, 34, 2424–2430 CrossRef CAS.
- R. A. Moats, S. E. Fraser and T. J. Meade, Angew. Chem., Int. Ed. Engl., 1997, 36, 726–728 CrossRef CAS.
- S. Zhang, M. Merritt, D. E. Woessner, R. E. Lenkinski and A. D. Sherry, Acc. Chem. Res., 2003, 36, 783–790 CrossRef CAS.
- J. Folkman, Eur. J. Cancer, 1996, 32A, 2534–2539 CrossRef CAS.
- P. S. Tofts, G. Brix, D. L. Buckley, J. L. Evelhoch, E. Henderson, M. V. Knopp, H. B. Larsson, T. Y. Lee, N. A. Mayr, G. J. Parker, R. E. Port, J. Taylor and R. M. Weisskoff, J. Magn. Reson. Imaging, 1999, 10, 223–232 CrossRef CAS.
- P. L. Choyke, A. J. Dwyer and M. V. Knopp, J. Magn. Reson Imaging, 2003, 17, 509–520 CrossRef.
- S. B. Wedam, J. A. Low, S. X. Yang, C. K. Chow, P. Choyke, D. Danforth, S. M. Hewitt, A. Berman, S. M. Steinberg, D. J. Liewehr, J. Plehn, A. Doshi, D. Thomasson, N. McCarthy, H. Koeppen, M. Sherman, J. Zujewski, K. Camphausen, H. Chen and S. M. Swain, J. Clin. Oncol., 2006, 24, 769–777 CrossRef CAS.
- L. Lee, S. Sharma, B. Morgan, P. Allegrini, C. Schnell, J. Brueggen, R. Cozens, M. Horsfield, C. Guenther, W. P. Steward, J. Drevs, D. Lebwohl, J. Wood and P. M. McSheehy, Cancer Chemother Pharmacol., 2006, 57, 761–771 CrossRef CAS.
- H. Q. Xiong, R. Herbst, S. C. Faria, C. Scholz, D. Davis, E. F. Jackson, T. Madden, D. McConkey, M. Hicks, K. Hess, C. A. Charnsangavej and J. L. Abbruzzese, Invest. New Drugs., 2004, 22, 459–466 CrossRef CAS.
- T. Barrett, M. Brechbiel, M. Bernardo and P. L. Choyke, J. Magn. Reson. Imaging, 2007, 26, 235–249 CrossRef.
- A. Luciani, E. Itti, A. Rahmouni, M. Meignan and O. Clement, Eur. J. Radiol., 2006, 58, 338–344 CrossRef.
- A. Lopez-Guillermo, L. Colomo, M. Jimenez, F. Bosch, N. Villamor, L. Arenillas, A. Muntanola, S. Montoto, E. Gine, D. Colomer, S. Bea, E. Campo and E. Montserrat, J. Clin. Oncol., 2004, 23, 2797–2804 CrossRef.
- E. Sivridis, A. Giatromanolaki, G. Galazios and M. I. Koukourakis, The Breast, 2006, 15, 382–389 Search PubMed.
- M. F. Bellin, L. Lebleu and J. B. Meric, Abdom. Imaging, 2003, 28, 155–163 CrossRef CAS.
- A. S. Feldman, W. S. McDougal and M. G. Harisinghani, Urol. Oncol., 2008, 26, 65–73 Search PubMed.
- A. G. Rockall, S. A. Sohaib, M. G. Harisinghani, S. A. Babar, N. Singh, A. R. Jeyarajah, D. H. Oram, I. J. Jacobs, J. H. Shepherd and R. H. Reznek, J. Clin. Oncol., 2004, 23, 2813–2821 CrossRef.
- M. G. Harisinghani, S. Saini, G. J. Slater, M. D. Schnall and M. D. Rifkin, J. Magn. Reson. Imaging, 1997, 7, 161–163 CrossRef CAS.
- T. Barrett, P. L. Choyke and H. Kobayashi, Contrast Media Mol. Imaging, 2006, 1, 230–245 Search PubMed.
- H. Kobayashi, S. Kawamoto, Y. Sakai, P. L. Choyke, R. A. Star, M. W. Brechbiel, N. Sato, Y. Tagaya, J. C. Morris and T. A. Waldmann, J. Natl. Cancer Inst., 2004, 96, 703–708 CrossRef CAS.
- T. Schaeffter, Progress Drug Res., 2005, 62, 15–81 Search PubMed.
- Y. Koyama, V. S. Talanov, M. Bernardo, Y. Hama, C. A. Regino, M. W. Brechbiel, P. L. Choyke and H. Kobayashi, J. Magn. Reson. Imaging, 2007, 25, 866–871 CrossRef.
- W. J. Mulder, G. J. Strijkers, J. W. Habets, E. J. Bleeker, D. W. van der Schaft, G. Storm, G. A. Koning, A. W. Griffioen and K. Nicolay, FASEB J., 2005, 19, 2008–2010 CAS.
- W. Cai, K. Chen, Z. B. Li, S. S. Gambhir and X. Chen, J. Nucl. Med., 2007, 48, 1862–1870 CrossRef CAS.
- H. Y. Lee, Z. Li, K. Chen, A. R. Hsu, C. Xu, J. Xie, S. Sun and X. Chen, J. Nucl. Med., 2008, 49, 1371–1379 CrossRef CAS.
- H. Kobayashi, Y. Koyama, T. Barrett, Y. Hama, C. A. Regino, I. S. Shin, B. S. Jang, N. Le, C. H. Paik, P. L. Choyke and Y. Urano, ACS Nano, 2007, 1, 258–264 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |