Amino acid transport in thermophiles : characterization of an arginine-binding protein in Thermotoga maritima†
Matthew S.
Luchansky
a,
Bryan S.
Der
a,
Sabato
D’Auria
b,
Gabriella
Pocsfalvi
b,
Luisa
Iozzino
b,
Daniela
Marasco
c and
Jonathan D.
Dattelbaum
*a
aDepartment of Chemistry, University of Richmond, Gottwald Center for the Sciences, 28 Westhampton Way, Richmond, VA 23173, USA. E-mail: jdattelb@richmond.edu
bInstitute of Protein Biochemistry, CNR, Naples, Italy
cUniversity of Naples Federico II, Naples, Italy
First published on 22nd September 2009
Abstract
Members of the periplasmic binding protein superfamily are involved in the selective passage of ligands through bacterial cell membranes. The hyperthermophilic eubacterium Thermotoga maritima was found to encode a highly stable and specific periplasmic arginine-binding protein (TM0593). Following signal sequence removal and overexpression in Escherichia coli, TM0593 was purified by thermoprecipitation and affinity chromatography. The ultra-stable protein with a monomeric molecular weight of 27.7 kDa was found to exist as both a homodimer and homotrimer at appreciable concentrations even under strongly denaturing conditions, with an estimated transition temperature of 116 °C. Its multimeric structure may provide further evidence of the importance of quaternary structure in the movement of nutrients across bacterial membranes. Purified and refolded TM0593 was further characterized by fluorescence spectroscopy, mass spectrometry, and circular dichroism to demonstrate the specificity of the protein for arginine and to elucidate structural changes associated with arginine binding. The protein binds arginine with a dissociation constant of 20 μM as determined by surface plasmon resonance measurements. Due to its high thermodynamic stability, TM0593 may serve as a scaffold for the creation of a robust fluorescent biosensor .
Introduction
Thermotoga maritima is a Gram-negative, rod-shaped hyperthermophilic bacterium originally isolated from geothermal vents on the ocean floor near Italy.1 Though a eubacterium, T. maritima contains a large number of Archaeal gene homologues. Based on RNA phylogeny and comparative genomics , this bacterial species has exhibited slow evolution that provides evidence that the first eubacteria may have been thermophiles .2 Comparing the genomes of T. maritima and Archaea revealed evidence of lateral gene transfer between Archaea and Eubacteria.3T. maritima contains 81 Archaea-like genes and demonstrates one of the highest levels of similarity to Archaea of any Eubacterium, with 24% of its open reading frames strongly related to Archaeal gene sequences.4 Insights into the structural and functional characteristics of thermophilic proteins may hold value in developing further evolutionary connections between organisms.As in many organisms, the T. maritimagenome contains a diverse range of ATP-binding cassettes, which couple the hydrolysis of ATP to the passage of ligands through the cell membrane.5 The expressed ATP-binding cassette consists of two transmembrane domains and two ATP-binding domains for transporting a specific ligand across the cell membrane.6 Each cassette relies on a soluble periplasmic binding protein (PBP) capable of interacting with a specific cognate ligand necessary for bacterial metabolic and nutrient gathering processes. T. maritima exhibits a high level of redundancy in its transport proteins, and the duplicated function provides augmented nutrient gathering and metabolic capabilities that represent a selective advantage.7 Combined with its position as a link between Archaea and Eubacteria, the wide array of transport systems that arose from operon duplication or horizontal gene transfer makes T. maritima an interesting and valuable subject for phylogenetic studies.
The PBPs that are associated with the ABC and other transport systems are also of interest for the construction of protein-based biosensors . Due to the variety of binding specificities, PBPs from Escherichia coli have been utilized as design platforms for fluorescent proteinbiosensors capable of targeting many naturally-occurring ligands, including sugars, anions, and amino acids.8–13 While diverse in ligand affinity, this family of proteins contains a highly conserved structural organization. PBPs are formed from a single polypeptide chain composed of two easily distinguishable domains that are connected by a hinge region which undergoes a large rotational-bending movement upon ligand binding.14,15 Using spectroscopic techniques, such as environmental-sensitivity, FRET, or plasmonic interactions, rational placement of fluorescent probes allows transduction of the binding event into a quantifiable optical signal that varies as a function of ligand concentration.16–18 In addition to detecting the natural set of ligands associated with PBPs, re-engineering the PBP binding pocket has significantly expanded the number of small-molecule analytes for which sensors may be constructed.19–21 However, there is a considerable cost in thermal stability for these re-engineered biosensors as a result of the large number of mutations required to alter the binding specificity.22 For this reason, putative thermophilic PBPs are being isolated and characterized because of their potential value in the design of new biosensing technology that features enhanced stability23 and the ability to monitor metabolite trafficking in vivo.24 Since T. maritima thrives at temperatures near the optimal growth temperature of 80 °C,1 its thermostable proteins are capable of retaining functional stability necessary to augment the current array of mesophilic scaffolds available for biosensing applications.
Here, we investigate a putative amino acid transport system in the T. maritimagenome .4 A periplasmic binding protein designated tm0593 at position chr: 627![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 081–627821 exhibits high homology to several E. coli amino acid-binding PBPs, particularly those for glutamine and histidine. As a hyperthermophilic protein, the gene product may be of interest for the creation of fluorescent biosensors based on a periplasmic binding protein scaffold, as well as the study of Archaea-like proteins in Eubacteria. In this study, we present the purification and spectroscopic characterization of the tm0593 gene product. Based on mass spectrometry, circular dichroism, fluorescence spectroscopy, and surface plasmon resonance, tm0593 was found to encode a thermostable periplasmic arginine-binding protein.
081–627821 exhibits high homology to several E. coli amino acid-binding PBPs, particularly those for glutamine and histidine. As a hyperthermophilic protein, the gene product may be of interest for the creation of fluorescent biosensors based on a periplasmic binding protein scaffold, as well as the study of Archaea-like proteins in Eubacteria. In this study, we present the purification and spectroscopic characterization of the tm0593 gene product. Based on mass spectrometry, circular dichroism, fluorescence spectroscopy, and surface plasmon resonance, tm0593 was found to encode a thermostable periplasmic arginine-binding protein.
Results
Cloning the tm0593 gene product
Isolation of purified tm0593 gene product was facilitated by the E. coli Rosetta strain, a BL-21 derived expression strain carrying the pRARE plasmid, which encodes several minor tRNA molecules normally underutilized in bacterial host strains and proteins. This compatible plasmid helped to overcome low expression problems caused by the 15 rare codons present in the tm0593 sequence and was required for acquiring the protein in good yield (see Experimental for full details). Based upon the translated DNA sequence, TM0593 is predicted to be a 27.7 kDa periplasmic binding protein. SignalP25 was used to identify the likely 19 amino acid periplasmic signal sequence at the N-terminus, and constructs with and without this region were cloned into pET21a. SDS-PAGE of purified TM0593 without the signal sequence showed a new band of the correct molecular weight present in the cytoplasmic fraction. However, a clone retaining the N-terminal signal sequence resulted in a new band corresponding to TM0593 found in the periplasmic extract (data not shown). Protein bands corresponding to dimer and trimer were also observed in the purified extract, leading to questions about the oligomeric state of this protein. Using gel filtration chromatography, the native molecular weight was determined to be ∼81 kDa, which is consistent with the formation of a homotrimer. Oligomers of greater size were also detected in these experiments (data not shown), which is consistent with recent observations made by Hellinga and colleagues on another thermophilic periplasmic protein (TM0322).26To serve as a starting point for determining the cognate ligand of TM0593, we measured the absorption and emission spectra in order to search for an altered spectroscopic response based on changes in Trp microenvironment polarity.27 The emission spectrum of the single Trp residue in TM0593 displays an unstructured peak with a maximum occurring at ∼320 nm (Fig. 1). This indicates an environment of low polarity similar to that observed for the common reference proteinRibonuclease T1 which is known to possess a buried Trp residue.28 Since TM0593 displays sequence similarity to many periplasmic amino acid-binding proteins, we tested for changes in Trp intensity and spectral shape in the presence of potential amino acid and sugar ligands, including arginine, glutamine, histidine, asparagine, glucose, ribose, and fructose (data not shown). Trp emission intensity did not change following prolonged incubations with any of these potential ligands. Furthermore, increasing the temperature for this thermophilic protein did not alter these observations, suggesting that the wild-type Trp residue location is not optimal for direct detection of ligand binding.
 | ||
| Fig. 1 Absorption and emission spectral properties of TM0593. Excitation of the single Trp residue was performed at 295 nm in 5 mM phosphate buffer, pH 7.0. | ||
TM0593 is an arginine-binding protein
Circular dichroism was used to characterize the secondary structure and thermal stability of TM0593. Far-UV CD spectra measured at 20 °C and 90 °C displayed identical traces consistent with a folded protein containing a mix of helices and β-sheets. To begin the characterization of TM0593 stability, we measured the thermal unfolding of the purified protein in the presence of increasing concentrations of Gdn-HCl (Fig. 2, top). Calculation of the transition temperature of TM0593 at each denaturant concentration allowed for extrapolation to 0 M Gdn-HCl. Based on these data, the natural transition temperature for the protein was estimated to be approximately 116 °C. This value represents the thermal stability of the ligand bound form of TM0593, which co-purifies with the cognate ligand attached and requires extensive dialysis or protein refolding to obtain the free protein.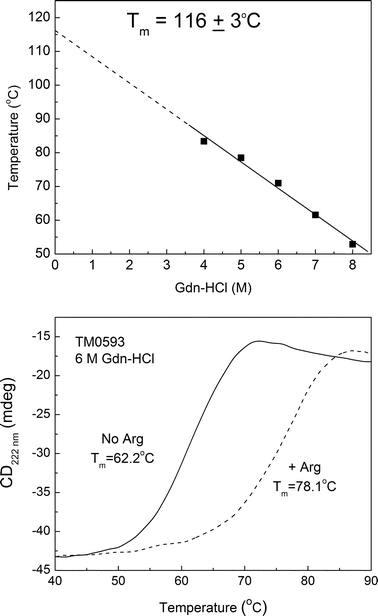 | ||
| Fig. 2 Thermal unfolding of TM0593 measured by circular dichroism in the presence of Gdn-HCl (top). The extrapolated unfolding temperature was found to be 116 ± 3 °C. In the presence of 6 M Gdn-HCl, the addition of 1 mM arginine produced a significant 16 °C increase in the melting temperature (bottom). | ||
Determination of the cognate ligand binding partner was accomplished using CD thermal unfolding experiments in the presence and absence of substrates as described previously.23,29,30 An increase in thermal stability in the presence of a particular ligand indicates a strong preference for binding. To test for binding, we performed CD thermal melting experiments on dialyzed TM0593 in the presence of 6 M Gdn-HCl and 1.0 mM of ligand: aspartate, glutamine, histidine, asparagine, lysine, or arginine (Table 1). Equilibration of each sample was complete following 24 h of incubation at room temperature. Of the ligands tested in this experiment, arginine alone produces a significant 16 °C increase in the TM0593 melting temperature (Fig. 2, bottom).
| Protein a | Unfolding temperature/°C | Standard error/°C |
|---|---|---|
| a All CD spectra were obtained using 1 μM protein equilibrated in 6 M Gdn-HCl for 24 h prior to taking measurements. b Ligand was added to 1 mM. | ||
| Native TM0593 (bound form) | 78.4 | 0.7 |
| TM0593 no ligand | 62.2 | 1.8 |
| TM0593 + Aspb | 62.7 | 0.9 |
| TM0593 + Glu | 63.4 | 0.9 |
| TM0593 + Asn | 62.8 | 0.8 |
| TM0593 + Lys | 62.5 | 0.2 |
| TM0593 + His | 62.8 | 0.5 |
| TM0593 + Arg | 78.1 | 1.9 |
Non-denaturing nano-ESI-MS was also used to study the selectivity of TM0593 for various amino acid ligands. Low concentration volatile buffer (10 mM ammonium formate) was used to maintain the native protein conformation in the solution phase, and carefully controlled low energy ionization conditions were applied to transfer weak molecular associations from the solution into the gas phase. Purified TM0593 protein (10 μM) was incubated with arginine (20 μM) and resulted in three distinct charge distributions in the nano-ESI mass spectrum corresponding to native TM0593, denatured TM0593, and native TM0593 plus ligand (Fig. 3). The most abundant distribution was observed in the mass range of m/z 1400–2300 which comprises the protonated molecular ion peaks of TM0593 with 19 to 12 positive charges. These peaks are indicative of the native non-denatured form of the protein. The denatured form, on the other hand, yields a second distribution of highly charged (34 to 24 positive charges) molecular ion peaks in the lower mass range m/z 800–1400. These two charge distributions were used to calculate the average molecular mass for the intact protein and yielded identical values of 27701.13 Da, which is the expected mass of TM0593 expressed with a linker to a hexahistidine peptide tag sequence.
 | ||
| Fig. 3 Nano-ESI mass spectrum of purified TM0593 (10 μM) incubated with 20 μM arginine in 10 mM ammonium formate. The three distinct charge distributions in the spectrum correspond to native TM0593 (circles), denatured TM0593 (shaded), and native TM0593 plus Arg (triangles). | ||
The third distribution observed in Fig. 3 occurs at slightly higher m/z than the non-denatured protein distribution. The molecular mass at this peak was determined to be 27875.6 Da, which corresponds to TM0593 in complex with the amino acid arginine (mw = 174.2 g mol−1) in a 1 : 1 protein–ligand stoichiometry. This indicates that only the native form of the protein takes part in the protein–ligand association, as would be predicted. Similar experiments performed with Asn and His did not lead to the formation of protein–ligand complex under the conditions used.
To quantitate the binding affinity and to further characterize the binding specificity of this protein for arginine, surface plasmon resonance (SPR) experiments were performed on TM0593. Two model peptides were designed using the crystallographic data for the E. coliglutamine bindingprotein in complex with glutamine (pdb code: 1WDN), which is a protein with high sequence homology to TM0593. The glutamine ligand is bound in a cleft between two protein domains and stabilized by hydrogen bonds and ionic interactions with Asp10, Gly68, Thr70, Ala67, Asp157, Arg75, Lys115, Gly119 and His156.31Peptides were constructed with either an N-terminal arginine or glutamine residue with sequences of (Arg-Gly)2-Lys-(Gly)3-Cys-NH2 and (Gln-Gly)2-Lys-(Gly)3-Cys-NH2. Five glycine residues were introduced as spacers to displace the N-terminal bindingamino acid from the C-terminus, whereas the cysteines were inserted into the peptide for conjugation with thiol-reactive dyes in future fluorescence studies. The branched end structure was added to produce peptides with a suitable molecular size for SPR binding studies. TM0593 was efficiently immobilized on the chip surface, and an overall 3500 RU immobilization level was achieved under the reported conditions. SPR analyses were performed by injecting increasing concentrations of each peptide: (Arg-Gly)2-Lys-(Gly)3-Cys-NH2 (0 to 400 μM ) and (Gln-Gly)2-Lys-(Gly)3-Cys-NH2 (0 to 700 μM ). RUmax values from each SPR binding experiment were plotted, and the EC50 values were evaluated to be 23.4 ± 1.1 μM and 160 ± 1.0 μM for (Arg-Gly)2-Lys-(Gly)3-Cys-NH2 and (Gln-Gly)2-Lys-(Gly)3-Cys-NH2, respectively (Fig. 4). These data demonstrate a highly preferential binding affinity of TM0593 for the polar amino acidarginine.
 | ||
| Fig. 4 SPR binding data for immobilized TM0593 with (Arg-Gly)2-Lys-(Gly)3-Cys-NH2 (top) and (Gln-Gly)2-Lys-(Gly)3-Cys-NH2 (bottom). The line drawn represents the best fit by non-linear regression analysis and was used to determine the EC50 values reported. | ||
TM0593 undergoes a conformational change in the presence of arginine
Having established Arg as the cognate ligand of TM0593, we investigated the ability of this protein to undergo conformational changes similar to other PBP family members. Trp emission was monitored to report on the global folded state as a function of increasing temperature in the presence and absence of Arg. In support of the CD unfolding data, the binding of Arg resulted in an increase in the thermal unfolding temperature above that of the unbound protein (Fig. 5). The presence of other amino acids resulted in no change to the TM, which suggests that only Arg binding induces an increase in protein stability. Next, we performed Stern–Volmer quenching analysis of the Trp emission to determine if a conformational change in the Trp microenvironment may be detected upon binding Arg (Fig. 6). The average ratio of fluorescence quenching (n = 3) at each iodide concentration was determined in the presence and absence of Arg. Using linear regression, the slope corresponding to the Stern–Volmer quenching constant (Ksv) was determined. The increase in iodide quenching of the Trp emission following the addition of Arg is significant (p = 0.048), which suggests that the Trp residue becomes more solvent accessible following ligand binding. Taken together, these experiments demonstrate that Arg binding provides an increase in protein stability and that the binding event involves some limited conformational changes, even at room temperature.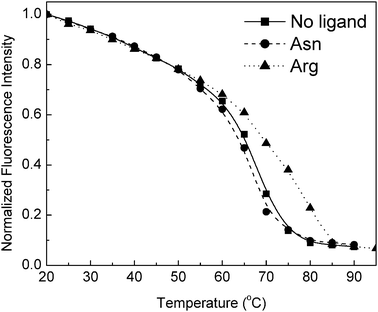 | ||
| Fig. 5 Representative data for TM0593 thermal unfolding monitored by a decrease in Trp emission in the presence and absence of 1 mM arginine or asparagine. Fluorescence measurements were obtained at an excitation wavelength of 280 nm in 5 mM phosphate buffer, pH 7.0. | ||
 | ||
| Fig. 6 TM0593 conformational change is identified by Stern–Volmer quenching with iodide in the presence and absence of 1 mM Arg. Error bars represent the standard deviation associated with the average of three independent trials. | ||
Conclusion
A novel thermostable binding protein for arginine was identified using preliminary genomic sequencing assignments and was experimentally characterized using mass spectrometry, CD, SPR, and Trp fluorescence. While the translated gene product of tm0593 has sequence similarity to several polar amino acid-binding proteins, this homology alone does not allow assignment of the specific cognate ligand for this PBP. Under the conditions used here, we observed exclusive binding to the basic amino acid arginine. We determined that TM0593 is exported to the periplasm and binds arginine with a significant increase in overall stability, suggesting that this protein exhibits a conformational change following ligand binding similar to other members of the PBP superfamily. The conformational change accompanying ligand binding in mesophilic PBPs provides melting temperatures typically in the range of 65–70 °C.22 The increased stability of thermophilic proteins like TM0593 (Tm = 116 °C) may have significant advantages for use as a protein design scaffold.23One of the major findings of this work is that it represents the first use of nano-ESI-MS to aid in the identification of the cognate ligand of a PBP. While sequence similarity searches are a good approach to narrow the pool of potential cognate ligands, we believe that the MS experiments presented here represent a more rapid alternative to traditional methodologies that involve screening libraries of ligands using fluorescence, CD, or radioactive binding assays . By combining the structural characterization capabilities of mass spectrometry with more traditional fluorescence and CD-based spectroscopic methods, it is possible to convincingly identify specific protein–ligand interactions.
This study also provides evidence that the arginine-binding PBP in T. maritima exists as both a homodimer and homotrimer at appreciable concentrations. Two previous studies, using the periplasmic binding proteins TM032226 and the Rhodobacter shpaeroides α-keto acid-binding protein,32 detected the presence of higher order oligomers in the native state. It is speculated that ligand-induced conformational changes lead to multimerization and that these changes are necessary for ligand transport.26,32 Additionally, the arginine–agmatineexchange transporter, a component of the arginine-dependent acid-resistance system in E. coli, has been demonstrated to exist as a homodimer within lipid membranes.33 Though increasingly observed in a variety of bacterial systems, the specific function of binding protein multimeric association is not well understood, as it is possible that either each monomer acts independently or that there exists significant cross-talk or cooperation between the monomers. The dimeric and trimeric nature of TM0593, even in strongly denaturing conditions, provides further evidence that quaternary structure and multimerization are significant elements of metabolic and homeostatic processes that involve arginine. Taken together, these results support a pattern of binding protein multimerization that is hypothesized to be important for the translocation of some ligands across the bacterial membrane.
Comprehensive phylogenetic analyses of T. maritima have been performed, and notable lateral gene transfer events between T. maritima and Archaeal species living in the same environment have been widely observed.34–36 TM0593 was originally assigned as a glutamine bindingprotein belonging to the 4th largest protein family, consisting of putative response regulators and ABC transport proteins. In E. coli, there are 4 well-characterized transport systems for polar amino acids, including glutamine37 (glnHPQ), glutamate–aspartate38,39 (gltIJKL), arginine40 (artPIQM artJ), and lysine–arginine–ornithine–histidine41,42 (argT hisJQMP). The confirmation of function for putative proteinsvia biochemical analysis is time-consuming and requires utilization of a broad array of experimental tools,36 so the ability to draw phylogenetic connections is particularly valuable. The ABC uptake system described here is organized from TM0593 (soluble periplasmic binding protein), TM0592 (transmembrane permease), and TM0591 (ATP-binding protein). Although functionally more similar to the arginine transport system, this gene organization is most similar to the glutamine permease operon (glnHPQ) where dimers of the permease and ATP-binding protein are necessary for activity.5 TM0592 displays 30% amino acid sequence identity (50% similarity) to the E. coli ArtQ transmembraneortholog, and TM0591 displays 43% identity (66% similarity) to E. coli ArtP ATP-binding protein. Based on the functional assignment of TM0593 as an arginine-binding protein and to reflect the nomenclature used to describe genes of ABC transport operons, we propose to rename the annotated T. maritimagenes (TM0593, TM0592, TM0591) to reflect an operon organization denoted artJQP.
Each of the ABC operons is dependent upon a periplasmic binding protein for transport specificity. A neighbor-joining phylogenetic analysis comparing TM0593 with PBPs from a variety of mesophilic and thermophilic organisms is shown in Fig. 7. Each of the proteins used has been experimentally characterized for ligand binding and for three-dimensional structure determination. While many PBP family members possess high levels of amino acid sequence similarity and overall three-dimensional structures reflecting their common origin, there are two distinct structural classes of PBPs based on the number and arrangement of β-strands present among these proteins.43 Class I proteins (red in Fig. 7) contain six β-strands ordered 213456, while class II proteins (blue in Fig. 7), which were derived later in evolutionary time, have five β-strands organized 21354 and also possess a more complex pattern of connectivity between upper and lower domains.43 Experiments analyzing the evolutionary significance of this structural differentiation have shown distinct differences in folding pathways; class I proteins typically follow simple two-state pathways, while more complex folding is observed in class II PBPs.44,45 However, caution in interpreting folding data on PBPs is important because additional data on GBP show that folding proceeds along a complex route of intermediates prior to the addition of glucose, and then a two-state mechanism of complementary folding is observed only after ligand binding.46
 | ||
| Fig. 7 Unrooted neighbor-joining tree of PBPs with experimentally established 3D structure and ligand specificity. Proteins representing class I (red) and class II (blue) structures are shown.43 Phylogenetic analysis of aligned sequences was performed using PAUP*, and the tree was constructed using 500 bootstrap replicates employing a distance optimality criterion.52 Cognate ligand designations: ABP, arabinose; ArtJ_EC, Arg; ArtJ_GST, Arg–His; DPBP, dipeptide; GBP, glucose; HBP, His; LBP, Leu; LIVBP, Leu–Iso–Val; LAO, Lys–Arg–Orn; MBP, maltose; NiBP, nickel(II); OPBP, oligopeptide; PhBP, phosphate; QBP, Gln RBP, ribose; SBP, sulfate. Abbreviations used for organisms: EC, E. coli; GST, G. stearothermophilus; ST, S. typhimurium; TM, T. maritima; TT, T. thermophilus. | ||
In the tree presented in Fig. 7, three distinct branches are evident, with the two structural classes grouping to a significant extent. On the TM0593 branch, binding proteins for the polar amino acidshistidine, glutamine, ornithine, and lysine, as well as Arg only, are found. The Geobacillus stearothermophiluslysine-, arginine-, histidine-binding protein (ArtJ_GST) which was recently characterized47 was the closest thermophilic protein neighbor in the diagram. Located on the same branch as TM0593, one of the most well-characterized periplasmic binding proteins is the Salmonella typhimurium LAO binding protein (LAO_ST).41 By performing X-ray crystallography and binding assays on LAO_ST, sub-micromolar dissociation constants were found for lysine, arginine, and ornithine, as well as for histidine.42,48 While broad ligand binding capabilities are found in many binding proteins with arginine affinity, our CD and SPR data indicate that TM0593 binds exclusively to Arg with micromolar affinity.
It is possible to use this phylogenetic analysis as a predictive tool for structural classification of newly characterized PBPs, and these results lead us to propose that TM0593 should crystallize as a class II PBP. These data suggest that the ability to acquire polar amino acids from the environment may have evolved later than transport systems for sugars or nonpolar amino acids based on the structural distinctions used here as well as by others. Our work represents the continuing advancement of biochemical comparisons among amino acid transport systems in mesophiles and thermophiles .
Additionally, this work succeeds in the characterization of a highly thermostable protein which may be utilized as a scaffold for biosensor design. In its native form, TM0593 is an ultra-stable protein that has the potential to provide the basis for a durable and highly specific sensor for arginine. Employing techniques such as FRET would allow for detection of arginine and its quantitation in unknown samples. Furthermore, through gene shuffling into less stable PBP family members specific for other analytes or by computationally-derived site-directed mutagenesis of the binding pocket, it may be possible to alter the specificity of TM0593 for other small-molecule targets. Ideally, this would allow access to a broad array of sensing targets while still maintaining the high structural stability characteristic of the native arginine-binding protein.
Experimental
Cloning and overexpression of TM0593 gene product
An 8 Kb fragment surrounding TM0593 was provided as a kind gift from The Institute of Genomic Research (Craig Venter Institute, Rockville, MD). E. coli Top10 was used as the background strain for cloning the TM0593 gene. An approximately 740 bp fragment containing the entire TM0593 gene was amplified using forward and reverse DNAoligonucleotide primers and was cloned into the NdeI-BamHI site of pET21a (Novagen, Madison, WI). Since a putative periplasmic signal sequence was identified using SignalP,25 two separate forward primers were used to either include or remove the first 19 amino acids of the TM0593 gene product. Additionally, the downstream primer included two extra bases to create a translational fusion with a 6×His tag when ligated into pET21a. Isolation of the correct cloning constructs was confirmed by restriction digestion and by the DNA Sequencing Core Facility at VCU Health Systems (Richmond, VA).Purification of TM0593-6×His
Clones verified to contain the correct insert sequences were transformed into CaCl2 competent E. coli Rosetta cells (Novagen) and were selected on LB agar plates containing ampicillin and chloramphenicol. Single colonies were incubated overnight at 37 °C with shaking in 2×YT broth containing ampicillin (100 μg mL−1) and chloramphenicol (30 μg mL−1). Fresh media was inoculated 1 : 100 for protein overexpression at 37 °C. After an OD600 ≈ 0.6 was reached, cells were induced with IPTG (0.2 mM) for 12 h. The cells were collected by centrifugation (3500 rpm, 10 min), re-suspended in 30 mL lysis buffer (50 mM phosphate, pH 8.0, 200 mM NaCl, 10 mM imidazole), and frozen at least overnight at −20 °C.Cells were thawed at room temperature and incubated with lysozyme (2 mg) and DNase (0.3 mg) for 20 min. The re-suspended cells were lysed by sonication, and the cell debris was removed by centrifugation (13000 rpm, 30 min). The protein supernatant was initially purified by thermoprecipitation (68 °C water bath, 35 min) and the heat-denatured proteins were removed by centrifugation (13000 rpm, 30 min). The remaining soluble protein extract was added to a Ni-NTA column for purification by affinity chromatography. TM0593 was bound to the column via a C-terminal 6×His tag and was extensively washed with 20 mM imidazole prior to elution with 250 mM imidazole. At this point, the protein was buffer exchanged into 5 mM phosphate, pH 7.0, and could be stored for more than 4 months at 4 °C. The protein concentration was determined by UV absorbance at 280 nm by a Nanodrop Spectrophotometer (Thermo Scientific, Wilmington, DE) using 14400 M−1 cm−1 as a predicted extinction coefficient.49
To remove any bound ligand remaining following purification, extensive dialysis or chemical denaturation/refolding was performed. For the latter method, isolated protein was unfolded by incubating the protein overnight at 68 °C in 7.2 M Gdn-HCl. The denatured protein was bound to a Ni-NTA column, washed, and refolded by incremental addition of decreasing Gdn-HCl concentrations. The refolded protein was eluted with 250 mM imidazole and buffer exchanged into 5 mM phosphate pH = 7.0 using a PD-10 column (Bio-Rad, Hercules, CA) to yield approximately 8 mg of TM0593 per litre of cells. Alternatively, isolated protein was dialyzed against 5 mM phosphate buffer for 3 days with twice daily buffer changes. Both methods yielded purified TM0593 with identical spectroscopic and chromatographic properties. Though TM0593 containing the signal sequence was isolatable from the periplasm using sucrose shock,11 higher levels of total protein without the signal sequence were obtained from the cytoplasm from an equivalent number of cells. The molecular weight of purified TM0593 was characterized using 15% SDS-PAGE and visualized using coomassie brilliant blue.
Circular dichroism
All CD measurements were performed using a Jasco (Cambridge, MD) J-720 spectrometer equipped with a thermal peltier temperature control module, and data were analyzed using the spectra manager software (ver. 5.1.0.0) supplied with the instrument. All measurements were performed using 2 mm square quartz cuvettes and protein solutions of approximately 0.5 μM in 5 mM phosphate buffer (pH 7.0), unless otherwise specified.Mass spectrometry
MALDI-TOF mass spectrometry (Bruker Daltonics) was used to determine the monomeric molecular weight of the purified protein. An aliquot of protein solution was diluted with matrix (10 mg mL−1hydroxycinnamic acid in 50% acetonitrile, 0.1% trifluoroacetic acid). Lysozyme (Mr = 14400 g mol−1) and bovine serum albumin (Mr = 66000 g mol−1) were added as internal reference protein standards.
Nano-ESI-MS experiments were performed on a quadrupole time-of-flight (Q-TOF) instrument, QSTAR Elite, from Applied Biosystems (Foster City, CA). Sample (10 μM) was introduced via a distal coated silica PicoTip (od 150 μm, id 20 μm, tip id 10 μm; New Objective) by infusion at 300 nL min−1. For MS analysis under native conditions, an aliquot of the TM05953 solution was buffer exchanged into 10 mM ammonium formate solution using a Millipore microcon column (10 kDa cut-off) immediately prior to injection. For analysis under denaturing conditions, a 10 μM sample was diluted twice in 50% acetonitrile, 0.1% formic acid. The following experimental parameters were used: ion spray voltage, 1.8 kV; curtain gas, 15 L h−1; declustering potential 1 and 2 were set to 40 V and 15 V, respectively; focusing potential, 280 V.
Surface plasmon resonance
A BIAcore 3000 SPR system for real-time kinetic analysis and related reagents were obtained from GE Healthcare (Milano, Italy). All other reagents and chemicals were commercially available from Sigma-Aldrich or Fluka (Steinheim, Germany). TM0593 was immobilized in 10 mM acetate buffer pH 4.5 (flow rate 5 μL min−1, 7 min injection time) on a CM5 Biacore sensor chip using EDC/NHS chemistry in accordance with the manufacturer’s instructions.50 Residual reactive groups were deactivated by treatment with 1 M ethanolamine hydrochloride, pH 8.5. The reference channel was prepared by activating with EDC/NHS and deactivating with ethanolamine. Peptides were diluted in HBS buffer (10 mM Hepes, 150 mM NaCl, 3 mM EDTA, pH 7.4) containing 0.1 mM TCEP at a flow rate of 20 μL min−1. Analyte injections of 90 μL were performed at the indicated concentrations. BIAevaluation analysis package (version 4.1, GE Healthcare, Milano, Italy) implemented by the instrument software was used to subtract the signal of the reference channel, while RUmax data for each peptide concentration were fit with GraphPad Prism, ver. 4.00, GraphPad Software (San Diego, California).Peptide synthesis
Peptides for SPR analysis were prepared by solid phase synthesis on a 50 μmol scale following the Fmoc strategy and using standard Fmoc-derivatized amino acids.51 Reagents for peptide synthesis (Fmoc-protected amino acids and resins, activation and deprotection reagents) were from Novabiochem (Laufelfingen, Switzerland) and InBios (Napoli, Italy). Solid phase peptide syntheses were performed on a fully automated multi-channel peptide synthesizer Syro I (Multisynthech, Germany). RINK AMIDE resin (substitution 0.5 mmol g−1) was used as solid support. Activation of amino acids was achieved using HBTU–HOBt–DIEA (1 : 1 : 2), whereas Fmoc deprotection was carried out using a 20% (v/v) piperidine solution in DMF. All couplings were performed for 15 min, and deprotections were performed for 10 min. Peptides were removed from the resin by treatment with a TFA–TIS–H2O (90 : 5 : 5, v/v/v) mixture, then they were precipitated in cold diethyl ether and lyophilized. Peptides were incubated in aqueous 1 mM TCEP for 20 min, and then purified using preparative RP-HPLC on a Shimadzu LC-8A, equipped with a SPD-M10 AV detector and with a Phenomenex C18 Jupiter column (50 × 22 mm id; 10 μm). Solvents for peptide synthesis and HPLC analyses were from Romil (Dublin, Ireland); reversed phase columns for peptide analysis and the LC-MS system were from ThermoFisher (Milan, Italy). LC-MS analyses were carried out on an LCQ DECA XP ion trap mass spectrometer equipped with an OPTON ESI source, operating at 4.2 kV needle voltage and 320 °C with a complete Surveyor HPLC system, composed of MS pump, an auto-sampler and a photo diode array (PDA). Experimental molecular weights were determined to be 845.44 ± 0.19 amu for (Arg-Gly)2-Lys-(Gly)3-Cys-NH2 and 789.36 ± 0.15 amu for (Gln-Gly)2-Lys-(Gly)3-Cys-NH2 in good agreement with those calculated (846.9 amu and 790.8 amu, respectively). Purified peptides were lyophilized and stored at −20 °C until use.Tryptophan emission
TM0593 fluorescence emission spectra were obtained with a Varian Cary Eclipse spectrofluorometer and were collected using excitation at 295 nm in 5 mM phosphate buffer, pH 7.0. Chemical denaturation of TM0593 (3 μM in 5 mM phosphate buffer, pH 7.0) with increasing concentrations of Gdn-HCl (0 to 6 M) was performed at room temperature and at 85 °C. Thermal denaturation using a Varian single cell peltier heating apparatus also provided information about protein stability and ligand binding. Quenching of tryptophan emission at 325 nm was carried out using purified TM0593 (3 μM) containing increasing concentrations of NaI (0 to 0.4 M) in phosphate buffer with NaCl added to achieve a consistent ionic strength of 0.5 M. To prevent iodine formation, 1 mM Na2S2O3 was added. The data were analyzed using the Stern–Volmer equation: F/F0 = 1 + Ksv[Q], where Ksv is the Stern–Volmer quenching constant, Q is the concentration of quencher , and F and F0 are the Trp emission intensities in the presence and absence of quencher , respectively.Abbreviations
| PBP | bacterial periplasmic binding protein |
| TM0593 | Thermotoga maritima arginine-binding protein |
| Gdn-HCl | guanidine hydrochloride |
| T M | melting temperature |
| CD | circular dichroism |
| Trp | tryptophan |
| Arg | arginine |
| MALDI-TOF | matrix assisted laser desorption ionization–time-of-flight |
| nano-ESI-MS | nanoflow electrospray ionization mass spectrometry |
| TIS | triisopropylsilane |
| TFA | trifluoroacetic acid |
| DMF | dimethylformamide |
| DCM | dichloromethane |
| HBTU | 1-H-benzotriazolium, 1-[bis(dimethylamino)methylene]-hexafluorophosphate(1-),3-oxide |
| HOBt | N-hydroxybenzotriazole |
| DIEA | diisopropylethylamine |
| Fmoc | fluorenylmethoxycarbonyl |
| TCEP | tris(2-carboxyethyl)phosphine |
| LC-MS | liquid chromatography–mass spectrometry |
| EDC | 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride |
| NHS | N-hydroxysuccinimide |
Acknowledgements
The authors would like to thank Research Corporation and the Jeffress Memorial Trust for their generous support of this research and EnEn Jang for his help with the gel filtration experiments. With great appreciation, the authors thank Dr Malcolm Hill for his help with the phylogenetic analysis. B. Der would like to thank the Howard Hughes Medical Institute for funding support.References
- R. Huber, T. A. Langworthy, H. Konig, M. Thomm, C. R. Woese, U. B. Sleytr and K. O. Stetter, Thermotoga maritima sp. nov. represents a new genus of unique extremely thermophilic eubacteria growing up to 90 °C, Arch. Microbiol., 1986, 144, 324–333 CrossRef CAS.
- L. Achenbach-Richter, R. Gupta, K. O. Stetter and C. R. Woese, Were the original Eubacteria thermophiles, Syst. Appl. Microbiol., 1987, 9, 34–39 Search PubMed.
- P. Worning, L. J. Jensen, K. E. Nelson, S. Brunak and D. W. Ussery, Structural analysis of DNA sequence: evidence for lateral gene transfer in Thermotoga maritima, Nucleic Acids Res., 2000, 28, 706–709 CrossRef CAS.
- K. E. Nelson, R. A. Clayton, S. R. Gill, M. L. Gwinn, R. J. Dodson, D. H. Haft, E. K. Hickey, J. D. Peterson, W. C. Nelson, K. A. Ketchum, L. McDonald, T. R. Utterback, J. A. Malek, K. D. Linher, M. M. Garrett, A. M. Stewart, M. D. Cotton, M. S. Pratt, C. A. Phillips, D. Richardson, J. Heidelberg, G. G. Sutton, R. D. Fleischmann, J. A. Eisen, O. White, S. L. Salzberg, H. O. Smith, J. C. Venter and C. M. Fraser, Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima, Nature, 1999, 399, 323–329 CrossRef CAS.
- C. F. Higgins, ABC transporters: from microorganisms to man, Annu. Rev. Cell Biol., 1992, 8, 67–113 Search PubMed.
- A. S. Ethayathulla, Y. Bessho, A. Shinkai, B. Padmanabhan, T. P. Singh, P. Kaur and S. Yokoyama, Purification, crystallization and preliminary X-ray diffraction analysis of the putative ABC transporter ATP-binding protein from Thermotoga maritima, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2008, 64, 498–500 Search PubMed.
- D. M. Nanavati, T. N. Nguyen and K. M. Noll, Substrate specificities and expression patterns reflect the evolutionary divergence of maltose ABC transporters in Thermotoga maritima, J. Bacteriol., 2005, 187, 2002–2009 CrossRef CAS.
- R. M. De Lorimier, J. J. Smith, M. A. Dwyer, L. L. Looger, K. M. Sali, C. D. Paavola, S. S. Rizk, S. Sadigov, D. W. Conrad, L. Loew and H. W. Hellinga, Construction of a fluorescent biosensor family, Protein Sci., 2002, 11, 2655–2675 CrossRef.
- J. S. Marvin, E. E. Corcoran, N. A. Hattangadi, J. V. Zhang, S. A. Gere and H. W. Hellinga, The rational design of allosteric interactions in a monomeric protein and its applications to the construction of biosensors, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4366–4371 CrossRef CAS.
- L. Tolosa, I. Gryczynski, L. R. Eichhorn, J. D. Dattelbaum, F. N. Castellano, G. Rao and J. R. Lakowicz, Glucose sensor for low-cost lifetime-based sensing using a genetically engineered protein, Anal. Biochem., 1999, 267, 114–120 CrossRef CAS.
- J. D. Dattelbaum and J. R. Lakowicz, Optical determination of glutamine using a genetically engineered protein, Anal. Biochem., 2001, 291, 89–95 CrossRef CAS.
- M. Brune, J. L. Hunter, J. E. T. Corrie and M. R. Webb, Direct, Real-Time Measurement of Rapid Inorganic Phosphate Release Using a Novel Fluorescent Probe and Its Application to Actomyosin Subfragment ATPase, Biochemistry, 1994, 33, 8262–8271 CrossRef CAS.
- L. L. Salins, E. S. Goldsmith, C. M. Ensor and S. Daunert, A fluorescence-based sensing system for the environmental monitoring of nickel using the nickel binding protein from Escherichia coli, Anal. Bioanal. Chem., 2002, 372, 174–180 CrossRef CAS.
- R. Tam and M. H. Saier, Jr., Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria, Microbiol. Rev., 1993, 57, 320–346 CAS.
- A. L. Davidson, E. Dassa, C. Orelle and J. Chen, Structure, function, and evolution of bacterial ATP-binding cassette systems, Microbiol. Mol. Biol. Rev., 2008, 72, 317–364 CrossRef CAS.
- M. A. Dwyer and H. W. Hellinga, Periplasmic binding proteins: a versatile superfamily for protein engineering, Curr. Opin. Struct. Biol., 2004, 14, 495–504 CrossRef CAS.
- B. S. Der and J. D. Dattelbaum, Construction of a reagentless glucose biosensor using molecular exciton luminescence, Anal. Biochem., 2008, 375, 132–140 CrossRef CAS.
- K. Deuschle, S. Okumoto, M. Fehr, L. L. Looger, L. Koszhukh and W. B. Frommer, Construction and optimization of a family of genetically encoded metabolite sensors by semirational protein engineering, Protein Sci., 2005, 14, 2304–2314 CrossRef CAS.
- M. Allert, S. S. Rizk, L. L. Looger and H. W. Hellinga, Computational design of receptors for an organophosphate surrogate of the nerve agent soman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7907–7912 CrossRef CAS.
- L. L. Looger, M. A. Dwyer, J. J. Smith and H. W. Hellinga, Computational design of receptor and sensor proteins with novel functions, Nature, 2003, 423, 185–190 CrossRef CAS.
- I. Lager, L. L. Looger, M. Hilpert, S. Lalonde and W. B. Frommer, Conversion of a putative Agrobacterium sugar-binding protein into a FRET sensor with high selectivity for sucrose, J. Biol. Chem., 2006, 281, 30875–30883 CrossRef CAS.
- N. C. Vercillo, K. J. Herald, J. F. Fox, B. S. Der and J. D. Dattelbaum, Analysis of Ligand Binding to a Ribose Biosensor using Site-directed mutagenesis and Fluorescence Spectroscopy, Protein Sci., 2007, 16, 362–368 CrossRef CAS.
- Y. Tian, M. J. Cuneo, A. Changela, B. Höcker, L. S. Beese and H. W. Hellinga, Structure-based design of robust glucose biosensors using a Thermotoga maritima periplasmic glucose-binding protein, Protein Sci., 2007, 16, 2240–2250 CrossRef CAS.
- H. Gu, S. Lalonde, S. Okumoto, L. L. Looger, A. M. Scharff-Poulsen, A. R. Grossman, J. Kossmann, I. Jakobsen and W. B. Frommer, A novel analytical method for in vivo phosphate tracking, FEBS Lett., 2006, 580, 5885–5893 CrossRef CAS.
- O. Emanuelsson, S. Brunak, G. von Heijne and H. Nielsen, Locating proteins in the cell using TargetP, SignalP, and related tools, Nat. Protocols, 2007, 2, 953–971 CAS.
- M. J. Cuneo, A. Changela, A. E. Miklos, L. S. Beese, J. K. Krueger and H. W. Hellinga, Structural analysis of a periplasmic binding protein in the tripartite ATP-independent transporter family reveals a tetrameric assembly that may have a role in ligand transport, J. Biol. Chem., 2008, 283, 32812–32820 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3 edn, 2006 Search PubMed.
- M. R. Eftink and C. A. Ghiron, Dynamics of a protein matrix revealed by fluorescence quenching, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3290–3294 CrossRef CAS.
- J. Schellman, Macromolecular binding, Biopolymers, 1975, 14, 999–1018 CrossRef CAS.
- H. D. Simpson, U. R. Haufler and R. M. Daniel, An extremely thermostable xylanase from the thermophilic eubacterium Thermotoga, Biochem. J., 1991, 277, 413–417 CAS.
- Y.-J. Sun, J. Rose, B.-C. Wang and C.-D. Hsiao, The Structure of Glutamine-binding Protein complexed with Glutamine at 1.94 A Resolution: comparisons with other Amino Acid Binding Proteins, J. Mol. Biol., 1998, 278, 219–229 CrossRef CAS.
- S. Gonin, P. Arnoux, B. Pierrru, J. Lavergne, B. Alonso, M. Sabaty and D. Pignol, Crystal structures of an Extracytoplasmic Solute Receptor from a TRAP transporter in its open and closed forms reveal a helix-swapped dimer requiring a cation for alpha-keto acid binding, BMC Struct. Biol., 2007, 7, 11 CrossRef.
- Y. Fang, L. Kolmakova-Partensky and C. Miller, A bacterial arginine-agmatine exchange transporter involved in extreme acid resistance, J. Biol. Chem., 2007, 282, 176–182 CAS.
- N. C. Kyrpides, C. A. Ouzounis, I. Iliopoulos, V. Vonstein and R. Overbeek, Analysis of the Thermotoga maritima genome combining a variety of sequence similarity and genome context tools, Nucleic Acids Res., 2000, 28, 4573–4576 CrossRef CAS.
- R. Huber and M. Hanning, Thermotogales, Springer-Verlag, New York, 2004 Search PubMed.
- S. B. Conners, E. F. Mongodin, M. R. Johnson, C. I. Montero, K. E. Nelson and R. M. Kelly, Microbial biochemistry, physiology, and biotechnology of hyperthermophilic Thermotoga species, FEMS Microbiol. Rev., 2006, 30, 872–905 CAS.
- T. Nohno, T. Saito and J. S. Hong, Cloning and complete nucleotide sequence of the Escherichia coli glutamine permease operon (glnHPQ), Mol. Gen. Genet., 1986, 205, 260–269 CrossRef CAS.
- M. Marcus and Y. S. Halpern, Genetic analysis of the glutamate permease in Escherichia coli K-12, J. Bacteriol., 1969, 97, 1118–1128 CAS.
- K. Marin and R. Kramer, in Amino Acid Biosynthesis, ed. V. F. Wendisch, Springer-Verlag, Berlin, 2007, vol. 5 Search PubMed.
- U. Wissenbach, S. Six, J. Bongaerts, D. Ternes, S. Steinwachs and G. Unden, A third periplasmic transport system for L-arginine in Escherichia coli: molecular characterization of the artPIQMJ genes, arginine binding and transport, Mol. Microbiol., 1995, 17, 675–686 CrossRef CAS.
- G. F. Ames, Bacterial periplasmic transport systems: structure, mechanism, and evolution, Annu. Rev. Biochem., 1986, 55, 397–425 CrossRef CAS.
- K. Nikaido and G. F. Ames, Purification and characterization of the periplasmic lysine-, arginine-, ornithine-binding protein (LAO) from Salmonella typhimurium, J. Biol. Chem., 1992, 267, 20706–20712 CAS.
- K. Fukami-Kobayashi, Y. Tateno and K. Nishikawa, Domain dislocation: a change of core structure in periplasmic binding proteins in their evolutionary history, J. Mol. Biol., 1999, 286, 279–290 CrossRef CAS.
- K. Kashiwagi, K. Shiba, K. Fukami-Kobayashi, T. Noda, K. Nishikawa and H. Noguchi, Characterization of folding pathways of the type-1 and type-2 periplasmic binding proteins MglB and ArgT, J. Biochem. (Tokyo), 2003, 133, 371–376 Search PubMed.
- K. Kashiwagi, K. Fukami-Kobayashi, K. Shiba and K. Nishikawa, Construction and characterization of chimeric proteins composed of type-1 and type-2 periplasmic binding proteins MglB and ArgT, Biosci., Biotechnol., Biochem., 2004, 68, 808–813 CrossRef CAS.
- G. Piszczek, S. D’Auria, M. Staiano, M. Rossi and A. Ginsburg, Conformational stability and domain coupling in D-glucose/D-galactose-binding protein from Escherichia coli, Biochem. J., 2004, 381, 97–103 CrossRef CAS.
- A. Vahedi-Faridi, V. Eckey, F. Scheffel, C. Alings, H. Landmesser, E. Schneider and W. Saenger, Crystal structures and mutational analysis of the arginine-, lysine-, histidine-binding protein ArtJ from Geobacillus stearothermophilus. Implications for interactions of ArtJ with its cognate ATP-binding cassette transporter, Art(MP)2, J. Mol. Biol., 2008, 375, 448–459 CrossRef CAS.
- B. H. Oh, J. Pandit, C. H. Kang, K. Nikaido, S. Gokcen, G. F. Ames and S. H. Kim, Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand, J. Biol. Chem., 1993, 268, 11348–11355 CAS.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, Protein identification and analysis tools on the ExPASy server, in The Proteomics Protocols Handbook, ed. J. M. Walker, Humana Press, Totowa, NJ, 2005, pp. 571–607 Search PubMed.
- B. Johnsosson, S. Lofas and G. Lindquist, Immobilization of protein to a carboxymethyldextran modified gold surface for biospecific interaction analysis in SPR sensor, Anal. Biochem., 1991, 198, 268–277 CrossRef CAS.
- G. B. Fields and R. L. Noble, Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids, Int. J. Pept. Protein Res., 1990, 35, 161–214 CAS.
- D. Swofford, PAUP*. Phylogenetic analysis using parsimony (* and other methods). Version 4, 2002.
Footnote |
| † Electronic supplementary information (ESI) available: Structures of the peptides used for SPR measurements are provided. Links to view 3D visualisations of structures using FirstGlance. See DOI: 10.1039/b908412f |
| This journal is © The Royal Society of Chemistry 2009 |