 Open Access Article
Open Access ArticleProtein allostery , signal transmission and dynamics: a classification scheme of allosteric mechanisms
Chung-Jung
Tsai
a,
Antonio
del Sol†
*b and
Ruth
Nussinov
*ac
aBasic Research Program, SAIC-Frederick, Inc., Center for Cancer Research Nanobiology Program, NCI-Frederick, Frederick, MD 21702, USA. E-mail: ruthn@ncifcrf.gov
bBioinformatics Research Unit, Research and Development Division, Fujirebio Inc., 51 Komiya-cho, Hachioji-shi, 192-0031, Tokyo, Japan
cSackler Inst. of Molecular Medicine, Department of Human Genetics and Molecular Medicine, Sackler School of Medicine, Tel Aviv University, 69978, Tel Aviv, Israel
First published on 6th January 2009
Abstract
Allostery has come of age; the number, breadth and functional roles of documented proteinallostery cases are rising quickly. Since all dynamic proteins are potentially allosteric and allostery plays crucial roles in all cellular pathways, sorting and classifying allosteric mechanisms in proteins should be extremely useful in understanding and predicting how the signals are regulated and transmitted through the dynamic multi-molecular cellular organizations. Classification organizes the complex information thereby unraveling relationships and patterns in molecular activation and repression. In signaling, current classification schemes consider classes of molecules according to their functions; for example, epinephrine and norepinephrine secreted by the central nervous system are classified as neurotransmitters. Other schemes would account for epinephrine when secreted by the adrenal medulla to be hormone-like. Yet, such classifications account for the global function of the molecule; not for the molecular mechanism of how the signal transmission initiates and how it is transmitted. Here we provide a unified view of allostery and the first classification framework. We expect that a classification scheme would assist in comprehension of allosteric mechanisms, in prediction of signaling on the molecular level, in better comprehension of pathways and regulation of the complex signals, in translating them to the cascading events, and in allosteric drug design. We further provide a range of examples illustrating mechanisms in proteinallostery and their classification from the cellular functional standpoint.
Introduction
Allostery has been attracting increasing attention. This is not surprising: allostery is the key in regulation of cellular processes and a vehicle through which the environment affects function.1 All dynamic proteins are potentially allosteric.2Allostery regulates cellular assemblies and pathways. It is a universal phenomenon: a perturbation by an effector leads to a functional change at the substrate binding site through alteration of the shape and (or) dynamics.3 Allosteric perturbation arises from the binding of small and large molecules; from changes in pH, temperature, ionic strength, or concentration; and from covalent modifications such as tethering,4glycosylation, phosphorylation and ubiquination.5–7Allostery is a cooperative event,8 positive or negative; up- or down-regulating protein functions. Further, allosteric behavior was observed also in eglin c, a small globular protein, which is presumably a non-allosteric protein.9 Yet, despite this intense interest, how signals initiating at the perturbation site transmit through the residue network is still an open and debated question. Nonetheless, there is a general agreement that the energetic strain at the perturbation site dissipates, leading to a functional change in the substrate binding site.The broad recognition of allosteric effects often does not translate into consideration in practical applications. One example is drug design: traditional strategy seeks to block enzyme activity by designing inhibitors snugly fitting the enzyme binding site. Following this rationale, to block a certain cellular process, a strategy of choice considers a high affinity inhibitor mimicking the natural binding epitope. Yet, in practice, such rationale can enhance function rather than repress it. HIV-1 gp120 provides a striking example:10 the binding of gp120 to CD4 precedes gp120 binding to a co-receptor and viral entry into the cell. Unexpectedly, a CD4 mimetic designed to interrupt the cascading pathway led to the opposite effect: rather than repressing, it enhanced gp120 co-receptor binding, mimicking the CD4 allosteric consequences. Subsequent design targeted CD4 binding and its allosteric effects obtaining peptide conjugates that function as dual receptor site antagonists of HIV-1 gp120.
The increasing number of reports on allosteric effects in a broad range of proteins, functions, assembly sizes and environments argues for the usefulness of classification of allosteric mechanisms. Here we describe allosteric mechanisms within the framework of cellular pathways and transmission of information,11–15 and provide a classification scheme with some examples from a broad range of processes taking place in the cell illustrating the functional effects. Our description initiates by uniting the old and new mechanistic views of allostery ; this traditional division is a matter of definition.
Allostery : the old and the new views
(i) The old view
The classical allosteric models (Monod–Wyman–Changeux: MWC16 and Koshland–Némethy–Filmer: KNF17) described allostery as a binding event at one site affecting the activity at another via a conformational change. The MWC model described the transition as a concerted action between two co-existing, discrete states (R and T); the KNF model described it as a sequential, induced conformational change by the binding event at the first site. The classical dogma had two components: first, there are two distinct R and T conformations; in the absence of a ligand they exist in a ratio governed by an equilibrium constant; and second, allostery involves a change of shape. This view perpetuated over decades. It was fed by static crystallographic snapshots of allosterically-regulated proteins showing a clear difference in the shape of the substrate binding site between the active/inactive species. And, the change of shape was observed between effector-bound and free states. Hence, it was an all-or-none view: either the On conformation is present or the Off. Although not directly stated by the classical view, the two-state model implied that signal propagation from the effector to the substrate binding site proceeded along a single, specific, well-defined path. Distinct residues involved in the allosteric signal transmission that were identified by experiment were interpreted in this light.18 The so-called old view can then be described by three characteristics: (i) there are only two states; (ii) there is a conformational change in the substrate binding site; and (iii) the allosteric signal is transmitted via a single pathway.(ii) The new view
Current evidence indicates that the native state is a conformational ensemble, and that allosteric perturbation involves a shift of pre-existing conformations; some already with altered binding site shapes.2,19–21 The dissipating strain22 drives the shift, increasing the pre-existing (though undetected) functional state population.21 Moreover, recent experimental data3,23 confirms early prediction:24 while visual inspection of the active/inactive states may not reveal any difference in the substrate binding site shape, allosteric effects can be there, reflected purely in changes in dynamics. Allostery without a change in shape was not foreseen by the old view.Taken together, the new view expands the definition of allostery : (i) it emphasizes that rather than only two conformational states, proteins exist in ensembles; (ii) it recognizes that allostery is a thermodynamic phenomenon; thus, can be governed by enthalpy, enthalpy and entropy or entropy. There may or may not be a backbone conformational change. Hence, the absence of a conformational change in the substrate binding site does not imply that allostery is not at play; and finally, (iii) the existence of multiple conformational and dynamic states implies multiple pathways through which the strain energy is released from the allosteric site following a perturbation event.
Allostery : the unified view
We cast the three attributes of the old view in the terminology of the new view, illustrating that the two can be unified by definition. A unified view facilitates a description of allosteric mechanisms and their classification.(i) Protein states: two versus multiple conformational states
Fundamental to the old view is the presence of two (On/Off) states. The unified view interprets the allosteric switch as simply referring to the difference between an observable and non-observable functional effect, rather than all-or-none conformational states.3,25–27(ii) There may or may not be a conformational change
The hallmark of the old view was a conformational change at the binding site induced by the allosteric perturbation event; in the unified view, this is one of the scenarios.(iii) Pathways: single well-defined versus multiple
Allostery is a purely thermodynamic phenomenon
Two components are responsible for the behavior of the system: enthalpy and entropy. A binding event leads to loss of freedom of motion of the binding partners, including their internal motions; thus it is entropy-unfavorable. On the other hand, the added interactions are enthalpy-favorable. The contributions of the two terms are not independent: during binding the interactions get increasingly tighter (enthalpy-favorable); at the same time, this process is accompanied by loss of degrees of freedom, thus the system increasingly becomes entropy-unfavorable. The converse also holds: loosening the interactions decreases the enthalpy (i.e. it is enthalpy-unfavorable); however, the concomitantly increased mobility is entropy-favorable. Allostery involves (at least) two binding events; that of the effector and of the substrate. The effector binding can increase or decrease the affinity of the substrate: if it increases the affinity of the substrate it is said to be positive cooperativity; if it decreases the affinity it is negative cooperativity. Positive cooperativity can be dominated by enthalpy or entropy. Enthalpy-dominated positive cooperativity is driven by the favorable tightening of the interactions, with some entropy penalty caused by the disorder to order events at the substrate binding site; that is, part of the entropy penalty for the binding of the substrate is already pre-paid. Enthalpy-dominated positive cooperativity will present a conformational change at the substrate binding site. In entropy-dominated positive cooperativity, the overall entropy cost of the two binding events is lower than their sum. Entropy-dominated positive cooperativity can occur with or without conformational change. Negative cooperativity is typically governed by unfavorable entropy. Binding of the effector leads to an order to disorder event at the substrate binding site; thus higher entropy cost. Negative cooperativity is accompanied by a conformational change. Cooperativity is non-independent; that is, the overall behavior of the system is not the sum of its components.The classification scheme with examples
Classification scheme
Classification schemes rest on definitions. Since the new view states that absence of a change in shape does not imply there is no allostery , we define allostery as a change in the protein activity in cellular pathways, for example, signaling; viral entry; gene regulation; enzymatic reactions. We focus on function: we do not consider conformational allostery -related changes through agents unrelated to function. The classification considers the following properties:(i) Whether there is conformational change at the substrate site.
(ii) Does the effector perturbation lead to positive (or negative) cooperativity of the substrate binding?
(iii) Is the allosteric effect enthalpy- or entropy-driven?
(iv) Does the functional site coincide with the substrate site? For the first, classification is inferred visually; for the second and fourth properties, biochemical tests can provide answers; for the third, in some cases it is straightforward; allosteric effects without a conformational change are entropy-dominated; large conformational change is generally enthalpy-dominated and disorder-to-order events entropy-dominated. For others, calorimetry determines thermodynamic factor domination.
(v) Whether phosphorylation is involved and if so in what functional capacity? Phosphates play key roles in signal transduction.
(vi) The oligomeric state, e.g. a pre-formed dimer (example 4 below) or separated monomers associating to form a dimer (example 2). If it is an oligomer, allostery may be concerted (e.g. in the GroEL, or example 3) or not concerted (example 8).
Fig. 1 implicitly provides all classification descriptors. Table 1 summarizes the examples in these terms. Examples are detailed in Fig. 2–4.
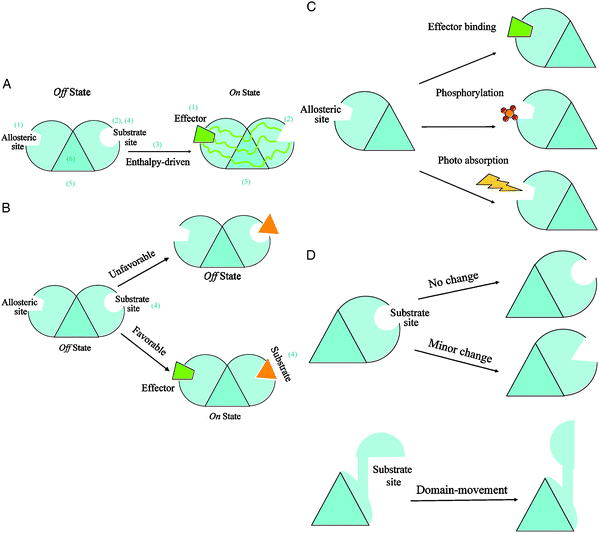 | ||
| Fig. 1 Simplified diagrams illustrating the scheme of classification of allosteric mechanisms. The scheme uses six descriptors to describe an allosteric reaction: (1) the type of perturbation event at the allosteric site; (2) the extent of conformational change at the substrate site; (3) the dominant thermodynamic factor; (4) the type of allosteric cooperativity; (5) the location of functional site (is it coincident with the substrate site?), and (6) the functional oligomeric state in action. To facilitate module design in the representation, the core of an allosteric monomer is represented by a triangle. Each triangle edge is occupied by one of three sites: the allosteric site, the substrate site, and the functional site. Fig. 1A depicts a typical allosteric case in which the perturbation by an effector binding at the allosteric site causes a minor conformational change at the substrate site. The shape changes from circle (Off state) to triangle (On state) via multiple propagation pathways. The absence of an attached module to the third edge of the core triangle indicates that in this case the substrate site is also the functional location of the monomeric protein. The positive binding cooperativity is implied in Fig. 1A since no substrate is attached in the Off state. This is an enthalpy-driven allosteric regulation in that the unfavorable substrate binding (Off state) becomes favorable (On state) due to an effector binding at the allosteric site (Fig. 1B). In this simplified modular scheme, a representation of a particular allosteric mechanism is just a combination of various stimuli at the allosteric site (depicted in Fig. 1C) and of conformational changes at the substrate site (illustrated in Fig. 1D). A stimulus is an event that directly influences protein function. The numbers in parentheses provide a visual guide corresponding to the six descriptors of the classification scheme. | ||
| 1. Perturbation event at the allosteric site | 2. Extent of conformational change at the substrate site | 3. Dominant thermodynamic factor | 4. Allosteric cooperativity | 5. Location of functional site | 6. Functional oligomer status in action | |
|---|---|---|---|---|---|---|
| Example 1 | Binding (ligands to extracellular pockets) | Minor change | Enthalpy-driven | Negative | At the substrate site | Hetero-oligomer |
| Example 2 | Binding (extracellularEGF binding) | Domain-movement | Enthalpy-driven | Positive | Not at the substrate site | Monomeric allostery followed by dimerization |
| Example 3 | Cytokine binding | Domain-movement | Enthalpy-driven | Positive | At the substrate site | Concerted homo-dimer |
| Example 4 | Binding (NotchIC RAM domain to CSL’s beta-trefoil domain) | Minor change | Enthalpy-driven | Positive | At the substrate site | Hetero-oligomer |
| Example 5 | Binding (gp120 to CD4) | Large change | Enthalpy-driven | Positive | At the substrate site | Monomer |
| Example 6 | Binding (c-AMP to CBD:β) | Large change | Enthalpy-driven | Positive | At the substrate site | Monomer |
| Example 7 | Binding (Spz molecules to Toll ectodomain) | Large change | Enthalpy-driven | Positive | Not at the substrate site | Concerted Homo-dimer |
| Example 8 | Binding (tryptophan to TRAP) | Minor change | Entropy-driven | Positive | At the substrate site | Non-concerted homo-oligomer |
| Example 9 | Binding (cAMP to CAP) | No change | Entropy-driven | Negative | At the substrate site | Homo-dimer |
| Example 10 | Binding (Ffw7/Sel10 to the phosphorylated site) | Minor change | Enthalpy-driven | Negative | At the substrate site | Hetero-oligomer |
To clarify this scheme, let us consider some scenarios. (i) Conformational change. If we exclude here allosteric cases governed by enthalpy and entropy, a population shift accompanied by conformational change could be dominated either by enthalpy or by entropy. On the other hand, allosteric free energy change without a conformational change is dominated solely by entropy. (ii) Cooperativity. In the case of enthalpy-dominated allosteric effect, the unfavorable active site conformation becomes substrate-favorable; thus, positive cooperativity. The opposite holds for negative cooperativity. For entropy-dominated conformational change, consider effector binding making a disordered substrate binding site become (partially) ordered. This is positive cooperativity with pre-paid entropy loss. (iii) Cooperativity and (absence of) conformational change. In the case of allostery without conformational change, there is an extra, non-additive entropy loss due to substrate binding. This is negative cooperativity with unfavorable extra entropy loss due to enhanced rigidification. (iv) Phosphorylation. This can lead to changes in protein activity either via direct interference, where the phosphate blocks the substrate binding site; through formation of binding sites; or the most common case, by a conformational change, acting as an allosteric effectorvia conformational perturbation.42
Example 1. Substrate (trimer) dissociation: the seven-helix receptors family (Fig. 2)
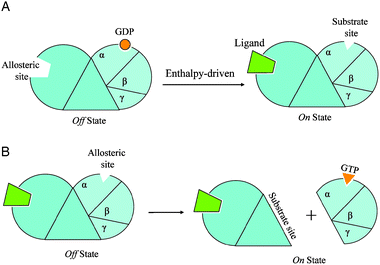 | ||
| Fig. 2 Two simplified diagrams to illustrate the allosteric mechanisms of the exchange of GTP to GDP in example 1 in the text. In Fig. 2A, an extracelluar ligand binding at the N-terminal domain of the receptor (the allosteric site) causes a conformational change at the GDP binding site (the substrate site) leading to the release of GDP. This is a hetero-oligomer system with negative binding cooperativity driven mainly by enthalpy. The empty GDP binding site then becomes an allosteric site for GTP binding in Fig. 2B. The GTP binding event causes the dissociation of trimeric G-protein from the seven-helix receptor. If we retain the classification scheme with the seven-helix receptor as the core protein, the substrate will be the entire trimetric G-protein and the allosteric site is located inside the substrate; however, if we view the G-protein as the core protein, then the GTP binding is a negative cooperative binding leading to dissociation of the substrate, now the seven-helix receptor. | ||
Downstream effectorproteins amplify the catalytic cascade, producing cAMP, which activates protein kinase A (PKA) via RII dissociation. PKA phosphorylatesphosphorylase kinases (PK). Phosphorylated calmodulin-phosphorylase binds Ca2+ which now phosphorylates and activates phosphorylases. The phosphorylase catalyzes the conversion of glycogen to glucose-6-phosphate. Thus, the mechanism of the seven-helix receptors family can be classified as conformational change, negative cooperativity, enthalpy-dominated (Table 1).
Example 2. Dimerization: epidermal growth factor (EGF), or its HER2/ErbB2 homolog (Fig. 3)
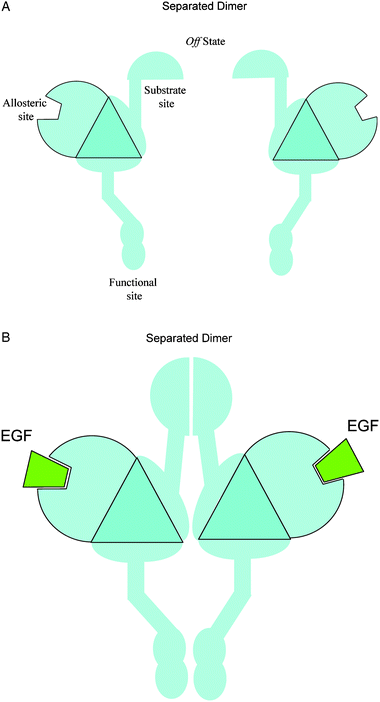 | ||
| Fig. 3 Simplified diagrams to illustrate the allosteric mechanism of dimerization in example 2. The EGF binding event causes a large-scale, domain-movement conformational change at the substrate site, which in turn facilitates the dimerization. The extracellular dimerization process leads to a contact between the two intracellular functional sites, the cytoplasmic kinase domains. This represents a homodimer system where the substrate site is not at the functional site. | ||
Example 3. Tightening dimeric interactions and association: cytokine binding (Fig. 4)
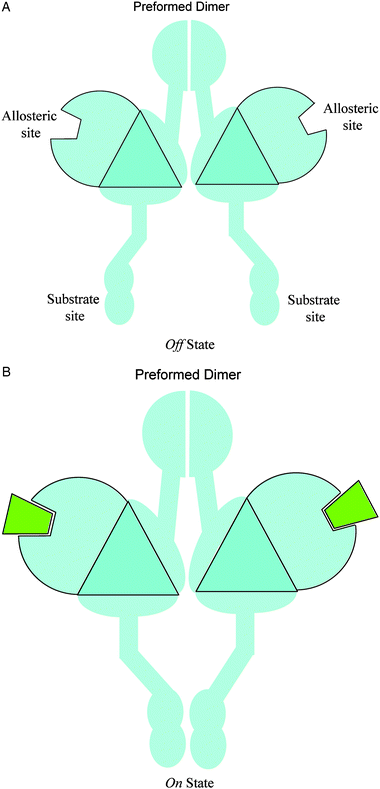 | ||
| Fig. 4 Simplified diagrams to illustrate the allosteric mechanisms of the pre-formed dimer in example 3. Effector binding at the allosteric sites causes a large-scale conformational change at the substrate site, which brings the two separated substrates (the two triangles) in close contact to each other. This is a concerted homodimer system: the two substrate sites become the functional site when they are in close contact. | ||
Example 4. Ternary complex formation: Notch receptors
Notch receptors regulate cell fate, and are key factors in cellular differentiation, proliferation, apoptosis and stem cell renewal.42,43Notch receptors consist of extracellular EGF-like domains and leucine-rich repeats, a transmembrane segment and intracellular ankyrin repeats. Transmembraneprotein DSL (Delta Serrate Lag-2) activates the Notch receptor of neighboring cells, freeing intracellular domains (NotchIC) which move back to the nucleus. Transcription factor CSL is bound to DNA. NotchIC RAM domain binds to CSL’s beta-trefoil domain (BTD) triggering conformational change in CSL’s N-terminal domain (NTD). As a result, a binding site for co-activator Mastermind is formed. NotchIC ankyrin domain (ANK) then interacts with the CSL’s C-terminal domain (CTD), creating an additional Mastermind interaction site. Thus, RAM’s binding to BTD increases the effective local concentration of ANK. Mastermind's binding (positive cooperativity) completes the enthalpy-dominated ternary complex formation, activating transcription initiation. The Notch receptor mechanism can thus be classified as conformational change, positive cooperativity, enthalpy-driven (Table 1).Example 5. Ternary complex formation: HIV-1 gp160
HIV-1 gp160 cleavage leads to envelopeproteins gp120 and gp41. gp120 binds to CD4, a T-cell surface glycoprotein. Binding leads to large conformational change in gp120, exposing a binding site and promoting co-receptor CCR5 or CXCR4 binding (positive cooperativity). The scorpion-toxin CD4-mimic, similarly elicits a conformational change in gp120.44 Thermodynamic data indicate an enthalpy-driven process. The gp120 co-receptor interaction results in large conformational changes in gp41, exposing a heptad repeat region and the leucine/isoleucine zipper region, assisting in gp41 relocation from the gp120–gp41 interior to host membrane proximity and subsequent viral nucleoprotein entry, initiating the infection cycle.45 We classify the mechanism of the HIV-1 gp160 as conformational change, positive cooperativity, enthalpy-driven (Table 1).Example 6. Salt-bridge cluster weakening and competing isomers: the cAMP-activated exchange protein
cAMP-activated exchange protein (EPAC) is a key ancient second messenger receptor in mammals. cAMP activates the guanine nucleotide-exchange factors in the small GTPases Rap1 and Rap2. A salt bridge cluster commonly called ionic latch (IL) exists between the catalytic core and the cAMP binding domain (CBD) helices α1/α2. In the IL-stabilized closed topology of the inactive state, the regulatory moiety occludes access to the catalytic domain. CBD:β (covalently bound to the Disheveled-Egl-10-Pleckstrin domain and to Ras Exchange Motif via the α6 hinge helix) interacts with the N-terminal helical bundle (NTHB) which interacts with the CDC25 homology domain via these salt bridges. cAMP binding to CBD:β leads to α6 hinge rotation and equilibrium shift toward the open state. Recent chemical shift mapping unraveled the likely allosteric mechanism: cAMP binding weakens the IL although no significant perturbation is observed.46 The weakened salt bridges facilitate α6 swiveling motions, allowing Rap1 and Rap2 binding (positive cooperativity). These GTPases control critical signaling pathways, including insulin secretion and cell adhesion. The authors argue that salt bridge weakening is due to increased entropic cost from the NHTB enhanced dynamics (without conformational change) following cAMP binding. Alternatively, we propose that this is an enthalpy-driven (conformational change) allostery , where the perturbation originating from cAMP binding propagates, leading to two competing states: salt bridges-favored state and a competing Rap-bound state. The fluctuations between these two isomers lead to salt bridge weakening. Thus, in Table 1, cAMP-activated exchange protein is classified as conformational change, positive cooperativity, enthalpy-driven.Example 7. Two ligand two receptor monomer (2 : 2) complex formation: the Toll and Toll-like receptors
Toll and Toll-like receptors (TLR), essential for immunity are activated by microbial pathogen-derived ligands which bind the extracellular Toll ectodomain. Drosophila Gram-positive bacteria-derived peptidoglycan stimulates Spätzle (Spz) synthesis, which binds to the ectodomain, initiating signal transduction. This domain consists of leucine-rich repeats (LRR) arranged as an N- and C-termini capped solenoid. There are two linker-connected terminal capped LRRs. A single transmembrane segment connects the ectodomain to an intracellular TIR signaling module. Previously it was believed that Toll activation involved a single Spz cross-linking two Toll ectodomains; however, recent cryo-EM and mass spectrometry data show that two Spz molecules bind to the N-termini of two Toll ectodomains triggering a more tightly curved ectodomain conformation (conformational change) and dimer formation (positive cooperativity, enthalpy-driven) through interaction at the N-terminal and the juxtamembrane sequences.47 Hence, this family provides an example of a classification of allostery with conformational change, positive cooperativity, enthalpy-driven (Table 1).Example 8. Symmetric multi-subunit ring: the trpRNA binding attenuation protein
trp RNA binding attenuation protein (TRAP) is a symmetric, 11-mer ring, involved in the regulation of intracellular levels of tryptophan in some bacteria.48 TRAP–tryptophan complex binds to trpmRNAleader sequence ; consequent hairpin formation prevents downstream transcription. Tryptophan-unbound TRAP cannot bind RNA. Bacillus stearothemophilus TRAPste NMR data indicate that in the absence of Trp, the RNA binding site is disordered;49Trp binding promotes disorder to order conformational change, prepaying the entropic cost. Trp binding to Bacillus subtilus TRAP (TRAPsub) is non-cooperative; thus, function does not require inter-subunit cooperativity. On the other hand, TRAPste presents weak positive cooperativity. While tryptophan-free TRAPste is unable to bind RNA, binding of one Trp to one of the eleven TRAP monomers is sufficient for high affinity RNA binding. Isothermal titration calorimetry explains the difference between the behavior of the two TRAPs: unlike TRAPste, in TRAPsub the bindings are non-cooperate events; however, a single TRAPsub (Ile to Leu) mutation leads to inter-subunit Trp binding cooperativity. The single mutation is sufficient for the weak positive cooperativitivity, presumably enthalpy-dominated. Hence, in Table 1 we classify this protein as conformational change, (weak) positive cooperativity, enthalpy-driven.Example 9. Homodimer: the CAP example
Catabolite activator protein (CAP) controls over 100 genes. CAP is a homodimeric transcriptional activator. Each subunit has an N-terminal ligand (cAMP) binding domain and C-terminal DNA binding domain. cAMP binding to CAP increases its affinity to DNA. CAP unwinds the double helix, allowing RNA polymerase binding and transcription. The two cAMPs bind CAP with negative cooperativity: despite the large spatial separation between the cAMP binding pockets (24 Å between the cAMP’s phosphates), binding of one cAMP reduces the binding affinity of the other by 2 orders of magnitude. NMR studies of N-terminal truncated CAP domains (CAPN) indicated that binding of the first cAMPdoes not lead to an observable conformational change in the binding site of the second cAMP; thus it is entropy-controlled. Popovych et al. proposed a mechanism where the first cAMP binding increases the entropy, forcing the second cAMP binding to pay higher cost.23 In the absence of cAMP, CAPN, including cAMP binding sites, is highly flexible as shown by hydrogen exchange rates. Binding of the first cAMP has little effect on the ps–ns time scales flexibility but it enhances the slow motions (μs–ms). This affects the conformation of the liganded but not the unliganded subunit. The second cAMP binding suppresses the fast and the slow motions, thus there is a lower favorable entropy change as compared to the first cAMP binding, and negative cooperativity. Alternatively,3 the first cAMP binding increases the overall rigidification; being symmetric occurrences, the second cAMP binding similarly increases the overall rigidification. Propagation of the rigidifications and path-meeting create extra unfavorable entropy terms.50 This is an example of no conformational change, negative cooperativity, entropy-driven (Table 1).Example 10. Ternary complex dissociation: the CSL/NotchIC/Mastermind organization
CSL, associates in the ternary complex with Notchintracellular (NotchIC) domains RAM, and ANK and Mastermind (see example 4).42,43 CDK8 phosphorylates the ANK domain, creating the Ffw7/Sel10 effector binding site. While no structural data are available, no conformational change is expected in phosphorylation-binding site formation. Ffw7/Sel10 binding to the phosphorylated site can compete for space with the CSL/NotchIC/Mastermind organization to avoid direct interference. The outcome is NotchIC and Mastermind dissociation with subsequent degradation by the E2 ubiquitin ligase. While no data are available, we can expect that effector binding leads to an enthalpy-driven conformational change. Dissociation implies negative cooperativity. Subsequent transcriptional co-repressor binding and histone deacetylase recruitment lead to repression. This is an example where phosphorylation creates a binding site, with conformational change, negative cooperativity, enthalpy-driven (Table 1).Discussion and conclusions
Classification assists not only in organizing information but also in making sense of observations: what are the differences between plants and animals; invertebrates and vertebrates; between cold and warm blooded animals; between mammals, birds, reptiles, fish, and amphibians; between classes of protein structures; drugs; types of interactions and chemical reactions. Sorting objects into distinct categories organizes the information, revealing patterns and relationships, and consequently provides insight. While the importance of classification is clear, how to classify and into which categories is less obvious. Relevant questions are (i) which properties to use; (ii) is the range of available cases sufficient; (iii) should categories lumped together require splitting; (iv) if we had started from different (to be discovered) examples, would the classification scheme be similar? A relevant classification scheme should relate to function and eventually must correlate with quantities allowing automated classification based on a set of analytical or empirical components. With these caveats in mind, we believe that the increase in the number of observed allostery cases, their breadth, the recent insight into allostery types and mechanisms, and their impact on cellular functions permits a first step in a classification scheme, which we expect to assist in organization, comprehension and in allosteric drug discovery.Here we provide a comprehensive description and a framework for classification of allosteric mechanisms with some examples abstracted from a range of cellular processes. This was made possible by first obtaining a unified mechanistic view of allostery . The classification is based on several properties: presence/absence of conformational change; cooperativity type; driving thermodynamic terms; whether the substrate binding site and the functional site coincide; and role of phosphorylation (if present). We observed numerous examples of disorder-to-order conformational change with the first binding event prepaying the entropy cost. These common allostery cases display positive cooperativity and are lumped into the same classification bin. At the same time, order-to-disorder cases entail high entropy cost and fall into the negative cooperativity with conformational change rubric.
Mutations were not treated here. Yet, mutations can lead to disease via allosteric effects. Consider for example the seven-helix transmembrane receptor. According to Pollard et al.42 over 600 mutations in more than 30 receptors of this family have been linked to disease. If the mutations occur at the ligand or the trimeric G-protein binding sites (including binding site truncation events, as for example at the N-terminal) these are not allosteric mutations. On the other hand, mutations occurring elsewhere and affecting binding are allosteric events. Mutations can lead to effector unfavorable N-terminal conformations; under such circumstances, there is no conformational change of the intracellular loops, thus no activation of the trimeric G-protein, which will remain in the receptor-bound state. These mutations lead to loss of function. In contrast, mutations leading to a conformational change at the G-protein binding site constitutively activate the reaction regardless of whether a ligand is bound. Mutations in tyrosinekinases can lead to disease. If these mutations are away from the binding sites, similar allosteric mechanisms may operate.51
Eventually, a systematic compilation and organization of available allostery cases, encompassing a range of effectors and environments within the functional signaling transduction context would be invaluable. Allostery is the vehicle through which function is exerted. Classifying allosteric cases transforms phenomenological descriptions to molecular mechanisms. To cite two examples, consider the text-book description of a GTP to GDP exchange in the seven-helix receptors case:42 here a classification scheme clarifies how the function is performed in this step of the catalytic phosphorylationsignaling cascade. Classification indicates that two allosteric steps are involved (Fig. 2; example 1) where a substrate site initially occupied by GDP becomes an allosteric site which, via negative cooperativity leads to the dissociation of the substrate, now the seven-helix receptor. Classifying the receptor tyrosine kinase case (example 2) clarifies that the functional site differs from the substrate binding site, a key mechanistic component which was unclear before (Fig. 3). Text books describe series of events; not mechanisms. They cite the event and its consequences: a certain gene knock-out will lead to a certain loss of function. Yet, in disease our goal is to be able to trace back to a particular signaling check-point; to identify the source of the functional loss. Further, a disease-related mutation does not have to be in the substrate or the functional sites; but it may block signal propagation.
Classification has proven immensely important in science. Here we provide the first classification framework for allostery based on six properties; this implies that a change in any of the six properties would affect function. A key question is which property would affect it the most. Hence, if the mechanism involves dimerization (example 2; property 6, Table 1) concentration and binding constants are crucial. If the substrate binding site differs from the functional site (example 3; property 5 Table 1) a mutation in the functional site can abolish substrate binding. If there is a hinge bending conformational change (examples 2 and 3; property 2) a mutation altering its extent can lead to functional loss; this property relates to propagation pathways and to dominant thermodynamic factors (property 3); mutations affecting perturbation (property 1) can have a similar effect to allosteric cooperativity (property 4): no perturbation, no cooperativity. Additional descriptors would surely be incorporated as the range of cases broadens. Clearly, figuring out allosteric mechanisms is crucial for the comprehension of how function is performed in the cell. Using a classification scheme along the lines proposed here should complement text-book descriptions; it should assist in the understanding of how the function is performed on the single molecule level within the framework of its complex cellular environment.
Acknowledgements
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.References
- N. M. Goodey and S. J. Benkovic, Nat. Chem. Biol., 2008, 4, 474–482 CrossRef CAS.
- K. Gunasekaran, B. Y. Ma and R. Nussinov, Proteins: Struct., Funct., Genet., 2004, 57, 433–443 Search PubMed.
- C. J. Tsai, A. del Sol and R. Nussinov, J. Mol. Biol., 2008, 378, 1–10 CrossRef CAS.
- J. A. Hardy, J. Lam, J. T. Nguyen, T. O’Brien and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12461–12466 CrossRef CAS.
- E. Ozkan, H. T. Yu and J. Deisenhofer, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18890–18895 CrossRef CAS.
- O. F. Lange, N. -A. Lakomek, C. Fares, G. F. Schroder, K. F. A. Walter, S. Becker, J. Meiler, H. Grubmuller, C. Griesinger and B. L. de Groot, Science, 2008, 320, 1471–1475 CrossRef CAS.
- D. D. Boehr and P. E. Wright, Science, 2008, 320, 1429–1430 CrossRef CAS.
- Q. Cui and M. Karplus, Protein Sci., 2008, 17, 1295–1307 CrossRef CAS.
- M. W. Clarkson, S. A. Gilmore, M. H. Edgell and A. L. Lee, Biochemistry, 2006, 45, 7693–7699 CrossRef CAS.
- H. Gopi, M. Umashankara, V. Pirrone, J. LaLonde, N. Madani, F. Tuzer, S. Baxter, I. Zentner, S. Cocklin, N. Jawanda, S. R. Miller, A. Schon, J. C. Klein, E. Freire, F. C. Krebs, A. B. Smith, J. Sodroski and I. Chaiken, J. Med. Chem., 2008, 51, 2638–2647 CrossRef CAS.
- C. Chennubhotla, Z. Yang and I. Bahar, Mol. BioSyst., 2008, 4, 287–292 RSC.
- L. W. Li, V. N. Uversky, A. K. Dunker and S. O. Meroueh, J. Am. Chem. Soc., 2007, 129, 15668–15676 CrossRef CAS.
- M. D. Daily, T. J. Upadhyaya and J. J. Gray, Proteins: Struct., Funct., Genet., 2008, 71, 455–466 Search PubMed.
- Y. W. Chen and N. V. Dokholyan, J. Biol. Chem., 2006, 281, 29148–29154 CrossRef CAS.
- C. N. Chi, L. Elfstrom, Y. Shi, T. Snall, A. Engstrom and P. Jemth, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4679–4684 CrossRef CAS.
- J. Monod, J. Wyman and J. P. Changeux, J. Mol. Biol., 1965, 12, 88–118 CrossRef CAS.
- D. E. Koshland, G. Nemethy and D. Filmer, Biochemistry, 1966, 5, 365–385 CrossRef CAS.
- G. M. Suel, S. W. Lockless, M. A. Wall and R. Ranganathan, Nat. Struct. Biol., 2003, 10, 59–69 CrossRef.
- M. S. Formaneck, L. Ma and Q. Cui, Proteins: Struct., Funct., Genet., 2006, 63, 846–867 Search PubMed.
- S. Kumar, B. Y. Ma, C. J. Tsai, N. Sinha and R. Nussinov, Protein Sci., 2000, 9, 10–19 CAS.
- B. Y. Ma, S. Kumar, C. J. Tsai and R. Nussinov, Protein Eng., 1999, 12, 713–720 CrossRef CAS.
- A. Ansari, J. Berendzen, S. F. Bowne, H. Frauenfelder, I. E. T. Iben, T. B. Sauke, E. Shyamsunder and R. D. Young, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 5000–5004 CrossRef CAS.
- N. Popovych, S. J. Sun, R. H. Ebright and C. G. Kalodimos, Nat. Struct. Mol. Biol., 2006, 13, 831–838 CrossRef CAS.
- A. Cooper and D. T. F. Dryden, Eur. Biophys. J., 1984, 11, 103–109 CrossRef CAS.
- D. Kern and E. R. P. Zuiderweg, Curr. Opin. Struct. Biol., 2003, 13, 748–757 CrossRef CAS.
- B. Y. Ma, M. Shatsky, H. J. Wolfson and R. Nussinov, Protein Sci., 2002, 11, 184–197 CrossRef CAS.
- A. Camara-Campos, C. A. Hunter and S. Tomas, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3034–3038 CrossRef CAS.
- S. W. Lockless and R. Ranganathan, Science, 1999, 286, 295–299 CrossRef CAS.
- A. I. Shulman, C. Larson, D. J. Mangelsdorf and R. Ranganathan, Cell, 2004, 116, 417–429 CrossRef CAS.
- X. G. Niu, Q. Chen, J. H. Zhang, W. Q. Shen, Y. Y. Shi and J. H. Wu, Biochemistry, 2007, 46, 15042–15053 CrossRef CAS.
- E. J. Fuentes, C. J. Der and A. L. Lee, J. Mol. Biol., 2004, 335, 1105–1115 CrossRef CAS.
- E. J. Fuentes, S. A. Gilmore, R. V. Mauldin and A. L. Lee, J. Mol. Biol., 2006, 364, 337–351 CrossRef CAS.
- A. Dhulesia, J. Gsponer and M. Vendruscolo, J. Am. Chem. Soc., 2008, 130, 8931–8939 CrossRef CAS.
- G. Schreiber and A. R. Fersht, J. Mol. Biol., 1995, 248, 478–486 CAS.
- P. J. Carter, G. Winter, A. J. Wilkinson and A. R. Fersht, Cell, 1984, 38, 835–840 CrossRef CAS.
- A. Horovitz and A. R. Fersht, J. Mol. Biol., 1990, 214, 613–617 CrossRef CAS.
- A. Horovitz and A. R. Fersht, J. Mol. Biol., 1992, 224, 733–740 CrossRef CAS.
- E. Sadovsky and O. Yifrach, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19813–19818 CrossRef CAS.
- J. A. Ubersax and J. E. Ferrell, Nat. Rev. Mol. Cell Biol., 2007, 8, 530–541 CrossRef CAS.
- K. Komurov and M. White, Mol. Syst. Biol., 2007, 3, 1–11.
- O. Keskin and R. Nussinov, Structure, 2007, 15, 341–354 CrossRef CAS.
- T. D. Pollard, W. C. Earnshaw and J. Lippincott-Schwartz, Cell Biology, W.B. Saunders Company, New York, 2nd edn, 2007 Search PubMed.
- D. R. Friedmann, J. J. Wilson and R. A. Kovall, J. Biol. Chem., 2008, 283, 14781–14791 CrossRef CAS.
- C. C. Huang, F. Stricher, L. Martin, J. M. Decker, S. Majeed, P. Barthe, W. A. Hendrickson, J. Robinson, C. Roumestand, J. Sodroski, R. Wyatt, G. M. Shaw, C. Vita and P. D. Kwong, Structure (London), 2005, 13, 755–768 Search PubMed.
- H. Garg and R. Blumenthal, Cell. Mol. Life Sci., 2008, 65(20), 3134–3144 CrossRef CAS.
- R. Das, M. T. Mazhab-Jafari, S. Chowdhury, S. Sildas, R. Selvaratnam and G. Melacini, J. Biol. Chem., 2008 Search PubMed.
- M. Gangloff, A. Murali, J. Xiong, C. J. Arnot, A. N. Weber, A. M. Sandercock, C. V. Robinson, R. Sarisky, A. Holzenburg, C. Kao and N. J. Gay, J. Biol. Chem., 2008, 283, 14629–14635 CrossRef CAS.
- J. G. Heddle, T. Okajima, D. J. Scott, S. Akashi, S. Y. Park and J. R. H. Tame, J. Mol. Biol., 2007, 371, 154–167 CrossRef CAS.
- C. McElroy, A. Manfredo, A. Wendt, P. Gollnick and M. Foster, J. Mol. Biol., 2002, 323, 463–473 CrossRef CAS.
- J. A. Boyer and A. L. Lee, Biochemistry, 2008, 47, 4876–4886 CrossRef CAS.
- J. Liu and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 901–906 CrossRef CAS.
Footnote |
| † Current address: Almendro 8, Madrid 28005, Spain. E-mail: antdelsol@gmail.com |
| This journal is © The Royal Society of Chemistry 2009 |