In search of small molecules blocking interactions between HIV proteins and intracellularcofactors
Katrien
Busschots
,
Jan
De Rijck
,
Frauke
Christ
and
Zeger
Debyser
*
Molecular Medicine, Molecular Virology and Gene Therapy, K.U.Leuven, Kapucijnenvoer 33, B-3000, Leuven, Belgium. E-mail: Zeger.Debyser@med.kuleuven.be; Fax: +32 16 33 63 36; Tel: +32 16 33 21 83
First published on 16th October 2008
Abstract
One of the major obstacles to pursue the discovery of small molecule inhibitors targeting protein–protein interactions is the flat nature of their interface. X-Ray structures have indeed shown that a large part of the interaction area is buried with atoms closely packed together, implying a lack of available cavities for small molecule binding. Yet, it has become clear that some protein–protein interfaces have a well-defined compact area, commonly referred to as a hot spot, that plays a major role in the affinity of the interaction. These hot spots define potential targets for the development of small molecule protein–protein interaction inhibitors (SMPPIIs). In this review we discuss the interactions between viral and hostproteins that have the potential for the future development of SMPPIIs. In light of the current anti-HIV therapy a short overview of protein–protein interactions that may serve as targets for novel drugs is provided. Our hypothesis will exemplify and discuss the interaction between HIV-1integrase and its cellular cofactorLEDGF/p75, which, as evidenced by crystallography and site directed mutagenesis, displays favourable properties needed for the development of interaction inhibitors.
 Katrien Busschots Katrien Busschots | Katrien Busschots obtained her master in Biomedical Sciences at the Katholieke Universiteit Leuven (K.U.Leuven), Belgium and is currently finishing a PhD in Medical Sciences in the group of Prof. Debyser. She has experience in biochemistry with a focus on protein–protein interactions between the viral integrase and host proteins. |
 Jan De Rijck Jan De Rijck | Jan De Rijck received his master in Biology at the Katholieke Universiteit Leuven (K.U.Leuven), Belgium and is currently continuing his PhD research as a postdoctoral fellow. His primary interest is the identification and validation of interactions between HIV-1 and its cellular environment during nuclear import and integration as a potential new antiviral strategy. |
 Frauke Christ Frauke Christ | Frauke Christ studied Biology at the Justus-Liebig University, Giessen, Germany and earned her PhD in Biochemistry studying the biochemistry of homing endonucleases. After a postdoc on recombination specific nucleases, she joined the lab of Prof. Debyser as a group leader in HIV co-factor identification and validation as well as HIV drug discovery. |
 Zeger Debyser Zeger Debyser | Zeger Debyser (1965) is Medical Doctor (K.U.Leuven, 1990) and he obtained his PhD at the K.U.Leuven (Flanders) in 1994. Dr Debyser currently heads the Division of Molecular Medicine. His research focuses on the molecular virology of HIV integration and the development of lentiviral vector technology. The link between both topics is the capacity of HIV to integrate in non-dividing cells enabling lentiviral vector (LV) mediated gene transfer into non-dividing cells. |
Protein–protein interactions in the light of academic and pharmaceutical research
Protein–protein interactions (PPIs) play a key role in most biological processes—from intracellular communication to programmed cell death—and therefore represent a large and important class of therapeutic targets.1,2 The wealth of information obtained from genome and proteome programs has increased the number of known protein–protein interactions involved in the pathogenesis of various human diseases. Nevertheless, these interactions were left aside for a long time, since development of drugs against the classical targets like enzymes and receptors has so far been more cost effective as discussed below. Since the expenses for developing these classical drugs is on the increase, new target proteins and/or protein–protein interactions have come within reach. The research on protein–protein interactions might boost the development of technologies to identify and validate interactions relevant for human disease and the development of assays to screen for inhibitors of protein–protein interactions. Taking into account the financial risk associated with this basic research, to date initial steps of target validation and hit identification might predominantly be taken by academia. However, further lead optimization and clinical development will have to be undertaken in collaboration with industry.Three classes of interaction inhibitors
The identification or design of SMPPIIs is considerably more difficult than the development of inhibitors targeting the active sites of enzymes or ligand binding pockets. Active sites or binding pockets of receptors are intrinsically shaped to bind a small molecule with extensive contacts with the protein. Furthermore, the availability of natural ligands and substrates provides an initial template for the de novo design of inhibitors. This stands in stark contrast to protein–protein interaction sites, which by definition do not bind small ligands that could serve as a template for the design of SMPPIIs.What are the options for blocking protein–protein interactions? A wide range of approaches have been used including antibodies, peptides and small molecules. Therapeutic antibodies (chimeric or humanized antibodies, antibody fragments or cocktails of different monoclonal antibodies) represent a large segment of the ongoing clinical trials for biopharmaceuticals.3 Although they have several excellent properties such as specific binding to their target with high affinity and the apparent stability in human serum, there are major pitfalls. Antibodies are not only difficult and expensive to produce, they also lack oral bioavailability. Furthermore, they are not well suited to target intracellularproteins because of their relatively large size and the reducing environment of the cell that destabilizes the disulfide bonded structure.
Although smaller peptideinhibitors are an attractive alternative to inhibit protein–protein interactions in vitro, they are not stable enough for in vivo applications. To overcome this problem, they can be designed as cross-linked interfacial peptides or helical mimetics. These reagents might have better pharmacokinetics than antibodies and may be more useful for targeting both intracellular and extracellular interactions. Examples of engineered peptides that are capable of blocking protein–protein interactions are summarized by Zhao et al.4 The only genuine example of an FDA approved peptideinhibitor in the field of HIV research is enfuvirtide or T20. After binding of gp120 to the CD4 cell surfacereceptor, this synthetic 36 amino acid peptidomimetic interferes with the conformational changes of the viral gp41 protein needed for membrane fusion and entry of the virus into the host cell.5 Still, this compound has to be administered by injection. As it is useful in multiple drug resistant HIV-1 patients, it gained FDA approval despite the disadvantages associated with peptide-based drugs.
The third and most attractive class consists of small molecule inhibitors. The vast majority of drugs making their way from bench to bedside belong to this group of compounds. As potential protein–protein interaction inhibitors, they benefit from several attractive properties such as the relatively easy and cheap production process, in comparison to that of peptide synthesis. Moreover, as many small molecule inhibitors are permeable to the cell membrane, they are useful as oral therapeutics. So far, several protein–protein interfaces have been identified as putative targets and a few SMPPIIs are currently under clinical trials as discussed below. Despite the fact that none of these have reached the pharmaceutical market, these compounds hold a promise for future therapies of human diseases, including cancer and viral infections.
SMPPIIs
One of the major challenges in the search of potent SMPPIIs is the flatness of most protein interfaces spanning a contact area of 750–1500 Å2. X-Ray structures have shown that a large part of the surface area of a typical protein–protein interface is buried with the atoms closely packed together, implying that only a few cavities are available for small molecule binding. However, it has become clear that for some protein–protein interfaces the affinity of the interaction is in large part governed by a small well-defined compact sub-area of a larger interface. These high-affinity areas are referred to as hot spots as nicely reviewed by Arkin and Wells.2 The main factors that contribute to protein–protein interactions are hydrogen bonds and hydrophobic and electrostatic interactions.6,7 Analysis of protein contacts has furthermore revealed that interface surfaces are quite complementary to each other. The degree of complementarity depends on the type of interaction. Permanent complexes, which may be considered as a continuation of protein folding, have the highest degree of complementarity, whereas temporary, short-living complexes are characterized by lower complementarity. Finally, the role of plasticity in protein–protein interfaces is becoming ever more apparent as additional crystal structures of related protein complexes are solved. Many protein-binding sites turn out to be structurally adaptive and some of these are also functionally adaptive and able to bind multiple diverse partners.8 Still, the bulk of the binding energy may reside in a relatively small area that can be occupied by a small molecule inhibitor. Therefore, it has become clear that it is possible to design SMPPIIs targeting defined hot spots.9 Nevertheless, as the field is young and the timeline from hit identification to the clinic spans more than a decade, none of such inhibitors has reached the market so far.An extra challenge in the design of SMPPIIs is the size and character of typical small molecule libraries. Available libraries are usually designed to span the characteristics of classical drug targets and are not specifically adapted to the characteristics of PPI. Nevertheless, several starting compounds were identified by high-throughput screening (HTS) involving small molecule libraries.9–12 Methods for unravelling protein interaction hot spots, for identification of structural motifs common to protein interaction interfaces and for mapping protein–protein contact surfaces have all contributed to the design of SMPPIs.4,13 The most recent advance is fragment-based drug discovery,14 in which very small molecules (fragments) are screened and the generally low potency hits are then linked to other low affinity binders to form the final high affinity molecule. This approach has generated a significant number of novel small molecules with high potency and improved pharmacokinetic properties.15
SMPPIIs targeting interactions between cellular proteins
Proof-of-concept for the development of small molecules has been obtained for different protein–protein interfaces of cellular interaction partners.9 The availability of the crystal structure of their interface is an indispensable tool to understand how a small molecule can directly compete with its natural protein partner. A typical example of a novel drug target in cancer treatment is the human protein double minute 2 (HDM2).16 Initially, it was found that the mouse homologue of HDM2 (known as MDM2) binds to the tumour-suppressor protein p53 and increases its degradation. Inhibition of this interaction can stimulate p53 activity in wild-type cancer cells driving them towards apoptosis.17X-Ray crystallography of the p53-MDM2 complex reveals that this protein–protein interaction is primarily mediated by a few key amino acids of p53 and a small but deep hydrophobic cleft in MDM2.18,19 Nutlins are a series of tetra-substituted imidazoles which can potently disrupt the MDM2–p53 interaction (Fig. 1).10 It was observed that nutlins mimic the binding of the helical region of p53 by interacting with the hydrophobic cleft of MDM2. Furthermore these compounds are active against tumour xenografts in vivo.10 A different promising class of HDM2 inhibitors are the benzodiazepinediones (Fig. 1).11,20,21 Although these compounds were initially selected for binding to HDM2 and not for functional disruption of the complex, they promote rapid dissociation of p53 from HDM2 in cells overexpressing HDM2. Recently, the compound MI-219 was positively evaluated in pre-clinical trials (Fig. 1).22 It binds to human MDM2 with a Ki value of 5 nM and is selectively toxic to tumours, leading to complete tumour growth inhibition. Ascenta Therapeutics (http://www.ascenta.com) is committed to advancing MI-219 (named AT-219) into clinical trials. | ||
| Fig. 1 Overview of several reported small molecule protein–protein interaction inhibitors (SMPPIIs). | ||
Another example of the interruption of a ligand-receptor interaction by a small molecule comes from interleukin studies. The cytokine interleukin-2 (IL-2), a mediator of the T-helper immune response, plays a role in the rejection of tissue grafts.23 A series of small molecules discovered through fragment assembly, that bind directly to IL-2, were shown to prevent interaction between IL-2 and the IL-2 receptor (Fig. 1).15,24–26 These preclinical studies also indicated that the binding surface on IL-2 is adaptive and can bind a small molecule with high affinity using the same hot spot residues.27
B-cell lymphoma-2 (BCL-2) and BCL-XL are anti-apoptotic proteins of which the function is regulated by the binding of anti- or pro-apoptotic factors such as BAK. The importance of BAK as target in the treatment of cancer has generated interest in developing an interaction inhibitor. Several laboratories have reported on the identification of small molecules binding BCL2 and/or BCL-XL. One of these molecules, ABT-737, exhibits monotherapy efficacy in xenograft models of small-cell lung cancer and lymphoma (Fig. 1).28 Another compound, A-385358, selectively binds to BCL-XL.29 These molecules could be useful in the complementation of existing chemotherapy and radiation. GeminX (http://www.geminx.com) and Ascenta Therapeutics (http://www.ascenta.com) have small-molecule compounds that might function as BCL-2 antagonists in early-phase clinical trials and a derivative of the Abbott Laboratories (http://www.abbott.com) compound ABT-737, ABT-263, has also progressed to phase I/II clinical testing (Fig. 1).
Why does anti-HIV therapy need SMPPIIs?
The insight has grown that the limited genomic capacity of viruses, such as HIV-1, requires multiple cellular partners to serve as cofactors for their replication.30 Although antiviral drugs typically target viral proteins to reach specificity, we propose to use our expanding knowledge on cellular cofactors to target protein–protein interactions between viral and cellular proteins. This novel paradigm is of the uttermost importance in HIV therapy. Because of its short replication cycle and the high-error rate of reverse transcriptase, HIV mutates very quickly, resulting in the rapid emergence of drug resistant HIV strains. Although the current highly active antiretroviral therapy (HAART) for HIV infection is based on combinations of effective antiretrovirals, drug resistant virus strains urge scientists to constantly look for second generation and novel inhibitors to outpace resistance development. To avoid cross-resistance, novel drugs preferentially target yet unexploited steps of the viral replication cycle. We put forward our thesis that the resistance development during treatment of chronic HIV infection is intrinsically linked to the selection of viral targets.The HIV-1 replication cycle is initiated by the attachment of the viral envelopeglycoprotein gp120 to the cellular CD4 receptor (Fig. 2). The interaction of gp120 to CD4 triggers a conformational change in the gp120 molecule, allowing the binding to the CXCR4 or CCR5 chemokine co-receptor. Another conformational change in the gp41 viral protein provokes fusion of the viral particle with the cellular membrane, which is followed by the viral uncoating. In the next step reverse transcription of the positive RNA strand is initiated. During reverse transcription the single stranded RNA is copied into double stranded cDNA, which will be imported into the nucleus as a preintegration complex (PIC). Apart from viral cDNA this complex contains viral matrix (MA), viral protein R (Vpr), reverse transcriptase (RT), integrase and several cellular proteins. In the nucleus the proviral DNA associates with the hostchromosomes, and the viral cDNA integration takes place. This step is catalyzed by the viral enzymeintegrase and is referred to as “the point of no return” since from that moment on the provirus will form part of the cellular genome. During the lifespan of the infected cell, the virus may either remain present in a latent reservoir or transcription of viral genes may initiate the formation of new virions. Viral proteins and genomic RNA are assembled at the cellular membrane carrying envelopeproteins. Immature viral particles bud from the membrane and mature into infectious virions through proteolytic processing of the viral polyproteins by the viral protease (for a detailed review on the viral replication cycle, see ref. 31).
 | ||
| Fig. 2 Replication cycle of HIV-1. After viral entry, the viral RNAgenome is reverse transcribed into double stranded viral DNA. The DNA embedded in the preintegration complex is transported into the nucleus, where it will be integrated in the hostDNA. Following transcription of the proviral DNA, the mRNA is transported to the cytoplasm where it is translated into viral proteins. After assembly and budding of the new virions, maturation will take place and a new round of infection can start. Highly active antiretroviral therapy is based on a cocktail of drugs targeting the viral enzymes protease, reverse transcriptase or integrase. The regimen can also be complemented with two inhibitors of the viral entry step. | ||
The standard therapy for HIV-1 infected patients (HAART) is based on cocktails of potent drugs targeting different steps of the replication cycle. There are three main classes of drugs: protease inhibitors,32nucleoside or nucleotidereverse transcriptase inhibitors (NRTIs) and non-nucleosidereverse transcriptase inhibitors (NNRTIs). Combinations of these drugs are given to minimise the risk of resistance development. Lately, novel drugs targeting other steps of the viral life cycle have entered the clinic and complement standard HAART therapy. Among these are Enfuvirtide (fusion inhibitor), Maraviroc (CCR5 entry inhibitor) and Raltegravir (integraseinhibitor).33
Proof-of-principle for development of small molecules targeting interactions between viral and cellular proteins
Although Enfuvirtide and Maraviroc are not protein–protein interaction inhibitorsper se, since they interfere with ligand–receptor interactions, these therapeutics illustrate the great potential of blocking the interaction between viral and cellular proteins for antiviral therapy. Enfuvirtide (T20, Fuzeon) binds to the viral gp41 protein and interferes with its ability to approximate virus and cell membrane. Therefore, it is also referred to as a fusion inhibitor.5 In September 2007, Maraviroc (Selzentry) was approved as an entry inhibitor and chemokine receptor antagonist (Fig. 3). It specifically blocks the chemokine receptor CCR5, preventing its interaction with gp120.34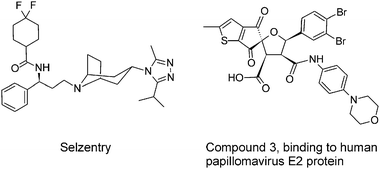 | ||
| Fig. 3 Small molecules blocking interactions between viral and/or cellular proteins. Maraviroc (Selzentry) specifically blocks the chemokine receptor CCR5, preventing its interaction with HIV-1 gp120 and subsequent viral entry. Compound 3 binds to human papillomavirus transcription factor E2, thereby preventing the interaction with the viral E1 helicase, which is necessary for viral replication. | ||
Several novel antivirals are under initial investigation or in clinical trials. The well known immunosuppressive agentcyclosporin A was originally proposed to suppress HIV-1 replication by interference with the binding of cyclophilin A to the viral Gag polyprotein.35,36 Although Bartz et al. searched for small molecules to block the cyclosporin target without significant immunosuppressive activity,37 this initial study was not followed up by more definitive studies. More recently it was shown that infection with hepatitis C virus can be reduced by inhibition of the interaction of viral NS5B polymerase with cyclophilin B. Clinical trials with oral cyclophilininhibitors are ongoing.38–41
The antiviral activity of protein–protein interaction inhibitors was also demonstrated with molecules targeting human papillomaviruses. These compounds consist of an indandione fused to a substituted tetrahydrofuran (Fig. 3).12 Characterization of their mechanism of action pointed to an interaction with the viral transcription factor E2, thereby preventing the interaction with the viral E1 helicase, necessary for viral replication.42X-Ray crystallography revealed that the inhibitors bind in a small molecule binding pocket on E2.43
The protein–protein interaction between HIV integrase and LEDGF/p75 exemplifies the concept
What makes a PPI an attractive target for the discovery and development of small molecules? The following points need to be addressed: (i) the target has to be validated as important for HIV replication, (ii) inhibition of the specific PPI should not be associated with toxicity, (iii) structural information on the PPI should be available and (iv) identification of genuine inhibitors would provide ultimate proof-of-concept.An example of a PPI that has the potential for the development of a SMPPII is the interaction between viral HIV-1integrase and the cellular protein lens epithelium-derived growth factor or transcriptional co-activator p75 (LEDGF/p75). After its initial identification by coimmunoprecipitation in 2003 as a high affinity binding partner of HIV integrase, it took several years to validate LEDGF/p75 as a genuine antiviral target.
Structural biology of HIV integrase and LEDGF/p75
The rational drug design of specific drugs targeting protein–protein interactions is greatly facilitated by structural data of the protein complex. In accordance with its ability to interact with HIV-1integrase, an evolutionary highly conserved integrase-binding domain (IBD) was identified in the C-terminus of LEDGF/p75 (amino acids 347–429) (Fig. 4).44 Structural analysis by NMR showed a compact right-handed bundle composed of five alpha helices that is topologically similar to a pair of HEAT repeats.45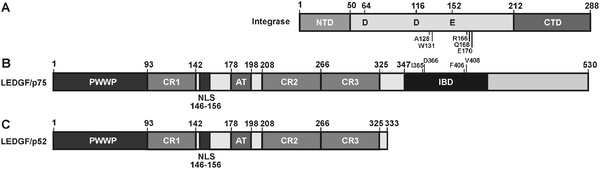 | ||
| Fig. 4 Domain organization of HIV-1integrase and LEDGFproteins. (A) Integrase consists of three distinct structural domains:46 the N-terminal zinc binding domain, the catalytic core domain (CCD) containing the catalytically active DDE motif and the positively charged C-terminal domain.47–49 The latter displays DNA binding activity, and is (like the other two domains) involved in multimerization.50 Amino acids in the catalytic core that are important for interaction with LEDGF/p75 are indicated.45,51–53 (B) The N-terminal part of LEDGF/p75, including the PWWP domain, two AT-hook motifs and charged regions (CR) one and two, contribute to chromatin binding.54–56 The PWWP domain most probably functions as the main chromatin tether. LEDGF/p75 has a functional nuclear localisation signal.57,58 In the C-terminal part of the protein an evolutionary conserved integrase binding domain (IBD) is present.44 (C) LEDGF/p52 is derived by alternative splicing from the LEDGFgene.59,60 The C-terminus of p52 differs in eight amino acids from its larger isoform LEDGF/p75. LEDGF/p52 lacks the integrase binding domain. | ||
Although a crystal structure of full length integrase or full length LEDGF/p75 is not available, a co-crystal of the interacting domains of integrase and LEDGF/p7561 provides the required structural information and knowledge on the interacting residues in integrase and LEDGF/p75 to embark on drug design. Confirmation of the biological relevance of the co-crystal was obtained by different mutagenesis studies.51–53,62 All data on the amino acids in integrase that contribute to the LEDGF/p75 binding are summarized in Table 1. The co-crystal revealed that two monomers of IBD interact with a dimer of the catalytic core domain (CCD) of integrase (Fig. 5). An interhelical loop of the IBD binds to a defined pocket at the interface of the two CCDs. The most critical interacting residues of the IBD are I365, D366 and F406.61 Based on the co-crystal, the principal structural features of integrase recognized by LEDGF/p75 are the backbone conformation of residues 168–171 (located in alpha helices 4 and 5) from one monomer and a hydrophobic patch (amino acids 102, 128–132) that is primarily comprised of alpha helices 1 and 3 in the second integrase monomer.61 Ideally, one could thus look for small molecules that enter this binding pocket in integrase, thereby preventing interaction with the cofactor.
 | ||
| Fig. 5 Structure of the HIV-1integraseCCD–IBD interface. (A) An integraseCCD dimer interacting with the LEDGF/p75IBD using Protein Data Bank crystal structure file 2BJ4. (B) Close-up view of the interface. The integraseCCD monomers are coloured purple and green and the IBD subunit is orange. The selected residues are shown as sticks, and hydrogen bonds are indicated by dotted lines. Integrase residues A128 and W131 are part of a hydrophobic patch which accommodates the side chains of the LEDGF/p75 residues I365, F406 and V408. Integrase residues 166–171 are part of the so-called α4/5 connector linking helices α4 and α5 of the second integrase monomer. LEDGF/p75 residue D366 forms hydrogen bonds with the backbone amides of integrase E170 and H171. | ||
| Integrase residue | Substitution | Ref. | Enzymatic activitya | LEDGF/p75 bindingb | HIV-1 replicationc | Main contact residuesd | ||
|---|---|---|---|---|---|---|---|---|
| 3′ processing | Strand transfer | LEDGF/p75 | Integrase | |||||
| a The enzymatic activity (including 3′-processing and strand transfer) of the different integrase mutants was compared to that of WTHIV-1integrase (denoted with +++). b Interaction profile of different integrase mutants with LEDGF/p75, compared to WTintegrase (denoted with +++). c Infectivity of mutant HIV strains was compared to WTHIV-1 replication (+++). Single-round luciferase-reporter virus was used in reference 52, NL4.3-based viruses were used in references 53 and 51, whereas reference 62 used HIV-1 Bru WT or mutant viruses. d Critical integrase-interacting residues with the IBD or integrase itself are depicted, based on the available co-crystal data.61 e na, data not applicable or not available. f The grey boxes highlight the integrase amino acids most important for interaction with LEDGF/p75 (see Fig. 4 and 5). | ||||||||
| WT | — | — | +++ | +++ | +++ | +++ | — | — |
| H12 | N | 63 | na e | na | − | na | — | — |
| E69 | R | 52 | +++ | na | + | − | — | — |
 f
f
|
T | 53 | + | + | ++ | ++ | I365, F406, V408 | — |
| Q | 52 | +++ | na | ++ | +++ | — | — | |
| A129 | Q | 52 | + | na | + | − | — | — |
| C130 | W | 62 | na | na | ++ | na | — | — |
| A | 51 | na | na | +++ | na | — | — | |

|
A | 51 | +++ | +++ | + | + | R405, F406, V408 | A128, W132 |
| D | 52 | +++ | na | ++ | + | — | — | |
| W132 | A | 52 | − | − | − | − | I365 | — |
| A | 51 | − | − | na | na | — | — | |
| I161 | A | 51 | +++ | +++ | + | na | — | — |
| Q164 | A | 51 | na | na | ++ | na | — | — |
| V165 | A | 52, 45 | ++ | na | − | − | — | — |
| A | 51 | + | + | + | na | — | — | |

|
A | 52 | +++ | na | − | − | — | E69 |
| A | 51 | ++++ | ++++ | − | na | — | — | |
| D167 | A | 52 | +++ | na | ++ | +++ | — | — |
| A | 51 | na | na | + | na | — | — | |
| K | 52 | +++ | na | ++ | − | — | — | |

|
A | 62, 51 | +++ | +++ | + | − | — | W132, M178 |
| A | 52 | +++ | na | ++ | − | — | — | |
| L | 62 | + | + | − | − | — | — | |

|
A | 51 | ++++ | ++++ | + | na | K364, D366 | — |
| G | 53 | ++ | ++ | + | + | — | — | |
| H171 | A | 52 | +++ | na | + | +++ | D366 | — |
| L172/K173 | A | 52, 45 | +++ | na | − | na | — | — |
Importance of LEDGF/p75 for HIV replication
After initial identification and validation of the interaction between LEDGF/p75 and integrase,63,64 a crucial role for LEDGF/p75 in HIV replication was evidenced through different mutagenesis, RNAi, transdominant and knock-out (KO) studies.51–52,62,65–68 Moreover, the structure of LEDGF/p75 with the IBD on the one end, the chromatin binding domain on the other end and the fact that overexpressed integrase is tethered to the chromatin by LEDGF/p75 all led to the hypothesis that LEDGF/p75 captures and guides incoming viral particles to the chromatin.57–58,62,63 This so-called “tethering” function has been corroborated by analysis of integration sites in human cells depleted for LEDGF/p75 or in embryonic fibroblasts derived from LEDGF/p75 KO mice, demonstrating a role for LEDGF/p75 during target site selection.68–70A separate approach to validate LEDGF/p75 as an important cofactor was undertaken by stably overexpressing the IBD, thereby blocking HIV-1 replication at the step of integration.67,71 Overexpression of the IBD apparently competes with endogenous LEDGF/p75 for binding to integrase. This result provides proof-of-concept for LEDGF/p75-integrase interaction as a novel target for antiviral therapy. Furthermore, by repeatedly passaging HIV in the cell lines overexpressing IBD, strains that could overcome the inhibition were selected.53 Although resistance developed, the resistant virus was severely crippled in its replication capacity. Analysis of the integrasegene revealed two amino acid mutations: A128T and E170G, located at the described interface between the integraseCCD dimer and the IBD. Although LEDGF/p75 could still interact with the mutant integrase, this occurred with a much lower affinity. Moreover, replication of the IBD-resistant virus was 10-fold more sensitive to depletion of LEDGF/p75 than WT virus, indicating that the resistant virus remained dependent on LEDGF/p75 for its replication. This study suggests an exclusive role of LEDGF/p75 during HIV integration.
Toxicity
When targeting a cellular protein, one needs to be concerned about cellular toxicity. So far, in all experiments that have used RNAi to knockdown cellular LEDGF/p75, the knockdown has not been associated with apparant toxicity in human cell lines. LEDGF appears to be important during embryonic development, since the majority of homozygous LEDGF KO mice die perinatally and the ones that survive display a range of abnormal phenotypes, compatible with defects in homeodomain proteins.72 However, both p75 and p52 splice variants are depleted in the KO mice, whereas an HIV therapeutic strategy would only target p75. In fact, HIV drug discovery aims at inhibiting protein–protein interaction with HIV integrase without affecting the cellular function of LEDGF/p75.Probably LEDGF/p75 plays a pleiotropic role in both cell survival and apoptosis-mediated cell death. On the one hand it can transcriptionally activate anti-apoptotic proteins in response to different kinds of stresses.73 On the other hand LEDGF/p75 is cleaved by main effector caspases in apoptosis, resulting in two cleavage fragments, which upon overexpression abrogate the pro-survival role of LEDGF/p75.74 In any case, future lead compounds will have to be carefully evaluated for teratogenicity and toxicity. Valuable information in this respect will come from cell biology studies on LEDGF/p75. An ongoing effort attempts to identify the cellular binding partners. Two groups independently identified JPO2 as a first cellular binding partner of the IBD.75,76 Competition assays showed a mutually exclusive binding of either JPO2 or HIV-1integrase to LEDGF/p75. However, while the binding regions overlap, differential interaction was proposed since JPO2 still interacts with LEDGF/p75 mutants that are totally defective for interaction with HIV-1integrase. The finding of differential interaction between integrase, LEDGF/75 and JPO2 suggests the feasibility of developing inhibitors specifically targeting the interaction between LEDGF/p75 and HIV-1integrase.
Proof-of-concept for the identification of genuine integrase-LEDGF/p75inhibitors
Finally, identification of the first small molecules that interfere with integrase-LEDGF/p75 interaction and block HIV replication will provide an enormous impetus to this field. In fact, prior to the identification of LEDGF/p75 as an integrase binding partner, evidence was obtained for the existence of a pocket in integrase that could accommodate a small molecule inhibitor.77 Molteni et al. used X-ray crystallography to identify compounds that bind at the dimer interface of the HIV-1integrasecatalytic domain, in a small cleft of about 5 Å deep. The two small molecules, tetraphenylarsonium and dihydroxyphenyltriphenylarsonium, are arsenic derivatives surrounded with four aromatic groups (Fig. 6). These compounds were defined as integraseinhibitors, although only the second compound weakly inhibited in vitro enzymatic activity. Nevertheless, the small molecule binding site from this study overlaps with the binding cleft for the IBD of LEDGF/p75. The tetraphenylarsonium showed a strong charge–charge interaction with the carbonyl O atom of Gln168 (subunit 1 of the integrase dimer), while Trp131 and Trp132 (subunit 2 of the integrase dimer) interacted with one of the phenyl rings of the compound. Using photoaffinity labeling and mass spectrometry, Al-Mawsawi et al. identified the same binding site in integrase in 2006.78 Their coumarin derivatives with a benzophenone moiety bound to the peptide backbone in the 128AACWWAGIK136 region of integrase (Fig. 6). Substitution of the Cys130 and Trp132 amino acids resulted in a marked resistance to the integraseinhibitors. Docking studies suggested a specific disruption of functional oligomeric integrase complex formation. The coumarin derivatives display in vitrointegrase inhibitory activity with IC50 values ranging from 7 to 20 μM. This study suggests that structure-based drug design could lead to inhibitors with a dual mode of action: disruption of HIV-1 integrase-LEDGF cofactor interaction and disruption of integrase multimer formation. In a more recent report from the same group it was shown that an LEDGF/p75 derived peptide (amino acid 355–377) which contains two integrase interacting residues (I365 and D366), could disrupt the integrase-LEDGF/p75 interaction with an IC50 value of 25 μM.79 The peptide furthermore inhibits in vitrointegrase enzymatic activity with IC50 values of 165 and 153 μM for WT 3′-processing and strand transfer, respectively. There are no data available yet on antiviral activity and toxicity in cell culture. | ||
| Fig. 6 Structure of small molecules binding in a pocket in HIV-1integrase which overlaps with the LEDGF/p75 binding cleft. | ||
Future perspectives: potential new targets for development of SMPPIIs targeting HIV infection
The insight that the cellular environment is of crucial importance for (retro)viral replication has opened a completely new field in virology. Novel methodologies to identify and validate new cofactors have been introduced, resulting in an exponential increase in the number of identified cellular co- and restriction factors.30,80–82 Proteomic-based approaches identify cellular proteins physically interacting with viral proteins and include coimmunoprecipitation and analysis of virion-associated proteins by mass spectrometry. Genomic screening techniques like yeast two-hybrid can also be used to identify direct interaction partners. Recently, however, functional genomic screens have emerged as a valuable tool for high-throughput identification of individual players in a pathway of interest.62,83–86 Using a large-scale siRNA screen, Brass et al. were able to identify more than 250 HIV-dependent factors.80 Further analysis will be required to validate these cofactors and evaluate their potential as future anti-HIV drug targets.Several protein–protein interactions involved in different stages of the HIV-1 replication cycle are currently investigated for their therapeutic potential. APOBEC3G (apolipoproteinB mRNA-editing enzymecatalyticpolypeptide-like 3G) was initially identified as an HIV restriction factor in target CD4 T cells.87 During reverse transcription, APOBEC carried within the incoming HIV particle, deaminates dC to dU in the nascent minus-strand viral DNA, resulting in G-to-A hypermutation in the plus strand DNA, and degradation of the viral DNA.88,89 The HIV-1 Vif protein counteracts this enzyme in producer cells by degradationvia the ubiquitin-proteasome pathway.90 Interference with the Vif-APOBEC3G interaction, raising intracellular or virion associated levels of APOBEC3G, or reducing intracellular levels of Vif, all hold promise as potential therapeutic approaches aimed at enhancing the cells innate antiviral activity. Still, incomplete suppression of Vif-APOBEC3G interaction holds the theoretical risk of inducing a mutator phenotype facilitating resistance development against other drugs present in the inevitable cocktails. Cellular screens are ongoing to identify small molecules that block Vif-APOBEC3G interaction and antagonize its proteolytic degradation.
A process during which perhaps the most cellular HIV cofactors are involved is virus assembly, budding and release of viral particles (recently reviewed by Goff in ref. 81). Potentially interesting for drug discovery might be CD137 (a membraneprotein called tetherin),91 tail-interacting protein of 47 kDa (TIP47)92 and Tsg101.93,94 The release and spread of retroviruses is inhibited in certain cell types by hostproteins called tetherins. Tetherins are counteracted by the action of the HIV-1 Vpu protein.91 Although no direct protein–protein interaction between Vpu and tetherin has been shown, better understanding of the mechanism of tetherin and the way by which Vpu circumvents tetherin, could lead in time to strategies to limit the spread of HIV-1. The hostprotein TIP47 is involved in virion assembly. The protein not only interacts with viral Env, but also binds to the matrix domain of HIV-1 Gag, resulting in a ternary complex formation.92,95 TIP47 is a cofactor that plays an essential role in Env incorporation, allowing HIV-1 Gag and Env proteins to encounter each other and physically associate during the viral assembly process and could therefore be considered as a potential target for drug discovery.92 Retrovirus budding also requires host machinery consisting of endosomal complexes named ESCRTs (endosomal sorting complex required for transport). In infected cells, Gag redirects the ESCRT machinery to the plasma membrane for budding out of the cell. This process involves the contact of viral Gag with many of the ESCORTs, for example interaction with Tsg101 through the Gag PTAP motif.96,97 It was furthermore demonstrated that this requirement for Gag-Tsg101 interaction is necessary for HIV-1 release.93,94 Although blocking of essential interactions between Gag and hostproteins constitutes an innovative strategy for impairing the production of infectious viruses and the spreading of HIV-1, drug development is jeopardized by toxicity. As sorting and trafficking of viral proteins is undistinguishable of normal cellular processes, it might be difficult to find a non-toxic inhibitor.
A different therapeutic strategy is represented by HIV integrase and reverse transcriptasedimerizationinhibitors.77–78,98,99 Although these protein interfaces are a promising alternative target for HIV drug discovery and could validate the potential of SMPPIIs, resistance against the molecules and/or cross-resistance against existing integrase and reverse transcriptase inhibitors will most likely develop rapidly, since only viral proteins are targeted.
Based on the lessons learnt from the studies on the integrase-LEDGF/p75 interaction, we would like to put forward the following requirements for the validation of a new PPI as an anti-HIV target: (i) the cellular protein functions as a cofactor rather than a restriction factor; (ii) the cofactor is unique, no alternative pathways are exploited by HIV; (iii) structural information on the interface is available; (iv) the role of the cofactor during HIV replication is different than its role in normal cellmetabolism; (v) the viral part of the interface is genetically conserved.
Summary
The development of a small molecule that specifically disrupts a particular protein–protein interface is a challenging endeavour. New insights point to protein–protein interfaces that might be potential targets for drug discovery. This is of particular interest for the field of virology that demands new antivirals preferably with a completely different mechanism of action to avoid resistance and cross-resistance. Although antiviral drugs typically target viral proteins, the expanding knowledge on interactions between viral and cellular proteins could open a new field of antiviral research targeting cellular cofactors. The interaction between the viral HIV-1integrase and its cellular cofactorLEDGF/p75 is one of the most promising targets at the moment. The interaction is important for virus replication. Furthermore, a cocrystal revealed a binding pocket which can accommodate a small molecule inhibitor. Development of an effective SMPPII as a new antiretroviral drug would considerably boost generic interest in protein–protein interaction inhibitors.Acknowledgements
We apologize to those authors whose papers were not cited. We are financially supported by the European commission (HEALTH-2007-2.3.2-1). KB is supported by a grant from the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT). FC is fellow of the IOF (Industrieel onderzoeksfonds, industrial oriented research).References
- D. P. Ryan and J. M. Matthews, Curr. Opin. Struct. Biol., 2005, 15, 441–446 CrossRef CAS.
- M. R. Arkin and J. A. Wells, Nat. Rev. Drug Discovery, 2004, 3, 301–317 CrossRef CAS.
- T. Logtenberg, Trends Biotechnol., 2007, 25, 390–394 CrossRef CAS.
- L. Zhao and J. Chmielewski, Curr. Opin. Struct. Biol., 2005, 15, 31–34 CrossRef CAS.
- J. M. Kilby, S. Hopkins, T. M. Venetta, B. DiMassimo, G. A. Cloud, J. Y. Lee, L. Alldredge, E. Hunter, D. Lambert, D. Bolognesi, T. Matthews, M. R. Johnson, M. A. Nowak, G. M. Shaw and M. S. Saag, Nat. Med., 1998, 4, 1302–1307 CrossRef CAS.
- A. I. Archakov, V. M. Govorun, A. V. Dubanov, Y. D. Ivanov, A. V. Veselovsky, P. Lewi and P. Janssen, Proteomics, 2003, 3, 380–391 CrossRef CAS.
- S. Jones and J. M. Thornton, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13–20 CrossRef CAS.
- W. L. DeLano, Curr. Opin. Struct. Biol., 2002, 12, 14–20 CrossRef CAS.
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef CAS.
- B. L. Grasberger, T. Lu, C. Schubert, D. J. Parks, T. E. Carver, H. K. Koblish, M. D. Cummings, L. V. LaFrance, K. L. Milkiewicz, R. R. Calvo, D. Maguire, J. Lattanze, C. F. Franks, S. Zhao, K. Ramachandren, G. R. Bylebyl, M. Zhang, C. L. Manthey, E. C. Petrella, M. W. Pantoliano, I. C. Deckman, J. C. Spurlino, A. C. Maroney, B. E. Tomczuk, C. J. Molloy and R. F. Bone, J. Med. Chem., 2005, 48, 909–912 CrossRef CAS.
- C. Yoakim, W. W. Ogilvie, N. Goudreau, J. Naud, B. Hache, J. A. O’Meara, M. G. Cordingley, J. Archambault and P. W. White, Bioorg. Med. Chem. Lett., 2003, 13, 2539–2541 CrossRef CAS.
- L. Pagliaro, J. Felding, K. Audouze, S. J. Nielsen, R. B. Terry, C. Krog-Jensen and S. Butcher, Curr. Opin. Chem. Biol., 2004, 8, 442–449 CrossRef CAS.
- D. A. Erlanson, R. S. McDowell and T. O’Brien, J. Med. Chem., 2004, 47, 3463–3482 CrossRef CAS.
- A. C. Braisted, J. D. Oslob, W. L. Delano, J. Hyde, R. S. McDowell, N. Waal, C. Yu, M. R. Arkin and B. C. Raimundo, J. Am. Chem. Soc., 2003, 125, 3714–3715 CrossRef CAS.
- L. T. Vassilev, Trends Mol. Med., 2007, 13, 23–31 CrossRef CAS.
- P. Chene, J. Fuchs, J. Bohn, C. Garcia-Echeverria, P. Furet and D. Fabbro, J. Mol. Biol., 2000, 299, 245–253 CrossRef CAS.
- S. W. Chi, S. H. Lee, D. H. Kim, M. J. Ahn, J. S. Kim, J. Y. Woo, T. Torizawa, M. Kainosho and K. H. Han, J. Biol. Chem., 2005, 280, 38795–38802 CrossRef CAS.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef CAS.
- D. J. Parks, L. V. Lafrance, R. R. Calvo, K. L. Milkiewicz, V. Gupta, J. Lattanze, K. Ramachandren, T. E. Carver, E. C. Petrella, M. D. Cummings, D. Maguire, B. L. Grasberger and T. Lu, Bioorg. Med. Chem. Lett., 2005, 15, 765–770 CrossRef CAS.
- H. K. Koblish, S. Zhao, C. F. Franks, R. R. Donatelli, R. M. Tominovich, L. V. LaFrance, K. A. Leonard, J. M. Gushue, D. J. Parks, R. R. Calvo, K. L. Milkiewicz, J. J. Marugan, P. Raboisson, M. D. Cummings, B. L. Grasberger, D. L. Johnson, T. Lu, C. J. Molloy and A. C. Maroney, Mol. Cancer Ther., 2006, 5, 160–169 CrossRef CAS.
- S. Shangary, D. Qin, D. McEachern, M. Liu, R. S. Miller, S. Qiu, Z. Nikolovska-Coleska, K. Ding, G. Wang, J. Chen, D. Bernard, J. Zhang, Y. Lu, Q. Gu, R. B. Shah, K. J. Pienta, X. Ling, S. Kang, M. Guo, Y. Sun, D. Yang and S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3933–3938 CrossRef CAS.
- B. H. Nelson and D. M. Willerford, Adv. Immunol., 1998, 70, 1–81 Search PubMed.
- M. R. Arkin, M. Randal, W. L. DeLano, J. Hyde, T. N. Luong, J. D. Oslob, D. R. Raphael, L. Taylor, J. Wang, R. S. McDowell, J. A. Wells and A. C. Braisted, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1603–1608 CrossRef CAS.
- J. W. Tilley, L. Chen, D. C. Fry, S. D. Emerson, G. D. Powers, D. Biondi, T. Varnell, R. Trilles, R. Guthrie, F. Mennona, G. Kaplan, R. A. LeMahieu, M. Carson, R. J. Han, C. M. Liu, R. Palermo and G. Ju, J. Am. Chem. Soc., 1997, 119, 7589–7590 CrossRef CAS.
- N. D. Waal, W. Yang, J. D. Oslob, M. R. Arkin, J. Hyde, W. Lu, R. S. McDowell, C. H. Yu and B. C. Raimundo, Bioorg. Med. Chem. Lett., 2005, 15, 983–987 CrossRef CAS.
- C. D. Thanos, W. L. DeLano and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15422–15427 CrossRef CAS.
- T. Oltersdorf, S. W. Elmore, A. R. Shoemaker, R. C. Armstrong, D. J. Augeri, B. A. Belli, M. Bruncko, T. L. Deckwerth, J. Dinges, P. J. Hajduk, M. K. Joseph, S. Kitada, S. J. Korsmeyer, A. R. Kunzer, A. Letai, C. Li, M. J. Mitten, D. G. Nettesheim, S. Ng, P. M. Nimmer, J. M. O’Connor, A. Oleksijew, A. M. Petros, J. C. Reed, W. Shen, S. K. Tahir, C. B. Thompson, K. J. Tomaselli, B. Wang, M. D. Wendt, H. Zhang, S. W. Fesik and S. H. Rosenberg, Nature, 2005, 435, 677–681 CrossRef.
- A. R. Shoemaker, A. Oleksijew, J. Bauch, B. A. Belli, T. Borre, M. Bruncko, T. Deckwirth, D. J. Frost, K. Jarvis, M. K. Joseph, K. Marsh, W. McClellan, H. Nellans, S. Ng, P. Nimmer, J. M. O'Connor, T. Oltersdorf, W. Qing, W. Shen, J. Stavropoulos, S. K. Tahir, B. Wang, R. Warner, H. Zhang, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, Cancer Res., 2006, 66, 8731–8739 CrossRef CAS.
- B. Van Maele, K. Busschots, L. Vandekerckhove, F. Christ and Z. Debyser, Trends Biochem. Sci., 2006, 31, 98–105 CrossRef CAS.
- P. K. Vogt, in Retroviruses, ed. J. M. Coffin, S. H. Hughes and H. E. Varmus, Cold Spring Harbor Laboratory Press, New York, 1997, pp. 1–25 Search PubMed.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS.
- T. H. Evering and M. Markowitz, Expert Opin. Investig. Drugs, 2008, 17, 413–422 Search PubMed.
- P. Dorr, M. Westby, S. Dobbs, P. Griffin, B. Irvine, M. Macartney, J. Mori, G. Rickett, C. Smith-Burchnell, C. Napier, R. Webster, D. Armour, D. Price, B. Stammen, A. Wood and M. Perros, Antimicrob. Agents Chemother., 2005, 49, 4721–4732 CrossRef CAS.
- E. K. Franke and J. Luban, Virology, 1996, 222, 279–282 CrossRef CAS.
- D. N. Streblow, M. Kitabwalla, M. Malkovsky and C. D. Pauza, Virology, 1998, 245, 197–202 CrossRef CAS.
- S. R. Bartz, E. Hohenwalter, M. K. Hu, D. H. Rich and M. Malkovsky, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5381–5385 CrossRef CAS.
- K. Watashi, N. Ishii, M. Hijikata, D. Inoue, T. Murata, Y. Miyanari and K. Shimotohno, Mol. Cell, 2005, 19, 111–122 CrossRef CAS.
- S. Ma, J. E. Boerner, C. TiongYip, B. Weidmann, N. S. Ryder, M. P. Cooreman and K. Lin, Antimicrob. Agents Chemother., 2006, 50, 2976–2982 CrossRef CAS.
- R. Flisiak, J. M. Dumont and R. Crabbe, Expert Opin. Investig. Drugs, 2007, 16, 1345–1354 Search PubMed.
- R. Flisiak, A. Horban, P. Gallay, M. Bobardt, S. Selvarajah, A. Wiercinska-Drapalo, E. Siwak, I. Cielniak, J. Higersberger, J. Kierkus, C. Aeschlimann, P. Grosgurin, V. Nicolas-Metral, J. M. Dumont, H. Porchet, R. Crabbe and P. Scalfaro, Hepatology, 2008, 47, 817–826 CrossRef CAS.
- P. W. White, S. Titolo, K. Brault, L. Thauvette, A. Pelletier, E. Welchner, L. Bourgon, L. Doyon, W. W. Ogilvie, C. Yoakim, M. G. Cordingley and J. Archambault, J. Biol. Chem., 2003, 278, 26765–26772 CrossRef CAS.
- Y. Wang, R. Coulombe, D. R. Cameron, L. Thauvette, M. J. Massariol, L. M. Amon, D. Fink, S. Titolo, E. Welchner, C. Yoakim, J. Archambault and P. W. White, J. Biol. Chem., 2004, 279, 6976–6985 CAS.
- P. Cherepanov, E. Devroe, P. A. Silver and A. Engelman, J. Biol. Chem., 2004, 279, 48883–48892 CrossRef CAS.
- P. Cherepanov, Z. Y. Sun, S. Rahman, G. Maertens, G. Wagner and A. Engelman, Nat. Struct. Mol. Biol., 2005, 12, 526–532 CrossRef CAS.
- E. Asante-Appiah and A. M. Skalka, Adv. Virus Res., 1999, 52, 351–369 CAS.
- A. Engelman and R. Craigie, J. Virol., 1992, 66, 6361–6369 CAS.
- A. Engelman, F. D. Bushman and R. Craigie, EMBO J., 1993, 12, 3269–3275 CAS.
- F. D. Bushman and B. Wang, J. Virol., 1994, 68, 2215–2223 CAS.
- T. M. Jenkins, A. Engelman, R. Ghirlando and R. Craigie, J. Biol. Chem., 1996, 271, 7712–7718 CrossRef CAS.
- K. Busschots, A. Voet, M. De Maeyer, J. C. Rain, S. Emiliani, R. Benarous, L. Desender, Z. Debyser and F. Christ, J. Mol. Biol., 2007, 365, 1480–1492 CrossRef CAS.
- S. Rahman, R. Lu, N. Vandegraaff, P. Cherepanov and A. Engelman, Virology, 2007, 357, 79–90 CrossRef CAS.
- A. Hombrouck, J. De Rijck, J. Hendrix, L. Vandekerckhove, A. Voet, M. De Maeyer, M. Witvrouw, Y. Engelborghs, F. Christ, R. Gijsbers and Z. Debyser, PLoS Pathog., 2007, 3, e47 Search PubMed.
- M. Llano, M. Vanegas, N. Hutchins, D. Thompson, S. Delgado and E. M. Poeschla, J. Mol. Biol., 2006, 360, 760–773 CrossRef CAS.
- Y. Botbol, N. K. Raghavendra, S. Rahman, A. Engelman and M. Lavigne, Nucleic Acids Res., 2008, 36, 1237–1246 CAS.
- F. Turlure, G. Maertens, S. Rahman, P. Cherepanov and A. Engelman, Nucleic Acids Res., 2006, 34, 1663–1675.
- G. Maertens, P. Cherepanov, Z. Debyser, Y. Engelborghs and A. Engelman, J. Biol. Chem., 2004, 279, 33421–33429 CrossRef CAS.
- M. Vanegas, M. Llano, S. Delgado, D. Thompson, M. Peretz and E. Poeschla, J. Cell Sci., 2005, 118, 1733–1743 CrossRef CAS.
- D. P. Singh, A. Kimura, L. T. Chylack, Jr., T. Shinohara, N. Ohguro, T. Kikuchi, T. Sueno, V. N. Reddy, K. Yuge and N. Fatma, Gene, 2000, 242, 265–273 CrossRef CAS.
- H. Ge, Y. Si and R. G. Roeder, EMBO J., 1998, 17, 6723–6729 CrossRef CAS.
- P. Cherepanov, A. L. Ambrosio, S. Rahman, T. Ellenberger and A. Engelman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17308–17313 CrossRef CAS.
- S. Emiliani, A. Mousnier, K. Busschots, M. Maroun, B. Van Maele, D. Tempe, L. Vandekerckhove, F. Moisant, L. Ben-Slama, M. Witvrouw, F. Christ, J. C. Rain, C. Dargemont, Z. Debyser and R. Benarous, J. Biol. Chem., 2005, 280, 25517–25523 CrossRef CAS.
- G. Maertens, P. Cherepanov, W. Pluymers, K. Busschots, E. De Clercq, Z. Debyser and Y. Engelborghs, J. Biol. Chem., 2003, 278, 33528–33539 CrossRef CAS.
- M. Llano, M. Vanegas, O. Fregoso, D. Saenz, S. Chung, M. Peretz and E. M. Poeschla, J. Virol., 2004, 78, 9524–9537 CrossRef CAS.
- M. C. Shun, J. E. Daigle, N. Vandegraaff and A. Engelman, J. Virol., 2007, 81, 166–172 CAS.
- L. Vandekerckhove, F. Christ, B. Van Maele, J. De Rijck, R. Gijsbers, C. Van den Haute, M. Witvrouw and Z. Debyser, J. Virol., 2006, 80, 1886–1896 CrossRef CAS.
- M. Llano, D. T. Saenz, A. Meehan, P. Wongthida, M. Peretz, W. H. Walker, W. Teo and E. M. Poeschla, Science, 2006, 314, 461–464 CrossRef CAS.
- H. M. Marshall, K. Ronen, C. Berry, M. Llano, H. Sutherland, D. Saenz, W. Bickmore, E. Poeschla and F. D. Bushman, PLoS ONE, 2007, 2, e1340 CrossRef.
- A. Ciuffi, M. Llano, E. Poeschla, C. Hoffmann, J. Leipzig, P. Shinn, J. R. Ecker and F. Bushman, Nat. Med., 2005, 11, 1287–1289 CrossRef CAS.
- M. C. Shun, N. K. Raghavendra, N. Vandegraaff, J. E. Daigle, S. Hughes, P. Kellam, P. Cherepanov and A. Engelman, Genes Dev., 2007, 21, 1767–1778 CrossRef CAS.
- J. De Rijck, L. Vandekerckhove, R. Gijsbers, A. Hombrouck, J. Hendrix, J. Vercammen, Y. Engelborghs, F. Christ and Z. Debyser, J. Virol., 2006, 80, 11498–11509 CrossRef CAS.
- H. G. Sutherland, K. Newton, D. G. Brownstein, M. C. Holmes, C. Kress, C. A. Semple and W. A. Bickmore, Mol. Cell. Biol., 2006, 26, 7201–7210 CrossRef CAS.
- T. Shinohara, D. P. Singh and N. Fatma, Prog. Retinal Eye Res., 2002, 21, 341–358 Search PubMed.
- X. Wu, T. Daniels, C. Molinaro, M. B. Lilly and C. A. Casiano, Cell Death Differ., 2002, 9, 915–925 CrossRef CAS.
- K. Bartholomeeusen, J. De Rijck, K. Busschots, L. Desender, R. Gijsbers, S. Emiliani, R. Benarous, Z. Debyser and F. Christ, J. Mol. Biol., 2007, 372, 407–421 CrossRef CAS.
- G. N. Maertens, P. Cherepanov and A. Engelman, J. Cell Sci., 2006, 119, 2563–2571 CrossRef CAS.
- V. Molteni, J. Greenwald, D. Rhodes, Y. Hwang, W. Kwiatkowski, F. D. Bushman, J. S. Siegel and S. Choe, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 536–544 CrossRef CAS.
- L. Q. Al-Mawsawi, V. Fikkert, R. Dayam, M. Witvrouw, T. R. Burke, Jr., C. H. Borchers and N. Neamati, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10080–10085 CrossRef CAS.
- L. Q. Al-Mawsawi, F. Christ, R. Dayam, Z. Debyser and N. Neamati, FEBS Lett., 2008, 582, 1425–1430 CrossRef CAS.
- A. L. Brass, D. M. Dykxhoorn, Y. Benita, N. Yan, A. Engelman, R. J. Xavier, J. Lieberman and S. J. Elledge, Science, 2008, 319, 921–926 CrossRef CAS.
- S. P. Goff, Nat. Rev. Microbiol., 2007, 5, 253–263 Search PubMed.
- L. Q. Al-Mawsawi and N. Neamati, Trends Pharmacol. Sci., 2007, 28, 526–535 CrossRef CAS.
- S. Hamamoto, H. Nishitsuji, T. Amagasa, M. Kannagi and T. Masuda, J. Virol., 2006, 80, 5670–5677 CrossRef CAS.
- D. G. Nguyen, K. C. Wolff, H. Yin, J. S. Caldwell and K. L. Kuhen, J. Virol., 2006, 80, 130–137 CrossRef CAS.
- D. G. Nguyen, H. Yin, Y. Zhou, K. C. Wolff, K. L. Kuhen and J. S. Caldwell, Virology, 2007, 362, 16–25 CrossRef CAS.
- G. V. Kalpana, S. Marmon, W. Wang, G. R. Crabtree and S. P. Goff, Science, 1994, 266, 2002–2006 CrossRef CAS.
- A. M. Sheehy, N. C. Gaddis, J. D. Choi and M. H. Malim, Nature, 2002, 418, 646–650 CrossRef.
- D. Lecossier, F. Bouchonnet, F. Clavel and A. J. Hance, Science, 2003, 300, 1112 CrossRef CAS.
- B. Mangeat, P. Turelli, G. Caron, M. Friedli, L. Perrin and D. Trono, Nature, 2003, 424, 99–103 CrossRef.
- A. M. Sheehy, N. C. Gaddis and M. H. Malim, Nat. Med., 2003, 9, 1404–1407 CrossRef CAS.
- S. J. Neil, T. Zang and P. D. Bieniasz, Nature, 2008, 451, 425–430 CrossRef CAS.
- S. Lopez-Verges, G. Camus, G. Blot, R. Beauvoir, R. Benarous and C. Berlioz-Torrent, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14947–14952 CrossRef CAS.
- J. Martin-Serrano, T. Zang and P. D. Bieniasz, Nat. Med., 2001, 7, 1313–1319 CrossRef CAS.
- D. G. Demirov, A. Ono, J. M. Orenstein and E. O. Freed, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 955–960 CrossRef CAS.
- G. Blot, K. Janvier, S. Le Panse, R. Benarous and C. Berlioz-Torrent, J. Virol., 2003, 77, 6931–6945 CrossRef CAS.
- L. VerPlank, F. Bouamr, T. J. LaGrassa, B. Agresta, A. Kikonyogo, J. Leis and C. A. Carter, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7724–7729 CrossRef CAS.
- J. E. Garrus, U. K. von Schwedler, O. W. Pornillos, S. G. Morham, K. H. Zavitz, H. E. Wang, D. A. Wettstein, K. M. Stray, M. Cote, R. L. Rich, D. G. Myszka and W. I. Sundquist, Cell, 2001, 107, 55–65 CrossRef CAS.
- Z. Hayouka, J. Rosenbluh, A. Levin, S. Loya, M. Lebendiker, D. Veprintsev, M. Kotler, A. Hizi, A. Loyter and A. Friedler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8316–8321 CrossRef CAS.
- D. Grohmann, V. Corradi, M. Elbasyouny, A. Baude, F. Horenkamp, S. D. Laufer, F. Manetti, M. Botta and T. Restle, Chembiochem, 2008, 9, 916–922 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |