Computational model and microfluidic platform for the investigation of paracrine and autocrine signaling in mouse embryonic stem cells†
David
Ellison
,
Alex
Munden
and
Andre
Levchenko
*
Department of Biomedical Engineering, Johns Hopkins University School of Engineering, Clark Hall 208C, 3400 North Charles Street, Baltimore, MD 21218, USA. E-mail: alev@jhu.edu; Fax: +1 410-516-6240; Tel: +1 410-516-5584
First published on 2nd July 2009
Abstract
Autocrine and paracrine signaling mechanisms are traditionally difficult to study due to the recursive nature of the process and the sub-micromolar concentrations involved. This has proven to be especially limiting in the study of embryonic stem cells that might rely on such signaling for viability, self-renewal, and proliferation. To better characterize possible effects of autocrine and paracrine signaling in the setting of expanding stem cells, we developed a computational model assuming a critical need for cell-secreted survival factors. This model suggested that the precise way in which the removal of putative survival factors could affect stem cell survival in culture. We experimentally tested the predictions in mouse embryonic stem cells by taking advantage of a novel microfluidic device allowing removal of the cell-conditioned medium at defined time intervals. Experimental results in both serum-containing and defined N2B27 media confirmed computational model predictions, suggested existence of unknown survival factors with distinct rates of diffusion, and revealed an adaptive/selective phase in mouse embryonic stem cell response to a lack of paracrine signaling. We suggest that the described computational/experimental platform can be used to identify and study specific factors and pathways involved in a wide variety of paracrine signaling systems.
Introduction
Embryonic stem cells (ESCs) are defined by their unique ability to both self-renew indefinitely and differentiate into multiple lineages. ESCs divide symmetrically, that is, the two daughter cells produced are identical.1 Additionally, these cells remain viable and mitotically active without the use of an immortalizing agent. These two characteristics, taken together, allow ESCs to undergo seemingly limitless passages, or complete self-renewal.2 The other defining characteristic of ESCs is their ability to differentiate into multiple lineages. Pluripotent ESCs have the ability, under proper culture conditions, to differentiate into the three different primary germ-layers: ectoderm, mesoderm, and endoderm.3 Thereafter, such cells can further be differentiated into end-type tissues, and thus have the ability to contribute to all somatic and germ line tissues of the organism.4 This differentiation capacity holds considerable promise for basic research into the mechanisms of cell differentiation and organism development, as well as the potential for facilitating therapeutic interventions involving tissue regeneration.First isolated over twenty years ago,5 mouse embryonic stem cells (mESCs) are the in vitro counterparts to the early embryo’s epiblast.6,7 Traditionally, these mESCs are cultured on top of a feeder-layer of inactivated primary mouse embryonic fibroblasts (pMEFs). By adding leukemia inhibitory factor (LIF) to a serum-containing media, the mESCs can be maintained in a self-renewing state indefinitely. More recently, groups have been developing protocols which eliminate the need for a feeder layer or serum-containing media. For instance, by complementing the serum-free growth medium with bone morphogenic proteins (BMP) and LIF, mESCs can be maintained in the undifferentiated state even without a feeder layer.8,9 While such efforts have shed light onto the exogenous soluble factors required for maintaining undifferentiating and proliferating mESCs, little is still understood about the paracrine and autocrine signaling that mESCs might engage in, including the signaling mechanisms underlying self-renewal. Knowledge of the mechanisms of cell–cell communications that might underlie cell survival and self-renewal is of importance for optimization of mESC growth in culture and for the better understanding of their behavior in vivo.
Methods of cell culture on a feeder layer, or in a monolayer on tissue-culture plastic, are subject to variable conditions due to the batch processing of typically daily media exchange.10 Microfluidic devices have become progressively more appealing as culturing systems due to their ability to precisely control the spatial and temporal cell micro-environment, including various aspects of signaling from soluble factors present in the media. Made from polydimethylsiloxane (PDMS), microfluidic devices or chips can be patterned by photolithography processes in order to house micro-scale cell culture chambers. PDMS offers many advantages for live cell work; it is biocompatible, optically transparent, and permeable to air. These characteristics allow for both long-term cell culture in a highly controlled fashion and high throughput real-time light-microscopy imaging. Flow can be controlled in these devices so that media exchange can be continuous or intermittent as the user desires. With these advantages in mind, microfluidic devices have been used to culture many different cell types in a wide variety of applications.11
In autocrine signaling, cells secrete soluble factors which then can bind to receptors on their own surface. Autocrine signaling is an important aspect of normal and abnormal cell biology12 and has been shown to be essential in the development of mESCs.13 Unfortunately, understanding autocrine signaling is severely limited due to the recursive nature of the process,14 and its action at sub-micron dimensions.15 Alternatively, instead of a cell signaling to itself, paracrine signaling occurs when the soluble factor secreted by one cell diffuses and binds to a cell that might be several cell diameters away, stimulating it to display the corresponding response. This allows for a wide variety of ensuing phenotypes, depending on cell proximity (or density) and number.13
Current mathematical and computational models of autocrine and paracrine signaling generally focus on the spatial signaling range. Such simulations are based on either single-cell approximations15 or compartmental models.16–18 Briefly, a cell or a confluent monolayer of cells is placed onto a two- or three-dimensional simulation grid. A simulated factor is released by the modeled cells and allowed to follow a Brownian walk until it is recaptured through binding to the respective receptor. This type of simulation is ideally suited to address questions of the spatial range of autocrine signaling, but is not useful for determining the overall effect that autocrine and paracrine signaling might have on the viability of mESCs.
In this report, we introduce both a computational and experimental model of autocrine and paracrine signaling in cell cultures. We intend for the modeling and experimental approaches to complement each other, so that the experimental platform can yield a sufficiently precise yet simple method of controlling the extracellular environment. Our particular emphasis in this study is on the ability to experimentally control the removal of molecular factors secreted by cells into the surrounding media, thus affecting the degree and efficiency of autocrine and paracrine regulation of cell functions. The model is explored to predict the effect of partial removal of the secreted signaling factors on cell survival, so that a close match between the modeling predictions and experimental validation can be achieved. We demonstrate that this method also allows controlled medium modification by addition of exogenous survival factors through the use of serum-free or serum-containing media. (see Fig. 1). Using this integrative biology approach, we provide evidence for the existence of a soluble autocrine/paracrine factor(s) secreted by mESCs that contributes to their viability in in vitro culture.
 | ||
| Fig. 1 Schematic of modeling and experimental system. Mouse embryonic stem cells experience autocrine and paracrine signaling as represented by the arrows labeled “Paracrine Survival Factors”. Two ways the signaling network can be perturbed is by differential wash rates and changes in media type. The media type may add exogenous survival factors to the system, whereas the wash rate can only remove the paracrine survival factors. This signaling network and its perturbations can be represented both through simulations and experimental design, which over time evolve and improve based on discoveries made both in vitro and in silico. | ||
Results
We first examined a set of mathematical and computational models, intended to assist in choosing informative conditions for the experimental model to be discussed later. This model examines the relative importance that synthesis, diffusion, and removal rate have on various aspects of paracrine signaling. Initially, as a guide, we considered the case in which there was a single spherical cell in suspension. The diffusion of soluble factors secreted by a cell in the radial geometry is described as the time dependant mass transport from this single cell as follows:19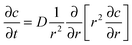 | (1) |
 | (2) |
 | (3) |
We examined the solutions of eqn (2) for various parameters D, F0, and krmv (Fig. 2A). The range of the diffusion coefficient values used was based on the experimentally observed values for signaling molecules in the 10–100 kDa range, whose coefficients range from 1.0 × 10−6 to 1.0 × 10−7 cm2 s−1.21 Another key parameter is the maximal secretion rate, F0. Our estimates for the range of this value were based on an in vivo analysis which showed secretion rates of approximately 2300–8000 molecules cell−1 s−1.22 The rate of basal ligand removal was based on simulated and experimental results of EGF accumulation in conditioned media in which cells “lost” 0.1 nM of EGF over an eight hour period, or about 0.02% of the total amount estimated to have been secreted each hour.23 Since this amount can vary based on receptor affinity, receptor concentration, cell density, and a number of other factors, we varied the removal rate by a factor of ten from 0.01% h−1 to 0.1% h−1 of the range that included the value of 0.02% h−1 cited above. Under low and medium synthesis rates, our model resulted in roughly the same effective width (see below), as that previously theoretically and experimentally verified, i.e., approximately 8–25 cell diameters.20,24 Under the maximal synthesis rates however, our estimates result in the effective width of up to 40 cell diameters.
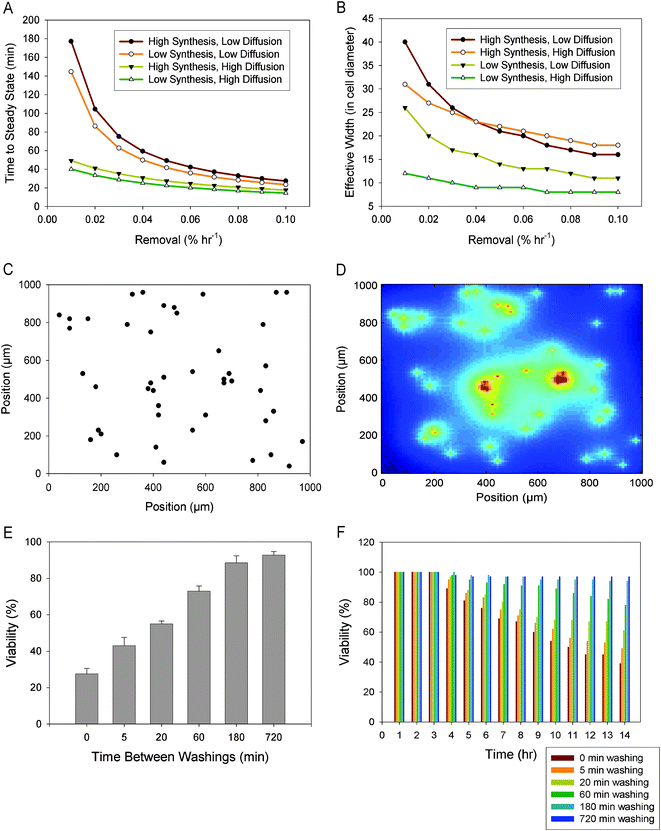 | ||
| Fig. 2 (A) Computational modeling of the time needed to reach a steady state in paracrine signaling for a single cell where synthesis rate, removal rate, and diffusion coefficient are varied. Synthesis rate is based on estimates from 2300–8000 molecules cell−1 s−1, removal on 0.02% h−1 lost ligands in conditioned media, and diffusion rates on estimates of 1.0 × 10−6 to 1.0 × 10−7 cm2 s−1. (B) Computational modeling of the distance, in cell diameters, that cells can communicate through paracrine signaling (the effective width). Model parameters are as in (A). (C) Modeling of cell distribution in a population. Cells are randomly seeded as single-cell colonies onto a 1200 μm × 3200 μm modeling area, a 1 mm × 1 mm subsection is shown here. (D) The cells that are seeded as in (C) are allowed to secrete ligands into the medium, creating a “cloud” of ligands based upon nominal parameters. Color coding: the local concentration varies from the lowest (blue) to the highest (red). (E) The dependence on the medium washout rate of the viability of model cell populations simulated as in (C–D), with the viability for individual cells calculated as a function of the time integral of the local ligand concentration, as described in the main text. Simulations are from five replicates, with the error bars representing the standard error of the mean. (F) Time course of mESC viability under specified washing rates. | ||
Within these parameter ranges, we explored the properties of the propagating diffusible signal. First, we examined the effect that these parameters had on the time it took for the diffusing molecule (also referred to here as the signaling factor) to reach the steady state distribution, a characteristic value of the potential cell–cell communication time scale. For ease of visualization, Fig. 2A only shows the modeling results that used the maximal and minimal values for the diffusion coefficient and secretion rates. Consequently, the simulation results with intermediate parameter values fell within the ranges established by the illustrated maximal and minimal parameter values. As expected, the slowly diffusing factors were predicted to take longer to reach a steady state distribution vs. highly diffusible ones. In addition slowly diffusing factors demonstrated higher sensitivity to ligand removal rate than quickly diffusing signals. We also observed that changes in the rate of secretion did not have a strong effect on the signal stabilization time for the parameter values assumed. Indeed, although in all cases considered it took longer for the system with high paracrine factor secretion rates to reach a steady state, this effect was comparatively minor. For the parameter ranges analyzed, the time required to reach a steady state varied between 20 and 200 min establishing the time scale for the experimental analysis, as described below.
Having explored the effects of the secretion, diffusion, and removal rates on the time required to reach a steady state signaling factor distribution, we investigated how far the secreted signal could be propagated from the secreting cell so that it was still present at a biologically relevant (or effective) concentration. This distance is referred to as the effective width. Francis and Palsson20 defined this effective width relative to the Kd, or the equilibrium dissociation constant, of a particular diffusing protein. In this definition, the effective width of a signal is the distance away from the cell in which the concentration, c, at that distance is equal to the Kd, mathematically expressed as follows,
 | (4) |
Having explored the temporal and spatial effects of diffusion and synthesis rates, and, in particular, the removal rate on paracrine signaling, we turned to the effects signaling factors could have on a population of cells. If cell survival is dependent upon the transmission of paracrine signaling, then timing and spatial ranges of action of diffusing factors become important considerations. More specifically, we wished to simulate the signaling environment created by a group of cells given a particular set of nominal parameters, paying particular attention to the effects of varying the removal of the paracrine factor. For simplicity, in the initial model, we focused on simulation of the effects of either a single paracrine ligand, or the aggregate average of a number of ligands, assuming that they had similar effects on cell survival. This is the simplest possible assumption, and in this study we tested whether predictions based on it would be supported by experimental results. However, the reality is likely more complex, with more than one factor likely mediating intercellular pro-survival communication. Fortunately, the computational model is easily extensible to consider multi-factor signaling systems, if suggested by experimental results.
For our analysis, therefore, we chose the case of a cell secreting a ligand with a relatively high affinity for its cognate receptor that has the rates of diffusion and synthesis that are intermediate in the sense of ranges explored in our modeling efforts so far. Therefore the baseline removal rate was set at a relatively low value 0.02% h−1, diffusion coefficient at 5 × 10−6 cm2 s−1, and synthesis rate at 4500 molecules cell−1 s−1. It is important to note that the primary focus of our analysis was to explore the role of the exogenously controlled removal of the ligand, and thus the effective removal rate was varied as described below. Thus, the model cells capable of a pro-survival factor secretion according to the assumptions stated above were then stochastically seeded onto a 1200 μm × 3200 μm simulation space (see Fig. 2C). Thereafter the cells were allowed to continuously secrete the diffusive paracrine factor, creating a “cloud” of the factor molecules around each cell (see Fig. 2D). As the factor “cloud” expanded, it might encounter another neighboring cell. This cell “recorded” the amount of the factor it was exposed to from this and other sources (other neighboring cells, as well as itself) and integrated the signal over a specified period of time. This graded input was then converted by the cell into a binary output (the life vs.death decision), as defined based upon a pre-determined threshold.25 The choice of this threshold is an important consideration, as placing it too high results in all cells uniformly dying, and placing the threshold too low results in almost no cells being affected. We therefore based the threshold and subsequent chance for cell death on colony-forming assays in which mESCs are plated from single-cell suspensions at low concentrations. In this situation, viability is reported at approximately 30%.26 For our simulations, we calculated the amount of ligand that a cell would receive if it had no close neighbors and set that amount as the threshold to undergo cell death. In other words, if the signal experienced by a cell fell below that threshold, the cell would only have a 30% chance to survive.
While diffusion and synthesis rate are dependent upon the ligand and cells themselves, the removal rate can be controlled through washing the cells. To explore this effect, the simulation spaces were “washed” of the signal at different intervals by periodically reducing the paracrine factor levels to zero. Continuous washing (0 min inter-washing interval) means that cells are largely deprived of their own autocrine signaling as well as the paracrine signaling. The 5 minute washing was chosen to allow some autocrine signaling, but very limited paracrine signaling. The 20, 60, and 180 minute washings coincide with the minimal and maximal times to steady states as illustrated in Fig. 2A. Finally, the 720 minute washing was chosen to be close to a control of unperturbed cell–cell communication.
The resultant simulated viability analysis of cells undergoing the various washing treatments (Fig. 2E) showed that under continuous flow conditions, roughly the same number of cells survive as when they are clonally seeded.26 At the other extreme, the 720 minute washing regime keeps the cell number approximately constant due to the low rate of death and low rate of proliferation. The viability for washing rates between these two extremes gradually varied, further suggesting that the selected inter-washing intervals might represent interesting experimental targets. In this analysis, as was later further justified by experimental results, we assumed that cells have negligible amount of proliferation. A number of considerations led to this assumption. First, the effects of seeding the cells initially retards growth, which, given the relatively short tracking time (twelve hours), is significant. Additionally the cells are cultured in supplementation-free media which is known to profoundly reduce the growth rate of mESCs.27 Therefore, we primarily concentrated on the effects soluble factors have on cell viability.
While computing the overall viability of the cells in the simulation we also tracked the cells over time to learn when death occurred. As can be seen in Fig. 2F, without any selective or adaptation mechanisms, the cell viability decreased approximately linearly with the increasing duration of the signaling factor removal before reaching a plateau, with the plateau survival levels dependent on the washing rates. Thus with 20 min inter-washing intervals, the plateau level was at approximately 65%. This effect related to a considerable graded decrease in the effective width with increasing signaling factor removal rate (Fig. 2B), further suggesting that, if the cell survival and proliferation depends on the presence of autocrine and paracrine signaling factors, periodic removal of the factors by washing the cells can reduce viable cell numbers as a function of the frequency of the washes. This prediction can be used to experimentally determine the presence of essential paracrine signaling, as shown below.
To validate the model predictions, we designed and fabricated a two-layer push down microfluidic device (Fig. 3A). This device has six cell culture areas in which the fluid flow can be separately controlled, allowing six different wash conditions in one experiment. Each culture area is 1200 μm × 3200 μm × 30 μm. One of the important concerns in microfluidic experiments involving flow over live cells is the degree of damage that might be caused by the shear stress. For the conditions present in our experiments, we calculated the predicted flow rate based on pressure difference and the calculated resistance of the chip, and then verified that flow rate experimentally using fluorescent beads. Images were taken at specific time intervals and the distance the bead had traveled was measured. Based on both calculated and measured flow rates, the shear stress experienced by the cells was estimated to be 1.29 × 10−1 dynes cm−2. This shear stress value was shown to be sufficiently low to successfully culture mESCs over a five day period (0.1–1 dynes cm−2)28 and was well below the 1.5–10 dynes cm−2,29 and 15 dynes cm−230 used by other groups for the culture of mESCs; strongly suggesting that the shear stress experienced by cells in our device did not adversely affect either their growth or survival.
 | ||
| Fig. 3 (A) Schematic diagram of the dual-layer microfluidic device with push-down valves used for experimentation. The device contains six cell-culture areas that are 1200 μm × 3200 μm × 30 μm and into which were seeded mESCs. Thereafter the pushdown devices regulated the differential washings of the cells in their respective chambers. The fluidic layer is shown in red and the push-down valves are shown in blue. (B) Phase contrast image of single-cell colonies seeded inside the microfluidic device taken at 40×. (C) The single-cell colonies where DAPI indicates the CellTracker™ and the Texas Red is Ethidium homodimer-1, a dead cell stain. The dead cells are indicated by the arrows. (D) Phase contrast image of a small multi-cellular colony inside the microfluidic device at 100×. (E) DAPI image of the multi-cellular colony stained with CellTracker™. | ||
We then fluorescently labeled mESCs with CellTracker™ Blue and seeded them evenly into each of the six chambers. Cells were seeded from a single-cell suspension so that roughly 250–350 cells were in each chamber and tracked for twelve hours after attachment. Given the low initial seeding, the lack of media supplementation, and relatively short tracking time, the effects of cell proliferation were minimal. The majority of cells were seeded as single-cell colonies, however a few small multi-cellular colonies were allowed as well. After a three hour period of attachment, the cells in each of the chambers were washed at the intervals identical to those in simulations described in Fig. 2E. During the course of the experiment, both single-cell colonies and multi-cellular colonies maintained a rounded morphology typical of undifferentiated mESCs attached to gelatinized glass (see Fig. 3B–E).
Since the model assumed no contribution of survival factors by the medium itself, our first experiments were done in the standard serum-free mESC medium, N2B27, whose composition is fully defined. Under these conditions (Fig. 4A), the cells undergoing the 720 minute washing regime largely survived. This suggests that the effects of seeding, the labeling agent, attachment to the glass, shear stress, and other potential complicating factors did not significantly affect cell viability. Additionally, as modeled, the cells experiencing continuous washings (0 minute washings) survived at approximately the same rate as clonally seeded cells. For the remaining conditions, the increase in viability was a function of the wash rate and closely matched the model predictions. This result suggests that mESCs indeed can secrete a critical survival factor or factors, not supplied by the N2B27 medium.
 | ||
| Fig. 4 (A) Experimental analysis of cell viability from the six-chamber microfluidic device where cells were washed at 0, 5, 20, 60, 180, and 720 minute intervals. Results include both those for cells cultured with the serum-containing medium and cells cultured with the defined, N2B27 serum-deficient medium. The data are presented as the means of three separate experiments, with the error bars representing the standard error of the mean. (B) Twelve hour time course of the viability of cells under each of the washing regimes, with a three hour initial attachment period (i.e., the cells are exposed to different washing regimes at 3 h and thereafter). | ||
After running the experiments with the N2B27 medium, we repeated them with a serum-containing medium. Serum contains a number of poorly defined growth and survival factors that are thought to influence signaling events in mESCs. If, as frequently assumed, some of these factors can boost cell survival, using serum-containing medium for mESCs culture in the presence of periodic washes can lead to a considerable decrease in cell death during frequent cell washings. Surprisingly however, cell viability of the cells cultured in the serum containing medium did not improve vs. the N2B27 medium, under all conditions tested. When the viability of cells in each washing regime is compared individually, only the 20 minute regime showed a significant difference between the two media types (t-test, P = 0.02). However, when the viability of cells in all washing regimes combined is compared across the two media, the N2B27 medium appears to have significantly increased cell viability (t-test, P = 0.03). This result suggested that serum cannot compensate for the loss of survival factors in the experiments described.
Next we examined the time course of cell viability in all chambers (Fig. 4B). During the initial three hour period of attachment all chambers remained approximately at 100% viability. This relative uniformity suggested the cells in all chambers adapted similarly to their environment inside the chip. After the first three hours, the exchange of media in all chambers was initiated and the variable washing conditions were simultaneously imposed. In the continuous (0 min washing period) and fast periodic (5 and 20 min washing periods) regimes, the majority of cell death occurred in the first three hours after the washings began, and thereafter viability remained relatively stable. However, the values of viability at which the cell populations stabilized varied: approximately 40% of the initial population in the continuous and 5 min periodic vs. roughly 60% in the 20 and 60 min periodic regimes. For the less frequent washing regimes, the decrease in cell viability was apparently linear and much slower. Therefore, the experimental results agreed with the model in some respects (a decrease in cell viability with time at rates dependent on the washing rates) and disagreed in others (plateau levels apparently clustering around 40% and 60% levels rather than being distributed in a more graded fashion as a function of the washing rate). The difference between the model and experiment suggests the potential presence of two distinct factors with different effective signaling widths and/or different survival thresholds, whose influence thus could be felt only under some but not all washing conditions.
Discussion
In this report, we describe a model accounting for the synthesis, diffusion, and removal rate of soluble cell-secreted factors and their effects on the viability of colonies of mESCs. From this model, we concluded that if soluble factors were responsible for the cell survival in mESCs, its periodic removal could lead to differential cell viability. Moreover, using parameter values available in the literature we determined that washing rates between 5 and 720 min might be interesting targets for experimental analysis. We verified this finding through the use of a six-chamber microfluidic device, which (a) confirmed that there is indeed likely to be a paracrine signaling factor (or several such factors) not supplied by the media; (b) that the dependency of cell viability on removal of this factor or factors was similar to that predicted by the model. The more precise kinetic measurements suggested that the cells are capable of partially withstanding the negative effects of the survival factor washout over the time scales of 20–60 minutes, and almost completely over 180 minutes and longer. This suggests that the cells can synthesize enough of the factors and that the factors can diffuse over sufficient effective signaling distances in the period of 1–3 hours. In fact, the apparent clustering of the plateau survival rates at two distinct values suggested that at least two distinct growth factors with different effective signaling widths or survival thresholds might be mediating paracrine signaling. It is of interest that the times to reach the different plateaus also varied, with the lower plateau reached much faster than the higher one. Assuming similar survival thresholds for the two putative survival factors, the modeling analysis addressing the rate of reaching the signaling steady state further suggests that one of the factors has higher diffusivity than the other one.Additionally, our observations suggested either an adaptive or selective process in the response of mESCs to removal of survival factors from the media. Indeed, cells that had been cultured in serum-free N2B27 media showed increased viability in comparison to those cells that were cultured in serum-containing medium. This suggests that the cells cultured in N2B27 media had already either adapted to the lower levels of ligand concentration in serum-free media, or that those cells more dependent upon ligand concentration had been selected out of the population. Therefore when the cells were deprived of full paracrine signaling by washings, they were better able to survive. This analysis further suggests that the paracrine factors that mESCs are responsive to are not present at all, or are not present in sufficient quantities, in the serum. These results differ from static culture conditions in which serum did increase viability.27 However in these studies the paracrine and autocrine signaling system was not disturbed as in the present study.
The identity of the putative survival factors remains a mystery at this stage of analysis. A number of candidate autocrine and paracrine factors can contribute to the increased viability of mESCs. Leukemia inhibitory factor (LIF) and other cytokines from the interleukin-6 (IL-6) family are known to not only promote self-renewal, but also serve an anti-apoptotic function.31 Additionally, mESCs secrete LIF,32 and respond to LIF in a dosage-dependant fashion.33 Another major factor to consider is Wnt, which binds a member of the frizzled family of receptors and regulates, among many other responses, apoptosis.34 Other factors that have been added exogenously to the cell culture leading to an increased cell survival include insulin-like growth factor (IGF1) and fibroblast growth factor (FGF4).35 However, this function of IGF1 and FGF4 apparently only occurs in a combinatorial manner.27 In the preceding study these two factors were added to the medium due to their known influence on stem cells and presumed presence in serum. As noted above however, our study suggests that if they are present in serum, then they are not present in sufficient quantities to elicit a strong anti-apoptotic effect. We propose that our experimental platform can be used for identification of the survival factors through back-addition of the potential candidate factors into the washing media.
Conclusion
This model and experimental system presented in this study allows for an expanded study of the paracrine signaling in mESCs and, potentially, other cell types. We demonstrated that mESC viability is dependent on soluble factors secreted by mESCs and not present in the supplied cell media, and that the viability can therefore be varied as a function of the medium replacement (wash) rate. Furthermore, we showed that mESCs undergo an adaptive and/or selective process in response to artificially decreased paracrine signaling. The described experimental platform is designed to allow further study of soluble factors and pathways in varied cell types, beyond the more specific application presented here. Addition of soluble factors to the media, blocking of pathways through pharmacological inhibitors, or use of RNA interference (RNAi) with expression of selected receptors in this device would allow a highly directed and specific evaluation of each of these factors on cellular response. With the use of a variety of extant live cell markers, a large array of additional cellular responses can be examined in a high throughput fashion.Materials and methods
mESC culture with the serum-containing medium
Undifferentiated ESD3 cells were maintained in a mixture of 15% fetal bovine serum (FBS) (Atlanta Biologicals), 100 μM β-mercaptoethanol (Gibco, Invitrogen), 2 mM L-glutamine, 0.1 mM non-essential amino acids (Gibco, Invitrogen), 1 mM sodium pyruvate (Gibco, Invitrogen), 50 μg mL−1penicillin/streptomycin in high glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Invitrogen), which was supplemented with 10 ng mL−1LIF (ESGRO, Millipore) on primary mouse embryonic fibroblast cells (ESGRO Millipore).mESC culture with the serum-free medium
ES cells had feeder layer subtractions performed and were plated onto gelatin-coated (0.01% w/v gelatin for one hour) petri dishes. N2B27 media36 supplemented with 10 ng mL−1LIF (ESGRO, Millipore) and 10 ng mL−1 BMP4 (R&D Systems) was used and the cells were passaged every 2–4 days using either enzyme-free cell dissociation buffer (Invitrogen) or 0.05% trypsin (Invitrogen).Feeder layer subtraction
Feeder layer subtraction was performed by washing the cells with PBS and then incubating them with 0.05% trypsin (Gibco, Invitrogen) for five minutes at 37 °C. The trypsin was then neutralized by the addition of culture media and the complete mixture was plated on uncoated plastic for one hour at 37 °C. Thereafter the supernatant was carefully removed and single-cell suspensions were created by gentle up-and-down pipetting of the cells.Microfluidic device fabrication
The two-layer polydimethylsiloxane (PDMS) device with push-down valves was created using standard soft photolithography procedures. The upper valve layer was fabricated by spin coating SU-8 2025 (Microchem, Newton, MA) onto a 3-inch-diameter wafer (Montco Silicon Technologies, Spring City, PA) at 1500 rpm for 70 seconds. The wafer was then soft baked at 95 °C for two hours and exposed to ∼15 mW cm−2 of UV light for 75 seconds through a high resolution transparency mask (In Tandem Design, Towson, MD). After exposure, the wafer was hard baked at 95 °C and finally baked at 200 °C for 30 min. The lower fluidic layer mold was fabricated by first spin coatingHMDS (hexamethyldisilazane, Sigma) at 1500 rpm for ten seconds and then spin coatingSPR 220–7.0 (Shipley, Marlborough, MA) at 730 rpm for 30 seconds onto a 3-inch-diameter silicon wafer, then soft baked at 115 °C for eight minutes, exposed to ∼15 mW cm−2UV light for 42 seconds using a high resolution transparency mask (In Tandem Design), developed in 453 developer (Clariant, Charlotte, NC), and then rounded by baking at 125 °C for 15 min. Both molds were coated with trimethylchlorosilane vapor for 30 minutes, then 20 g : 1 g parts A : B of RTV615 (GE Silicones, Wilton, CT) was spin-coated onto the fluidic layer mold at 1600 rpm for 60 seconds, and 35 g : 7 g RTV615 was poured onto the valve layer mold. The molds were baked at 80 °C for 45 minutes, and then the control layer mold was cut, hole-punched, cleaned, and aligned onto the fluidic layer mold. The layers were cured to each other at 95 °C for 90 minutes, and the final device was cut, hole-punched, cleaned, and reversibly bonded to clean 22 mm × 40 mm coverglasses (Fisher) by baking at 80 °C overnight.Use of the microfluidic device
Devices were coated with 0.1% w/v gelatin for one hour at room temperature. Cells were stained with CellTracker™ Blue CMAC (Invitrogen) for 30 minutes at 37 °C, and thereafter the cells were introduced into the device at a concentration of 6 × 106 cells mL−1 and allowed to attach for three hours at 37 °C 5% CO2. After the attachment phase, all chambers were washed to remove unattached cells. Thereafter, the differential washing regimes ran for the next twelve hours. All experiments were done in triplicate. During the experiment, dead cell stain was added to the media (live/dead kit for mammalian cells, Molecular Probes, Invitrogen) to continuously stain for dead cells. Flow was controlled by push-down valves that opened at specified intervals automated by a custom JAVA program.Image analysis
Using custom MATLAB (Math Works) programs, each of the images from all six chambers were montaged together in their respective positions. The positions of all cells were tracked over the twelve hour period based on centroid position of the cell projection areas.Implementation of the computational model
A custom JAVA program was written using J2SDK 1.4.2 Version 12 in JPadPro (ModelWorks). Code available upon request. Simulated chambers were created as a 1200 μm × 3200 μm space, onto which was overlaid a 10 μm grid. A user-defined value was then used to establish the percentage chance that any particular square in the grid would be occupied by a cell. The resultant chambers had approximately 250–350 cells per chamber.Statistical analysis
Two-tailed, Student t-tests assuming unequal variances were performed to compare the effects of different culture conditions. P-values < 0.05 were considered significant.Acknowledgements
The authors are grateful to the members of the Levchenko lab and Nicholas Christoforou for useful discussions and technical assistance. The research was partially supported by TEDCO RFAMA072 award from the Maryland Stem Cell Research Fund.References
- T. Burdon, A. Smith and P. Savatier, Trends Cell Biol., 2002, 12, 432–438 CrossRef CAS.
- Y. Suda, M. Suzuki, Y. Ikawa and S. Aizawa, J. Cell. Physiol., 1987, 133, 197–201 CrossRef CAS.
- A. Bradley, M. Evans, M. H. Kaufman and E. Robertson, Nature, 1984, 309, 255–256 CrossRef CAS.
- G. M. Keller, Curr. Opin. Cell Biol., 1995, 7, 862–869 CrossRef CAS.
- M. J. Evans and M. H. Kaufman, Nature, 1981, 292, 154–156 CrossRef.
- J. Nichols, Curr. Biol., 2001, 11, R503–R505 CrossRef CAS.
- R. L. Gardner and F. A. Brook, Int. J. Dev. Biol., 1997, 41, 235 CAS.
- Q. L. Ying, J. Nichols, I. Chambers and A. Smith, Cell, 2003, 115, 281–292 CrossRef CAS.
- I. Chambers, Cloning Stem Cells, 2004, 6, 386–391 CrossRef CAS.
- L. R. Castilho and R. A. Medronho, Adv. Biochem. Eng. Biotechnol., 2002, 74, 129 CAS.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS.
- M. B. Sporn and A. B. Roberts, Ann. Intern. Med., 1992, 117, 408 CAS.
- M. Freeman and J. B. Gurdon, Annu. Rev. Cell Dev. Biol., 2002, 18, 515–539 CrossRef CAS.
- D. A. Lauffenburger, G. T. Oehrtman, L. Walker and H. S. Wiley, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15368–15373 CrossRef CAS.
- S. Y. Shvartsman, H. S. Wiley, W. M. Deen and D. A. Lauffenburger, Biophys. J., 2001, 81, 1854–1867 CrossRef CAS.
- K. E. Forsten and D. A. Lauffenburger, Biophys. J., 1992, 61, 518–529 CrossRef CAS.
- G. T. Oehrtman, H. S. Wiley and D. A. Lauffenburger, Biotechnol. Bioeng., 1998, 57, 571–582 CrossRef CAS.
- A. E. DeWitt, J. Y. Dong, H. S. Wiley and D. A. Lauffenburger, J. Cell Sci., 2001, 114, 2301 CAS.
- R. B. Bird, W. E. Stewart and E. N. Lightfoot, Transport Phenomena, Wiley, New York, 1960 Search PubMed.
- K. Francis and B. O. Palsson, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12258–12262 CrossRef CAS.
- M. E. Young, P. A. Carroad and R. L. Bell, Biotechnol. Bioeng., 1980, 22, 947–955 CrossRef CAS.
- J. M. Savinell, G. M. Lee and B. O. Palsson, Bioprocess Eng., 1989, 4, 231–234 CrossRef CAS.
- M. I. Monine, A. M. Berezhkovskii, E. J. Joslin, H. S. Wiley, D. A. Lauffenburger and S. Y. Shvartsman, Biophys. J., 2005, 88, 2384–2390 CrossRef CAS.
- D. Mobest, E. Strobel, S. von Kleist, S. Ries, M. Dangel, R. Mertelsmann and R. Henschler, Blood, 1996, 88, 534.
- R. E. Davey, K. Onishi, A. Mahdavi and P. W. Zandstra, FASEB J., 2007, 21, 2020–2032 CrossRef CAS.
- S. J. Abbondanzo, I. Gadi and C. L. Stewart, Methods Enzymol., 1993, 225, 803–823 CAS.
- S. Viswanathan, T. Benatar, M. Mileikovsky, D. A. Lauffenburger, A. Nagy and P. W. Zandstra, Biotechnol. Bioeng., 2003, 84, 505–517 CrossRef CAS.
- L. Kim, M. D. Vahey, H. Y. Lee and J. Voldman, Lab Chip, 2006, 6, 394–406 RSC.
- K. Yamamoto, T. Sokabe, T. Watabe, K. Miyazono, J. K. Yamashita, S. Obi, N. Ohura, A. Matsushita, A. Kamiya and J. Ando, Am. J. Physiol. Heart Circ. Physiol., 2005, 288, H1915–H1924 CAS.
- H. Wang, G. M. Riha, S. Yan, M. Li, H. Chai, H. Yang, Q. Yao and C. Chen, Arterioscler., Thromb., Vasc. Biol., 2005, 25, 1817–1823 Search PubMed.
- H. Boeuf, K. Merienne, S. Jacquot, D. Duval, M. Zeniou, C. Hauss, B. Reinhardt, Y. Huss-Garcia, A. Dierich, D. A. Frank, A. Hanauer and C. Kedinger, J. Biol. Chem., 2001, 276, 46204–46211 CrossRef CAS.
- P. D. Rathjen, S. Toth, A. Willis, J. K. Heath and A. G. Smith, Cell, 1990, 62, 1105–1114 CrossRef CAS.
- P. W. Zandstra, H. V. Le, G. Q. Daley, L. G. Griffith and D. A. Lauffenburger, Biotechnol. Bioeng., 2000, 69, 607–617 CrossRef CAS.
- M. Kleber and L. Sommer, Curr. Opin. Cell Biol., 2004, 16, 681–687 CrossRef CAS.
- L. Powell-Braxton, P. Hollingshead, C. Warburton, M. Dowd, S. Pitts-Meek, D. Dalton, N. Gillett and T. A. Stewart, Genes Dev., 1993, 7, 2609–2617 CrossRef CAS.
- Q. L. Ying and A. G. Smith, Methods Enzymol., 2003, 365, 327–341 CAS.
Footnote |
| † This article is part of the 2009 Molecular BioSystems ‘Emerging Investigators' issue: highlighting the work of outstanding young scientists at the chemical- and systems-biology interfaces. |
| This journal is © The Royal Society of Chemistry 2009 |