Mirror image phage display—a method to generate D-peptide ligands for use in diagnostic or therapeutical applications
Susanne Aileen
Funke
a and
Dieter
Willbold
*ab
aForschungszentrum Jülich, ISB-3, Jülich, Germany
bInstitut für Physikalische Biologie, Heinrich-Heine-Universität Düsseldorf, Germany. E-mail: dieter.willbold@uni-duesseldorf.de; Fax: +49 2461 61 2023; Tel: +49 2461 61 2100
First published on 12th June 2009
Abstract
Mirror image phage display is a straightforward approach to identify new potentially therapeutically active D-enantiomeric peptides . Such D-peptides are more resistant to proteolytic degradation compared to L-peptides . In this review, several examples of mirror image phage display derived D-peptides with therapeutical potential are introduced and discussed.
 Susanne Aileen Funke | Dr Susanne Aileen Funke is group leader of the Alzheimer’s research team of the Institute of Structural Biology and Biophysics of the Research Centre Jülich. Her main interests are therapy and early diagnosis of Alzheimer’s disease. She majored in Biology in Bochum, Germany. She received a PhD in Biology at the University of Düsseldorf, Germany, after performing studies in Groningen, the Netherlands, and Marseille, France. She performed postdoctoral studies at the Institute for Physical Biology, Heinrich-Heine-Universität Düsseldorf, Germany. |
 Dieter Willbold | Dr Dieter Willbold has been full professor for physical biology at the Heinrich-Heine-Universität Düsseldorf since 2004. In parallel, he is director of the Institute of Structural Biology and Biophysics of the Research Centre Jülich. His main research interest is the structural basis for affinity and specificity of protein ligand interactions, focusing on viral proteins and proteins that are relevant for function and dysfunction of neurons. He majored in Biochemistry in Tübingen, Bayreuth and Boulder, CO. He received a PhD in Biophysical Chemistry at the University of Bayreuth, then performed postdoctoral studies there and at Tel-Aviv University, and headed a junior research group in Jena, Germany. In 2001, he accepted a professorship in Düsseldorf. |
Peptides are key mediators of biological functions. Their high biological activity associated with low toxicity and high specificity renders them attractive for therapeutic use. Today, hundreds of peptide therapeutics are being developed and some dozens are under study in clinical trials. Some can be expected to play a significant role in the treatment of cancer, AIDS, Alzheimer’s disease, malaria and as antimicrobials. New technologies like the establishment of large biological and synthetic peptide libraries, high throughput screening or selection, and innovative ways for drug administration have paved the way for the increased interest in therapeutically active peptides.1 In spite of this progress, the development of peptide drugs can be severely hampered by their short half-life in vivo.1 In general, peptides are rapidly degraded by proteases. This problem can at least partially be overcome by the use of D-enantiomeric peptides .
In general, natural proteins and peptides are composed of L-enantiomeric amino acids, usually providing binding sites that often specifically recognize only one enantiomer of a given ligand . In 1992, Milton and co-workers showed that the D-enantiomer of human immunodeficiency virus type 1 (HIV-1) protease only hydrolyses the D-enantiomeric form of its substrate, not the L-form.2 Very similarly, the S-proteinribonuclease forms an active complex only with the L-form of the S-peptide .3 In some other cases, protein–protein or protein–peptide interactions do not exert strong chiral preferences. Wilkemeyer and co-workers reported on ethanolantagonistpeptides , which show structural specificity but lack stereospecificity.4 Different amyloid-β (Aβ) bindingpeptides , supposed to act as inhibitors of fibrillogenesis, were unexpectedly more potent after chiral transversal to D-enantiomers .5
D-Peptides have several advantages over L-enantiomeric peptides . Most importantly, they are resistant to proteases,2,5,6 which can dramatically increase serum7 and saliva8 half-life. Additionally, D-peptides can be absorbed systemically after oral administration9 (with the efficiency depending on the sequence), in contrast to L-peptides , which have to be injected to avoid digestion. Increased stability, however, can eventually turn into a disadvantage if the D-peptide has toxic side effects. D-Peptides have long shelf-lives and because they are chemically synthesized, they can easily be modified. The need for synthetic preparation, however, turns into a disadvantage, as larger proteins are difficult to synthesize.
It is still not clarified yet (or it is case-dependent) if D-peptides are more or less immunogenic in contrast to L-peptides , but this will not be the topic of this review (see van Regenmortel and Muller10). In general, peptides composed of L-amino acids can be processed for major histocompatibility complex class II-restricted presentation to T-helper cells which can induce a vigorous humoral immune response.11 There are at least some examples in which immunogenicity upon D-peptide application was reduced.12
Because of the advantages described above, D-peptides have recently been investigated for their application as therapeutics. There are, however, diverse ways to identify D-peptides suitable for a specific application. An easy way might be direct translation and synthesis from already known L-peptide sequences into D-enantiomeric forms, which might be applicable if the protein–petide interactions do not have chiral preferences, see above.4,5 Another way is to use the retro-inverso form of an already known L-peptide , as shown by different groups.13 Retro-inverso peptides have the identical side chain topology but the opposite chain direction as compared to the respective L-peptides .14 The “one bead one compound method”15 is the direct chemical equivalent to mirror image phage display. Chemical compounds are synthesized on beads, followed by ligand binding and decoding, to obtain the sequences of the binding library peptides . Screening of combinatorial libraries consisting of D-peptides has been done,16 but seems to be elaborate as lots of synthesis steps have to be performed. In 2008, Esteras-Chopo and co-workers reported on the screening of a combinatorial library for selection of specific D-peptide amyloid inhibitors and presented a new strategy for their generation, extracting a set of empirical rules.17 Additionally, peptide array approaches,18 suitable for selection of L, D, and L–D-peptides , can be performed. Peptides are spotted on a glass chip and detection of binders can be performed easily and rapidly using acceptor molecules coupled to an enzyme or a fluorescent dye. High density peptide arrays are available by now.19 For a review on high density arrays, also mentioning phage display and the “one bead one compound method”, see ref. 20.
A very elegant and attractive way to obtain D-peptides that bind to specific targets is mirror image phage display. During a common phage display approach, as invented in 1990,21 an L-peptide library is presented on the surface of phages and selected for those L-peptides (e.g. by biopanning) that bind to a given target protein. In mirror image phage display, the selection is carried out against the mirror image of the original target, which in the case of proteins is a protein with the same amino acid sequence but fully composed of D-enantiomeric amino acids (see Fig. 1). This D-target protein is then used as the bait in a simple phage display procedure, thereby selecting L-enantiomeric ligands for the D-enantiomeric target.
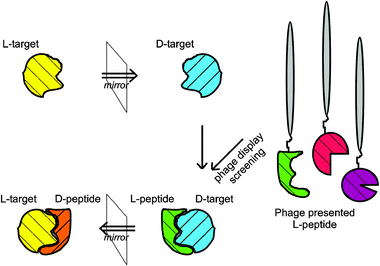 | ||
| Fig. 1 The principle of mirror image phage display. The D-enantiomeric form of any peptide-target is synthesized and used for phage display. A L-peptide , which binds to the D-enantiomeric target, is selected. The D-enantiomeric form of the selected L-peptide is synthesized, which will bind to the L-enantiomeric form of the target. | ||
The ultimately selected peptide of interest is translated into its D-enantiomeric form, which is expected to interact with the original target protein composed of natural L-amino acids. The finally obtained D-enantiomeric peptide is a specific ligand for the L-enantiomeric target.
Therefore, mirror image phage display uses the full advantages of phage display, allowing the relatively easy and straightforward selection of ligands out of a huge biologically encoded library that consists of up to 1013 different peptide or protein variants, thereby also delivering a rich source of structural diversity. There is not even a need for any structural information about the target. One has to admit that mirror image phage display might also have disadvantages. The selection of false positives may occur if the selection conditions are not perfectly suited to the target protein and library. In contrast to the “one bead one compound method”, which can directly select for D-, L- or L–D-peptides , mirror image phage display can only select full D-enantiomeric peptides . Both methods allow the generation of huge libraries with limited efforts, but have a slow readout in comparison to peptide array techniques. In contrast, spotting many different peptides is difficult, laborious and expensive.20
Mirror image phage display was first described by Schumacher and colleagues in 1996. They identified a cyclic D-peptide interacting with the c-Src homology 3 (SH3) domain of chicken-Src kinase. For that, a 10 residue random peptide library, flanked by Ser or Cys residues, was expressed and presented on the surface of bacteriophage fd by fusion of the peptide library with the N-terminus of the pIII protein. The library was constructed to include a large number of sequences prone to disulfide bond formation. Different to formerly investigated L-peptide binders, all identified D-enantiomeric ligands contained a pair of Cys residues and there was no obvious sequence similarity to any L-peptide binder described before. This led the authors to the conclusion that there should be a totally different mode of binding. Interestingly, nuclear magnetic resonance studies indicated that the binding site of the D-peptides partially overlaps with the binding site of the L-peptide ligands . The dissociation constant (Kd) of one D-peptide denoted Pep-D1 was determined to be 63 μM by competition binding experiments, which is well in the range of most proposed physiological ligands for SH3 domains. The reduced linear form of Pep-D1 did not show any detectable binding affinity, indicating that the disulfide bond is needed for binding.22
After the first description of the mirror image phage display method, the same group came up with a more therapeutically motivated application in 1999. The HIV-1 gp41 protein promotes viral entry by mediating the fusion of viral and cellular membranes. The authors constructed a peptide that was expected to represent a prominent pocket of gp41. This pocket was identified in earlier studies as a potential target for drugs, inhibiting HIV entry. The D-enantiomeric form of the pocket-mimicking peptide was used for the discovery of compounds that inhibit HIV entry by mirror image phage display, using the mirror image phage display system described before. Several cyclic D-peptides were identified which inhibited cell–cell fusion, as assayed by using cells expressing HIV-1 envelope glycoprotein mixed with cells expressing CD4 and coreceptor. These D-peptides also inhibited HIV-1 infection, as assayed by a recombinant luciferase-based HIV-1 infection assay . The authors stated that those peptides could serve as a starting point for the development of therapeutic and prophylactic agents inhibiting HIV entry.23
After the first direct proof that pocket-specific binding is sufficient to block HIV-1 entry by D-peptides , which bind to the N-trimer pocket and inhibit HIV-entry with a potency of IC50 ≈ 1 mM, in 2007, Welch et al.24 presented a study in which modified mirror image phage display and structure assisted design was used to discover “D-peptide pocket-specific inhibitors of entry”, called PIE, with up to 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold improved antiviral potency over the previously reported D-peptides . Those are potent enough for preclinical studies. The consensus sequence
000-fold improved antiviral potency over the previously reported D-peptides . Those are potent enough for preclinical studies. The consensus sequence ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) X5
X5![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif)
![[W with combining low line]](https://www.rsc.org/images/entities/char_0057_0332.gif) X
X![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) , identified by Eckert and co-workers,23 was used to develop a constrained library in which the consensus residues were fixed (underlined), while the other six positions were randomized. Accidentally, an 8-mer peptide, missing two of the randomized residues, was found to be most potent. Mirror image phage display of an 8-mer library CX4WXWLC, using a soluble competitor (L-peptide version of 2K-PIE2, the most successful binding peptide at the time), revealed that PIE7 was even 15-fold more potent than the most effective first-generation peptide (IC50 = 620 nM). The PEG crosslinked trimeric version of PIE7 (native gp41 has a trimeric nature) has an IC50 value of 250 pM. The authors stated that it will be beneficial to “overengineer” future D-peptides to further improve affinity.
, identified by Eckert and co-workers,23 was used to develop a constrained library in which the consensus residues were fixed (underlined), while the other six positions were randomized. Accidentally, an 8-mer peptide, missing two of the randomized residues, was found to be most potent. Mirror image phage display of an 8-mer library CX4WXWLC, using a soluble competitor (L-peptide version of 2K-PIE2, the most successful binding peptide at the time), revealed that PIE7 was even 15-fold more potent than the most effective first-generation peptide (IC50 = 620 nM). The PEG crosslinked trimeric version of PIE7 (native gp41 has a trimeric nature) has an IC50 value of 250 pM. The authors stated that it will be beneficial to “overengineer” future D-peptides to further improve affinity.
Therewith, it was demonstrated that D-peptides can form specific and high affinity interactions with natural protein targets, additionally addressing limitations associated with current L-peptide entry inhibitors like delivery by injection and high dose requirements due to protease degradation.24
A number of further mirror image phage display approaches were used for the selection of D-peptides that selectively bind to Alzheimer’s amyloid-β (Aβ) peptide. Such peptides are thought to be potentially helpful for early diagnosis and therapy of the disease. Aβ is the major component of the amyloid plaques found in the brains of patients with Alzheimer’s disease (AD). It consists of 39 to 43 amino acid residues, Aβ(1–42) being the most dominant form in the so-called senile plaques.25 To improve diagnosis and treatment evaluation, neuroimaging tools, making use of Aβ-bindingligands to visualize amyloid plaques, are developed. To select for an Aβ-bindingD-peptide which binds especially to aggregated Aβ species, aggregated D-enantiomeric Aβ was used as a target screening a fully randomized 12-mer peptide library presented on M13 phages. The most representative peptide, denoted D1, was shown to have a binding constant in the submicromolar range. It stains the amyloid plaques in the brain tissue sections derived from AD patients, whereas other fibrillar deposits were not stained.26 A study was done to investigate the influence of D1 to Aβ polymerization and toxicity. Using cell toxicity assays, thioflavin fluorescence, fluorescence correlation spectroscopy and electron microscopy, a significant influence of D1 on both Aβ aggregation and cytotoxicity was shown. The presence of D1 reduced the average size of the aggregates, but increased their number. Aβ cytotoxicity on PC12 cells was reduced in the presence of D1.27 The development of amyloid specific ligands is highly desirable, but many substances failed because of unspecific binding and poor distribution in the brain. D1 was tested for its in vivo binding characteristics in transgenic APP/PS1 transgenic mice, which develop AD-like plaque pathology at a very early age. Upon direct injection into the brain, D1 bound very specifically to Aβ(1–42), staining all dense deposits in the brain but not diffuse plaques, which contain mainly Aβ(1–40) and are not AD specific. D1 was not taken up by astrocytes or microglia, nor did it cause any inflammation. This demonstrated that D1 might be suitable for further development into a molecular probe to monitor Aβ(1–42) plaque load in the living brain.28 To what extent D1 crosses the blood–brain barrier is currently a matter of investigation.
In a similar mirror image phage display approach, the D-enantiomeric peptide D3 was selected, using predominantly monomeric and small oligomeric D-Aβ species as target. D3 exerted interesting therapeutical properties. The peptide had a strong influence on Aβ aggregation, disaggregation and cytotoxicity in vitro. In vivo, it reduced amyloid plaque load and cerebral damage of transgenic mouse models of AD to an outstanding extent in comparison to other Aβ anti-aggregation agents, therefore providing a basis for a new therapeutic approach.29 As D-peptides can be absorbed systemically after oral administration9 in contrast to L-peptides , D3 might be well suited for use in oral treatment.
The selection of D-enantiomeric “spiegelmers” (German “Spiegel” means mirror) is not restricted to genetically encoded peptide libraries because ribonucleides and deoxyribonucleides also contain chiral centres. For example, nuclease-resistant inhibitory RNA aptamers have been developed against a variety of targets implicated in disease (for a review, see ref. 30).
Elkin and co-workers presented the computational analogue for mirror image display, generating mutant sequences for the selection of a D-peptide binder for the hepatitis delta virus “hepatitis delta antigen” in silico.31
Conclusions
In conclusion, it must be stated that so far, only a few examples are reported for successful application of mirror image phage display to generate new therapeutic lead compounds. Nevertheless, the few existing examples seem to be very interesting and promising. If D-peptides selected by mirror image phage display have the expected properties with respect to stability and oral availability, the advantages of the method are quite obvious. It is possible to start the selection for a new lead compound with a biological library that can easily be purchased or constructed. Using a selection process, no time consuming and costly high throughput screening methods have to be used. One disadvantage is the need for a given target molecule to be synthetically available, so far limiting the method to smaller targets. This problem, however, can partially be overcome by the isolation of separate domains as target molecules.22Acknowledgements
This work has been supported by a grant of the Volkswagen-Stiftung to D.W. (I/82 649) and by a grant from the “Präsidentenfond der Helmholtzgemeinschaft” (HGF, “Virtual Institute of Structural Biology”) to D.W.Notes and references
- S. Lien and H. B. Lowmann, Trends Biotechnol., 2003, 21, 556–562 CrossRef CAS; A. Pini, C. Falciani and L. Bracci, Curr. Protein Pept. Sci., 2008, 9, 468–477 CrossRef CAS.
- R. C. Milton, S. C. Milton and S. B. Kent, Science, 1992, 256, 1445–1448 CAS.
- M. A. Corigliano-Murphy, L. A. Xun, C. Ponnamperuma, D. Dalzoppo, A. Fontana, T. Kanmera and I. M. Chaiken, Int. J. Pept. Protein Res., 1985, 25, 225–231 CAS.
- M. F. Wilkemeyer, S. Chen, C. E. Menkari, K. K. Sulik and M. E. Charness, J. Pharmacol. Exp. Ther., 2004, 309, 1183–1189 CrossRef CAS.
- R. J. Chalifour, R. W. McLaughlin, L. Lavoie, C. Morissette, N. Tremblay, M. Boulé, P. Sarazin, D. Stéa, D. Lacombe, P. Tremblay and F. Gervais, J. Biol. Chem., 2003, 278, 34874–34881 CrossRef CAS.
- S. Pujals, E. Sabidó, T. Tarragó and E. Giralt, Biochem. Soc. Trans., 2007, 35, 794–796 CrossRef CAS.
- M. Sadowski, J. Pankiewitz, H. Scholtzova, J. A. Ripellino, Y. Li, S. D. Schmidt, P. M. Mathews, J. D. Fryer, D. M. Holtzman, E. M. Sigurdsson and T. Wisniewski, Am. J. Pathol., 2004, 165, 937–948 Search PubMed.
- G.-X. Wei and L. A. Bobek, Antimicrob. Agents Chemother., 2005, 49, 2336–2342 CrossRef CAS.
- J. R. Pappenheimer, C. E. Dahl, M. L. Karnovsky and J. E. Maggio, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 1942–1945 CrossRef CAS; J. R. Pappenheimer, M. L. Karnovsky and J. E. Maggio, J. Pharmacol. Exp. Ther., 1997, 280, 292–300 CAS.
- M. H. V. Van Regenmortel and S. Muller, Curr. Opin. Biotechnol., 1998, 9, 377–382 CrossRef CAS.
- C. T. Dooley, N. N. Chung, B. C. Wilkes, P. W. Schiller, J. M. Bidlack, G. W. Pasternak and R. A. Houghten, Science, 1994, 266, 2019–2022 CrossRef CAS; T. J. Gill, H. J. Gould and P. Doty, Nature, 1963, 197, 746–747 CrossRef CAS.
- P. Chong, C. Sia, B. Tripet, O. James and M. Klein, Lett. Pept. Sci., 1996, 3, 99–106 CrossRef CAS; H. M. Dintzis, D. E. Symer, R. Z. Dintzis, L. E. Zawadzke and J. M. Berg, Proteins, 1993, 16, 306–308 CrossRef CAS.
- L. M. Higgins, I. Lambkin, G. Donnelly, D. Byrne, C. Wilson, J. Dee, M. Smith and D. J. O’Mahony, Pharm. Res., 2004, 21, 695–705 CrossRef CAS; C. Das, O. Berezovska, T. S. Diehl, C. Genet, I. Buldyrev, J. Y. Tsai, B. T. Hyman and M. S. Wolfe, J. Am. Chem. Soc., 2003, 125, 11794–11795 CrossRef CAS; K. Sakurei, H. S. Ching and D. Kahne, J. Am. Chem. Soc., 2004, 126, 16288–16289 CrossRef.
- M. Chorev and M. Goodman, Acc. Chem. Res., 1993, 26, 266–273 CrossRef CAS.
- K. S. Lam, S. E. Salmon, E. M. Hersh, V. J. Hruby, W. M. Kazmierski and R. J. Knapp, Nature, 1991, 354, 82–84 CrossRef CAS.
- B. C. Monk, K. Niimi, S. Lin, A. Knight, T. B. Kardos, R. D. Cannon, R. Parshot, A. King, D. Lun and D. R. Harding, Antimicrob. Agents Chemother., 2005, 49, 57–70 CrossRef CAS.
- A. Esteras-Chopo, M. T. Pastor, L. Serrano and M. Lopez de la Paz, J. Mol. Biol., 2008, 377, 1372–1381 CrossRef CAS.
- R. Frank, Tetrahedron, 1992, 48, 9217–9232 CrossRef CAS.
- M. Beyer, A. Nesterov, I. Block, K. König, T. Felgenhauer, S. Fernandez, K. Leibe, G. Torralba, M. Hausmann, U. Trunk, V. Lindenstruth, F. R. Bischoff, V. Stadler and F. Breitling, Science, 2007, 318, 1888 CrossRef CAS.
- F. Breitling, A. Nesterov, V. Stadler, T. Felgenhauer and F. R. Bischoff, Mol. BioSyst., 2009, 5, 224–234 RSC.
- J. K. Scott and G. P. Smith, Science, 1990, 249, 386–249 CrossRef CAS.
- T. N. M. Schumacher, L. M. Mayer, D. L. Minor Jr., M. A. Milhollen, M. W. Burgess and P. S. Kim, Science, 1996, 271, 1854–1857 CrossRef CAS.
- D. M. Eckert, V. N. Malashkevich, L. H. Hong, P. A. Carr and P. S. Kim, Cell (Cambridge, Mass.), 1999, 99, 103–115 CrossRef CAS.
- B. D. Welch, A. P. vanDenmark, A. Heroux, C. P. Hill and M. S. Kay, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16828–16833 CrossRef CAS.
- J. Hardy and D. J. Selkoe, Science, 2002, 197, 353–356 CrossRef.
- K. Wiesehan, K. Buder, R. P. Linke, S. Patt, M. Stoldt, E. Unger, B. Schmitt, E. Bucci and D. Willbold, ChemBioChem, 2003, 4, 748–753 CrossRef CAS.
- K. Wiesehan, J. Stöhr, L. Nagel-Steger, T. van Groen, D. Riesner and D. Willbold, Protein Eng. Des. Sel., 2008, 21, 241–246 CrossRef CAS.
- T. van Groen, I. Kadish, K. Wiesehan, S. A. Funke and D. Willbold, ChemMedChem, 2009, 4, 276–282 CrossRef CAS.
- T. Van Groen, K. Wiesehan, S. A. Funke, I. Kadish, L. Nagel-Steger and D. Willbold, ChemMedChem, 2008, 3, 1848–1852 CrossRef CAS.
- H. Ulrich, Handb. Exp. Pharmacol., 2006, 173, 305–326 Search PubMed.
- C. D. Elkin, H. J. Zuccola, J. M. Hogle and D. Joseph-McCarthy, J. Comput. Aided Mol. Des., 2000, 14, 705–718 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |